
Abstract
Deficient activities of multiple steroidogenic enzymes have been reported without and with Antley-Bixler syndrome (ABS), but mutations of corresponding cytochrome P450 enzymes have not been found. We identified mutations in POR, encoding P450 oxidoreductase, the obligate electron donor for these enzymes, in a woman with amenorrhea and three children with ABS, even though knock-out of POR is embryonically lethal in mice. Mutations of POR also affect drug-metabolizing P450 enzymes, explaining the association of ABS with maternal fluconazole ingestion.
Similar content being viewed by others

Main
Steroid hormones are required for reproduction, development and stress responses. Defects in each steroidogenic enzyme are well described, but reports of individuals with defects in both P450c17 (17α-hydroxylase/17,20 lyase) and P450c21 (21-hydroxylase), without1 and with2 associated Antley-Bixler syndrome (ABS; OMIM 207410), remain unexplained. Cholesterol biosynthetic P450 enzymes were implicated by reports that infants of mothers treated with fluconazole (an antifungal agent that interferes with yeast ergosterol synthesis by inhibiting lanosterol 14α-demethylase) had a pattern of congenital malformations that resembled ABS3,4. Mutations in human genes CYP17A1 (encoding P450c17; located at 10q24.3), CYP21A2 (encoding P450c21; located at 6p21.3) and CYP51A1 (encoding lanosterol 14α-demethylase; located at 7q21.2–q21.3) have been sought but not found2,4,5. We suggested that defects in P450c17 and P450c21 might be due to mutant POR, the flavoprotein that donates electrons to all microsomal P450 enzymes6, but the affected individuals lacked apparent disorders of bile acid synthesis or drug metabolism, which also require P450 enzymes, and knock-out of POR is embryonically lethal in mice7,8. Here we describe mutations in POR (located at 7q11.2) in four individuals with disordered steroidogenesis.
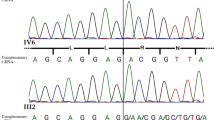
Individual 1 had a serum and urinary steroidal profile that suggested partially deficient activities of P450c17 and P450c21 (see Supplementary Note online). Her mother had become virilized during pregnancy, and this virilization had resolved after delivery, which suggested fetoplacental deficiency of steroid aromatase (microsomal P450aro), in which defective conversion of fetal adrenal C19 androgen precursors to estrogens leads to virilization of the mother and the 46,XX fetus. Decreased P450aro activity may be mistaken for spontaneously resolving luteomas of pregnancy, which have been reported in mothers of ABS infants9. Therefore, we sequenced POR, the product of which provides electrons to all three of these microsomal P450 enzymes. Individual 1 carried the missense mutation 1370G→A (leading to the amino acid substitution R457H) on the maternal allele and 731+1G→A in the first base of the splice donor site of intron 6 on the paternal allele; the corresponding minigene construct retained intron 6, leading to a premature stop codon (Fig. 1 and Supplementary Methods online). Individuals 2 and 4 were initially thought to have partial deficiencies of P450c17 activity, but complete sequencing of all exons and intron-exon boundaries of CYP17A1 detected no mutations. Sequencing of POR showed that individual 2 was heterozygous with respect to the missense mutation 1475T→A (leading to the amino acid substitution V492E; also found on the maternal allele) but detected no mutations in the exons or first 50 bp of splice donor-acceptor sites on the paternal allele. Individual 3 had genital ambiguity, typical findings of ABS and abnormal sterol metabolism, but no steroidal data are available5; he was apparently homozygous with respect to the substitution 859G→C (leading to the amino acid substitution A287P), and his mother was heterozygous (paternal DNA was not available). Individual 4, a phenotypically normal woman with amenorrhea and disordered steroidogenesis (see Supplementary Note and Supplementary Table 1 online), was compound heterozygous with respect to 1706G→A and 1822G→T (leading to the amino acid substitutions C569Y and V608F, respectively; Fig. 1); parental DNA was not available. We confirmed the mutations causing the amino acid substitutions A287P, V492E, C569Y and V608F by cleaving PCR-amplified DNA with diagnostic restriction endonucleases (see Supplementary Methods online). Mutations in the IgIIIa and IgIIIc domains of FGFR2 have been associated with the ABS phenotype10, but sequencing of exons 8 and 10 of FGFR2 encoding these domains in individuals 1 and 2 (data not shown)and 3 (ref. 5) detected no mutations.

(a) Genetic analysis. Individual 1 carried nucleotide change 731+1G→A in the first nucleotide of intron 6 on the paternal allele, and the nucleotide change 1370G→A on the maternal allele, resulting in the amino acid substitution R457H (parental sequences not shown). Individual 2 was heterozygous with respect to the nucleotide change 1475T→A on the maternal allele, resulting in the amino acid substitution V492E, but the paternal mutation was not found. Individual 3 was apparently homozygous with respect to the nucleotide change 859G→C, resulting in the amino acid substitution A287P; his mother was heterozygous but paternal DNA was not available. Individual 4 carried the nucleotide substitutions 1706G→A on one allele, causing the amino acid substitution C569Y, and 1822G→T on the other allele, resulting in the amino acid substitution V608F. Mutations are described as recommended (http://www.genomic.unimelb.edu.au/mdi/mutnomen/disc.html), using GenBank sequence accession number NM_000941.1 as the reference sequence; this increases the amino acid residue numbers by three compared with assignments for rat POR11. Each mutation is assigned to a diagram of the POR protein, based on the crystal structure of rat POR11 (92% amino acid identity). A287P, R457H and V492E are in the FAD binding domain, and C569Y and V608F are in the NADPH binding domain. M, mother. (b,c) Assays of cytochrome c reduction by human POR expressed in bacteria. Fitted plots of cytochrome c (Cyt c) reduction using cytochrome c (b) and NADPH (c) as variable substrates. (d–f) Steroidogenic activity of W(B) yeast cotransfected with expression vectors for human P450c17 and for wild-type or mutant POR. (d,e) Lineweaver-Burk analyses of 17α-hydroxylase activity (d) and 17,20 lyase activity (e). No significant steroid conversion was detected for R457H and V492E, and so no kinetic data are drawn. The calculated Km and Vmax values are shown in Table 1. (b–e) Wild-type, open triangles; A287P, filled circles; R457H, filled triangles; V492E, open squares; C569Y, filled squares; V608F, open circles. Data are mean ± s.e.m. (f) Representative thin layer chromatograms. Upper panel: 17α-hydroxylase activity assayed as the conversion of [14C]progesterone (Prog) to 17OH progesterone (17OHP) and 16OH progesterone (16OHP). Lower panel: 17,20 lyase activity assessed as conversion of [3H]17OH pregnenolone (17OHPreg) to dehydroepiandrosterone (DHEA). The vector control, which contains P450c17 but no POR, lacks significant activity as the W(B) yeast lack the gene CPR1 encoding yeast POR.
To assess the activities of the POR mutants, we expressed each in Escherichia coli and measured the ability of bacterial membrane-bound proteins to receive electrons from NADPH and donate them to cytochrome c (see Supplementary Methods online). All five of the missense mutations markedly reduced both the capacity to oxidize NADPH and the catalytic efficiency (Vmax/Km) of the reduction of cytochrome c (Table 1). Crystallographic analysis of rat POR showed that the residues Ala287, Arg457 and Val492, mutant in individuals with ABS, lie in the FAD-binding domain, and that two of the corresponding rat residues, Arg454 and Val489, directly participated in FAD binding11. Residues Cys569 and Val608, mutant in the woman with primary amenorrhea, correspond to Cys566 and Val605 in the rat, which lie at the ends of β-sheet 18 and helix Q in the NADP+-binding domain11.
POR binds NADPH, accepts a pair of electrons through its FAD moiety, transfers them to its FMN moiety and then donates them to a P450 (ref. 11). The FMN domain is characterized by acidic residues11, whereas the redox-partner binding sites of P450 enzymes are characterized by basic residues12. Mutations in the redox-partner binding site of P450c17 decrease electron flow and catalysis13,14, suggesting that POR missense mutations might retain partial activity. Yeast strain W(B), generated by targeted disruption of the gene CPR1 encoding the yeast homolog of POR15, permits assessment of human P450c17 activities without background yeast activity (see Supplementary Methods online). The A287P, R457H and V492E mutants from the individuals with ABS disrupted both the 17α-hydroxylase and the 17,20 lyase activities of P450c17, greatly reducing activity, whereas the C569Y and V608F mutants retained substantial activity (Table 1). Thus, the electron-transfer properties of POR correlated well with the phenotypes of affected individuals: severe mutations were found in individuals with apparent ABS, and milder mutations were found in an individual with disordered steroidogenesis but normal skeletal development.
The basis of ABS is controversial. Mutations of various receptors for the fibroblast growth factor cause the phenotypically related Apert, Crouzon, Pfeiffer and Jackson-Weiss syndromes, all characterized by craniosynostosis and skeletal anomalies, and about half of individuals with apparent ABS also have mutations in FGFR2 (refs. 2,5,10). But disorders of steroidogenesis and genital development are not seen in the craniosynostosis syndromes associated with FGFR mutations, and the FGFR2 mutations and genital ambiguity in ABS seem to segregate2. Thus, we propose that severe mutations in POR without associated disorders of FGF receptors are sufficient to cause the ABS phenotype. The grossly dysmorphic embryonic lethal phenotype of POR-knockout mice, with neural tube, cardiac, eye and limb anomalies7,8, suggests that severe disorders of POR may be sufficient to account for the skeletal findings in some individuals with ABS. By contrast, milder mutations in POR may present only with mild disorders of steroid synthesis; as such disorders are common, mutations in POR may be more common than is suggested by the low incidence of ABS.
Because POR is required for the activity of all microsomal P450 enzymes, environmental toxins and drugs that are usually metabolized to benign products by hepatic P450 enzymes may achieve teratogenic concentrations in the absence of POR. Fluconazole, which acts on a fungal cytochrome P450 system, has already been implicated in this fashion3. The toxicity of morphogens like retinoids may also be enhanced by POR variants; decreased maternal retinoic acid exposure partially ameliorates the POR-knockout mouse phenotype8. POR mutations would account for the decreased lanosterol 14α-demethylase (CYP51) activity described in ABS5. The identification of more POR mutations and analysis of correlations between the resulting electron-transfer activity and the clinical phenotype will be of substantial interest.
Note: Supplementary information is available on the Nature Genetics website.
Accession codes
References
Peterson, R.E., Imperato-McGinley, J., Gautier, T. & Shackleton, C. N. Engl. J. Med. 313, 1182–1191 (1985).
Reardon, W. et al. J. Med. Genet. 37, 26–32 (2000).
Aleck, K.A. & Bartley, D.L. Am. J. Med. Genet. 72, 253–256 (1997).
Adachi, M., Tachibana, K., Asakura, Y., Suwa, S. & Nishimura, G. Endocr. J. 46, 285–292 (1999).
Kelley, R.I. et al. Am. J. Med. Genet. 110, 95–102 (2002).
Miller, W.L. N. Engl. J. Med. 314, 1321–1322 (1986).
Shen, A.L., O'Leary, K.A. & Kasper, C.B. J. Biol. Chem. 277, 6536–6541 (2002).
Otto, D.M. et al. Mol. Cell. Biol. 23, 6103–6116 (2003).
Roth, C., Hinney, B., Peter, M., Steinberger, D. & Lakomek, M. Eur. J. Pediatr. 159, 189–192 (2000).
Chun, K., Siegel-Bartelt, J., Chitayat, D., Phillips, J. & Ray, P.N. Am. J. Med. Genet. 77, 219–224 (1998).
Wang, M. et al. Proc. Natl. Acad. Sci. USA 94, 8411–8416 (1997).
Auchus, R.J. & Miller, W.L. Mol. Endocrinol. 13, 1169–1182 (1999).
Geller, D.H., Auchus, R.J., Mendonca, B.B. & Miller, W.L. Nat. Genet. 17, 201–205 (1997).
Geller, D.H., Auchus, R.J. & Miller, W.L. Mol. Endocrinol. 13, 167–175 (1999).
Auchus, R.J., Lee, T.C. & Miller, W.L. J. Biol. Chem. 273, 3158–3165 (1998).
Acknowledgements
This work was supported by grants from the US National Institutes of Health (W.L.M.), from the Swiss National Research Foundation for Medical-Biological Grants and the Foundation Eugenio Litta (C.E.F.) and from the Deutsche Forschungsgemeinschaft (W.A.).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Rights and permissions
About this article
Cite this article
Flück, C., Tajima, T., Pandey, A. et al. Mutant P450 oxidoreductase causes disordered steroidogenesis with and without Antley-Bixler syndrome. Nat Genet 36, 228–230 (2004). https://doi.org/10.1038/ng1300
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ng1300
This article is cited by
-
Congenital adrenal hyperplasia, disorders of sex development, and infertility in patients with POR gene pathogenic variants: a systematic review of the literature
Journal of Endocrinological Investigation (2022)
-
Successful live birth in a Chinese woman with P450 oxidoreductase deficiency through frozen-thawed embryo transfer: a case report with review of the literature
Journal of Ovarian Research (2021)
-
POR polymorphisms are associated with 21 hydroxylase deficiency
Journal of Endocrinological Investigation (2021)
-
Cytochrome P450 oxidoreductase deficiency caused by a novel mutation in the POR gene in two siblings: case report and literature review
Hormones (2021)
-
Biased cytochrome P450-mediated metabolism via small-molecule ligands binding P450 oxidoreductase
Nature Communications (2021)
