
Abstract
Inhibitor of κB kinase (IκK) has historically been studied in the context of immune response and inflammation, but recent evidence demonstrates that IκK activity is necessary and sufficient for regulation of neuronal function. Chronic social defeat stress of mice increases IκK activity in the nucleus accumbens (NAc) and this increase is strongly correlated to depression-like behaviors. Inhibition of IκK signaling results in a reversal of chronic social defeat stress-induced social avoidance behavior. Here, we more completely define the role of IκK in anxiety and depressive-like behaviors. Mice underwent stereotaxic microinjection of a herpes simplex virus expressing either green fluorescent protein, a constitutively active form of IκK (IκKca), or a dominant negative form of IκK into the NAc. Of all three experimental groups, only mice expressing IκKca show a behavioral phenotype. Expression of IκKca results in a decrease in the time spent in the non-periphery zones of an open field arena and increased time spent immobile during a forced swim test. No baseline differences in sucrose preference were observed, but following the acute swim stress we noted a marked reduction in sucrose preference. To determine whether IκK activity alters responses to other acute stressors, we examined behavior and spine morphology in mice undergoing an acute social defeat stress. We found that IκKca enhanced social avoidance behavior and promoted thin spine formation. These data show that IκK in NAc is a critical regulator of both depressive- and anxiety-like states and may do so by promoting the formation of immature excitatory synapses.
Similar content being viewed by others

INTRODUCTION
The inhibitor of kappaB kinase (IκK)–nuclear factor kappaB (NFκB) pathway has a central role in inflammation and immune response, as well as cell growth and survival (Chen and Greene, 2004; Häcker and Karin, 2006). More recently, IκK and NFκB activity has been implicated as an important regulator of synaptic signaling and neuronal morphology both in vitro and in vivo (Christoffel et al, 2011a, 2011b; Kaltschmidt et al, 1993; Meffert et al, 2003; O’Mahony et al, 2006; Russo et al, 2009). The IκK signaling pathway is activated by extracellular cytokines (such as interleukin 1 beta (IL-1β) and IL-6), infectious agents, glutamate, and neurotrophins (Israel, 2010; O’Neill and Kaltschmidt, 1997; Schölzke et al, 2003). Downstream of IκK, many target genes of the NFκB transcriptional complex contribute significantly to synaptic plasticity, such as NCAM, BDNF, opioid receptors, glutamate receptors, and neuregulin (Bunting et al, 2007; Chen et al, 2006; Frensing et al, 2008; Richter et al, 2002; Saha et al, 2006). Indeed, c-Rel, a member of the NFκB transcriptional complex, is required for long-term potentiation in the hippocampus (Ahn et al, 2008). Thus, IκK is capable of integrating many extracellular signals to alter gene expression and cause long-lasting changes in neuronal function and ultimately behavior.
The nucleus accumbens (Nac) is a brain region critical in evaluating the salience of aversive and rewarding stimuli to direct behavior (Mogenson et al, 1980). This subcortical nuclei of the ventral striatum mediates neural communication between cortical regions and the limbic system in order to regulate emotion and cognition. The NAc acts as a central regulator of emotional behaviors, and its function is tightly modulated by a number of molecular mechanisms, including histone deactylases, IκK, and ΔFosB, among others (Christoffel et al, 2011a; Covington et al, 2009; Vialou et al, 2010). Importantly, the NAc is a relevant target for therapeutic exploration in major depressive disorder, as deep brain stimulation of the NAc reduces anxiety and depression in previously treatment-resistant major depressive disorder patients (Bewernick et al, 2010). These findings highlight the critical role of NAc in emotional behaviors and suggest that novel modulators of neuronal function in NAc may improve therapeutic outcomes for psychiatric patients.
Previously, we demonstrated that chronic social defeat stress induces IκK activity and the formation of immature dendritic spines on medium spiny neurons (MSNs) in NAc of susceptible mice. Expression of the susceptible phenotype is dependent upon IκK, as inhibition of IκK activity, through viral mediated gene transfer of a dominant negative mutant (IκKdn), reverses both social avoidance behavior and the immature dendritic spine formation that appears to drive this behavior (Christoffel et al, 2011a). However, it is still unknown whether IκK more generally regulates emotional behaviors, and whether increased IκK activity in mice is sufficient to promote the synaptic and behavioral changes associated with repeated stress exposure.
To evaluate the role of IκK in regulating emotional behavior, we utilized viral-mediate gene transfer of herpes simplex viruses (HSVs) expressing either green fluorescent protein (GFP), a constitutively active IκK (IκKca) or IκKdn into the NAc of C57BL/6J mice and performed a battery of behavioral tests (open field, forced swim, and sucrose preference). To assess whether IκK promotes susceptibility to acute stress, we exposed mice either to an acute swim stress or acute social defeat and examined sucrose preference (anhedonia), social interaction (avoidance), and synaptic adaptations. We found that elevation of IκK activity in the NAc increases baseline anxiety- and depression-like behaviors, as well as susceptibility to acute stress-induced anhedonia and social avoidance. Importantly, we found that IκKca is sufficient to promote immature spine synapse formation in mice vulnerable to acute social defeat stress. These findings, in conjunction with our previous work, highlight the critical nature of de novo synaptic formation in guiding emotional behavior.
METHODS
Animals
Eight-week-old C57BL/6J mice (Charles River Laboratories, Wilmington, Massachusetts) were used for all experiments. For acute social defeat studies, 4-month-old retired CD-1 breeders (Jackson Laboratories, Bar Harbor, Maine) were used as aggressors. One-week before the start of all experiments, mice were group housed and maintained on a 12 h light/dark cycle with ad libitum access to food and water. Behavioral assessments and tissue collection were performed during the animals’ light phase (0700–1900 hours). Mouse procedures were performed in accordance with the Institutional Animal Care and Use Committee guidelines of the Mount Sinai School of Medicine.
Stereotaxic Surgery and Viral Gene Transfer
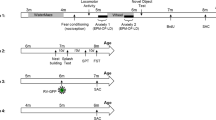
Eight-week old C57BL/6J mice (n=53 mice, 4–8 mice/group) were anesthetized with a mixture of ketamine (100 mg/kg) and xylazine (10 mg/kg) and positioned in a small-animal stereotaxic instrument (David Kopf Instruments, Tujunga, California), and the skull surface was exposed. Thirty-three-gauge syringe needles (Hamilton , Reno, Nevada) were used to bilaterally infuse 0.5 μl of HSV (1.5 × 108 infectious units/ml) expressing GFP, or IκKca and IκKdn mutants to activate or inhibit NFκB signaling, respectively, into the NAc (bregma coordinates: anteroposterior, 1.5 mm; mediolateral, 1.6 mm; dorsoventral, 4.4 mm; angle 10°; see Figure 1a for degree of infection just before behavior and after behavioral testing) at a rate of 0.1 μl/min. All of these viral vectors have been tested and validated in vivo and in vitro (Christoffel et al, 2011a; LaPlant et al, 2009; Russo et al, 2009).

(a) Representative images of viral expression in NAc at 72 and 120 h, just before and following behavioral testing, respectively. (b) Experimental timelines for each behavioral study.
Construction of Viral Vectors
Viral vectors were constructed as previously described (Russo et al, 2009). Briefly, for the IκKdn, using the coding sequence of the β form, lysine 44 was substituted with methionine (Mercurio et al, 1997) and subcloned into the bicistronic p1005+ HSV plasmid. For the IκKca mutant, serine 177 and 181 of the β form were mutated to glutamic acid (Mercurio et al, 1999), we designed primers with KpnI restriction sites (forward primer, CAAGGTACCARGAGCTGGTCACCTTCCC; and reverse primer, CAAGGTACCTCATGAGGCCTGCTCCA) and amplified fragments were subcloned into the bicistronic p1005+ HSV plasmid at Kpn1. As previously reported, both mutant vectors express IκK at similar levels to wildtype IκK in PC12 cells. IκKca expression increases levels of phospho-p65 and in vivo overexpression in the NAc of NFκB-LacZ reporter mice increases β-gal expression. Additionally, overexpression of HSV-IκKdn in NAc of the NFκB-LacZ reporter mice results in a 50% reduction in β-gal expression (Russo et al, 2009).
Experimental Timeline
Mice underwent stereotaxic surgery to deliver the virus and then were given 1 day to recover. During the recovery day, mice were habituated to two bottles for sucrose preference testing. Ninety-six hours after surgery, depressive behavior was assessed with the forced swim test. The last sucrose measurement was taken the morning after forced swim. This behavioral test constituted the acute swim stressor. In a separate cohort, 72 h after surgery, anxiety-like behavior was assessed with the open field test. All animals were perfused on day 5 for histological viral placement assessment (Figure 1b). Any mouse without viral expression or poor targeting outside of the NAc was removed from the analysis. Behavioral tests were timed to take place during peak HSV expression on day 2–4 post-surgery (Carlezon and Neve, 2003).
Behavioral Testing
Open field
Open field test was performed as previously described (Monteggia et al, 2007). Videotracking-based methods (Ethovision, Noldus Systems, Leesburg, Virginia) were used during 10-min trials to record the distance moved and time spent in the arena (72 cm in diameter) under dim lighting, along with a delineated ‘periphery zone’, a delineated ‘non-periphery zone’, and a delineated ‘center zone’ (34 cm × 34 cm) (Figure 2a).

IκKca expression in NAc is axiogenic. (a) Schematic of zones in open field arena and representative traces of mouse movement during an open field test. (b) Mice expressing IκKca spend significantly less time in the non-periphery and significantly more time in the periphery of the arena. There was also a trend for a decrease in time spent in the center. (c) IκKca-expressing mice also showed an increased latency to enter the center zone. (d) There were no differences in total distance traveled across all groups. Data are represented as group means. Error bars represent SEM (*p<0.05, tp=0.10, one-way ANOVA, n=8 mice/group).
Forced swim test
Forced swim test was performed as previously described (Monteggia et al, 2007). Mice were videotaped while in a 4 l Pyrex glass beaker containing 3 l of water at 24±1 °C for 6 min. Water in each beaker was changed after each trial. Two trained and blinded observers scored the videotape manually. Total immobility was measured as the time spent without any motion except for single limb paddling to maintain flotation. Latency to immobility was assessed as the time until the mouse first became immobile (>3 s).
Sucrose preference
Sucrose preference was performed as previously described (Monteggia et al, 2007). For sucrose-preference testing, a solution of 1% sucrose or diluent alone (drinking water) was placed into 50 ml tubes with ball-point sipper tubes (Ancare, Bellmore, NY). All animals were acclimatized to two-bottle choice conditions before testing conditions. Daily, the weight of each bottle was measured, and the positions of the tubes were interchanged to prevent preferential drinking based on location in the cage. Sucrose preference was calculated as a percentage (amount of sucrose consumed (in bottle A)/total amount consumed (bottles A and B)) across 3 days of testing.
Social defeat stress and social interaction
To measure increased susceptibility to stress, we adapted a subthreshold ‘microdefeat’ as previously described (Krishnan et al, 2007). Under these conditions, C57BL/6J mice were exposed to a novel CD1 aggressor for 5 min, followed by 5 min rest in the home cage. Exposure to the CD1 aggressor occurred three times with 5-min intervals between each exposure. Twenty-four hours later mice were assessed using the social interaction test. Under control conditions, this subthreshold microdefeat protocol does not induce social avoidance.
Social interaction was performed as previously described (Berton et al, 2006; Golden et al, 2011). Briefly, mice were placed into a novel arena with a small animal cage at one end. Their movement was monitored for 2.5 min in the absence of an aggressive CD1 mouse (used to determine baseline exploratory behavior), followed by 2.5 min in the presence of the caged aggressor. We measured the distance traveled (in centimeters), and the duration spent in the interaction zone and corner zones (in seconds) using Ethovision 3.0 software (Noldus Information Technology, Attleboro, Maine). We calculated social interaction as a ratio of the time spent in the interaction zone with an aggressive mouse present over the time spent with the aggressive mouse absent. All mice with a ratio >1 were classified as resilient and all mice with a ratio <1 were classified as susceptible.
Perfusion and Tissue Processing
On day 5 after viral expression, all mice were given a euthanizing dose of 15% chloral hydrate and transcardially perfused with cold 1% paraformaldehyde in phosphate-buffered saline (pH 7.4), followed by fixation with cold 4% paraformaldehyde in phosphate-buffered saline. Brains were dissected and postfixed for 18 h in the same fixative. The brain was cut into 50 μm coronal slices to assess viral placement or 150 μm for spine analysis.
Imaging and Spine Analysis
For spine analysis, dendritic segments 50–150 μm away from the soma were randomly chosen from HSV-infected cells that express GFP (Figure 5a). Images were acquired on a confocal LSM 710 (Zeiss, Oberkochen, Germany) for morphological analysis as described previously (Radley et al, 2006). Neurons were selected from the NAc shell. To qualify for spine analysis, dendritic segments had to satisfy the following requirements: (1) the segment had to be completely filled (all endings were excluded) and (2) segment must be at least 50 mm from the soma (Radley et al, 2006). Dendritic segments were imaged using a × 100 lens (NA 1.4, Zeiss) and a zoom of 2.5. Pixel size was 0.03 mm in the x–y plane and 0.01 mm in the z plane. Images were taken with a resolution of 1024 × ∼300 (the y dimension was adjusted to the particular dendritic segment to expedite imaging), pixel dwell time was 1.27 μm/s, and the line average was set to 4. An average of 2 dendrites per neuron on 5 neurons per animal (n=13 mice, n=4–5 mice/group) totaling approximately 1000 dendritic spines per experimental group were analyzed. For quantitative analysis of spine size, shape, and volume, NeuronStudio was used employing the rayburst algorithm previously described (Rodriguez et al, 2008). NeuronStudio classifies spines as thin, mushroom, or stubby based on the following values: (1) aspect ratio, (2) head to neck ratio, and (3) head diameter. Spines with a neck can be classified as either thin or mushroom; those without a significant neck are classified as stubby. Spines with a neck are labeled as thin or mushroom based upon head diameter. These parameters have been verified by comparison with trained human operators.
Statistics
All data are expressed as the mean±SEM. Mean differences between groups were determined using either a one- or two-way analysis of variance (ANOVA) followed by Newman Keuls post-hoc tests when the main effect was significant at p<0.05. Statistical analyses were performed using Prism 5.0 (GraphPad Software, La Jolla, California).
RESULTS
To assess anxiety and measure basal activity, we performed an open field test in mice following expression of IκKca, IκKdn, or GFP in NAc (Figure 2a). We found a main effect of viral expression for time spent in the non-periphery (one-way ANOVA: F(2, 23)=7.308, p<0.01) and periphery zones (one-way ANOVA: F(2, 23)=7.308, p<0.01; Figure 2b). Post-hoc analysis revealed that the IκKca group spent less time in the non-periphery zone and increased time spent in the periphery compared with GFP controls (p<0.01). We also observed a trend towards decreased time spent in center for the IκKca group (one-way ANOVA: F(2, 23)=1.918, p=0.10). ANOVA revealed a main effect of virus on latency to enter the center (one-way ANOVA: F(2, 23)=5.209, p=0.01), with mice expressing IκKca taking significantly longer to enter the center zone compared with mice expressing GFP or IκKdn (Figure 2c). We observed no differences in total distance traveled for any group, suggesting that any changes observed were not due to altered locomotor activity (Figure 2d). Together, these data suggest that IκKca expression in the NAc increases basal anxiety behavior as measured in this exploratory-based behavioral assay.
To inspect how manipulation of IκK activity alters baseline depression-like behaviors, we performed the forced swim test, an established measure of behavioral ‘despair’ (Porsolt et al, 1978). We found a main effect of virus on time spent immobile (one-way ANOVA: F(2, 17)=5.21, p<0.01), with post-hoc analysis revealing a significant increase in time spent immobile in the IκKca group compared with the GFP control group (Figure 3a). However, there were no significant differences in latency to immobility (p>0.05; Figure 3b).

IκKca expression is pro-depressant. During a forced swim test, mice expressing IκKca spend significantly more time (a) immobile. (b) No differences were observed in latency to immobility. Data are represented as group means. Error bars represent SEM (*p<0.05, one-way ANOVA, n=6–7 mice/group).
Next, to assess consummatory reward behavior, we measured sucrose preference, a standard assay of anhedonia (Papp et al, 1991). We found no effect of either IκK mutant on basal sucrose consumption or total liquid consumed (Figure 4a and b, respectively). However, 24 h after an acute swim stress, we found a main effect of virus on sucrose preference (one-way ANOVA: F(2, 20)=5.098, p<0.05). Post-hoc analysis revealed a significant reduction in sucrose preference in the IκKca group compared with both the GFP and IκKdn groups. In addition, no change in total liquid consumed was observed, indicating that viral expression did not affect normal drinking behavior (p<0.05; Figure 4c and d, respectively). In mice undergoing an acute social defeat stress paradigm, we confirmed our previous results, finding a significant interaction between stress and virus revealing that IκKca decreased time spent in the interaction zone. Likewise, there was an interaction effect observed in time spent in corner zones, with increased time only for the IκKca group (two-way ANOVA: F(2, 16)=9.94, p<0.001; Figure 5b). As would be expected, we found a significant difference in social interaction ratio, with post-hoc analysis revealing a significant decrease in the IκKca group (one-way ANOVA: F(2, 13)=11.23, p<0.01; post-hoc analysis p<0.01; Figure 5c). Previously, we reported that IκKdn reversed social avoidance and immature spine formation in NAc only in mice susceptible, but not resilient, to chronic social defeat stress. (Christoffel et al, 2011a). However, IκKdn expression did not have any significant effect after acute social defeat (two-way ANOVA: F(2, 12)=6.26, p<0.01; post-hoc analysis p<0.001; Figure 5a).

IκKca expression promotes anhedonia following acute swim stress. There was no effect of either IκK mutant on (a) basal sucrose consumption or (b) basal levels of liquid consumption. (c) There was a significant decrease in the IκKca group 24 h after undergoing an acute swim stress. (d) Swim stress did not result in any significant difference in total liquid consumed. Data are represented as group means. Error bars represent SEM (*p<0.05, one-way ANOVA, n=6–7 mice/group).

IκKca expression promotes social avoidance after acute social defeat stress. (a) Mice expressing IκKca show a decrease in time spent in the interaction zone with a target mouse present 24 h after acute social defeat. (b) IκKca-expressing mice spend more time in the corner zones. (c) IκKca mice have a significantly lower social interaction ratio. Data are represented as group means. Error bars represent SEM (*p<0.05, two-way ANOVA, one-way ANOVA, n=4–5 mice/group).
To further examine the interaction between IκK activity and stress-induced plasticity, we analyzed spine density and morphology in NAc MSNs in mice expressing HSV-GFP, HSV-IκKca, or HSV-IκKdn after an acute stressor. Expanding upon our previous finding, we observed that IκKca is sufficient to induce new immature spine formation, whereas IκKdn did not alter the spine morphology profile. Specifically, we found a significant increase in total spine density (one-way ANOVA: F(2, 13)=5.03, p<0.05: post-hoc analysis p<0.05; Figure 6a and b), driven predominantly by an increase in thin spine density (one-way ANOVA: F(2, 13)=8.79, p<0.01: post-hoc analysis p<0.01; Figure 6c) in mice expressing IκKca compared with GFP alone. We did not find significant differences in stubby or mushroom spine density (Figure 6d and e). Additionally, we see a trend in the correlation between spine density and social interaction, suggesting that these neuroadaptations may indeed drive social avoidance (r2=0.2967, p=0.06; Figure 6f). We observed a similar correlation between thin spin density and social interaction (r2=0.2848, p=0.07; Figure 6g). We further examined other parameters of spine size finding a significant decrease (one-way ANOVA: F(2, 13)=5.14, p<0.05: post-hoc analysis p<0.05) in average spine head volume only in mice expressing IκKca as compared with GFP controls (Figure 7a). This average decrease is due to a shift towards more spines with smaller head volume (Figure 7b). Consistent with the change in volume, there was a trend observed for a decrease in average spine head diameter only in mice expressing IκKca as compared with GFP controls (one-way ANOVA: F(2, 13)=3.42, p=0.07; Figure 7c) There was no change in average spine length for any group (Figure 7d).

(a) IκKca expression alters spine density and morphology on NAc MSNs after acute social defeat. There was a significant increase (b) in total, and (c) thin spine density in the IκKca group only. No changes in (d) stubby or (e) mushroom spine density was observed for either IκKca or IκKdn. (f) There is a trend for a correlation between social interaction ratio and total spine density. (g) A similar trend was observed between thin spine density and social interaction ratio. Data are represented as group means. Error bars represent SEM (*p<0.05, one-way ANOVA, n=4 mice/group).

IκKca affects spine size. (a) There was a significant decrease in the average spine head volume only in the IκKca group. (b) Cumulative frequency plot shows a greater frequency of small spines across a range of sizes for the IκKca group. A strong trend was observed for (c) spine head diameter. No changes were observed in (d) length for either IκKca or IκKdn. Data are represented as group means. Error bars represent SEM (*p<0.05, tp=0.07, one-way ANOVA, n=4 mice/group).
DISCUSSION
Our results demonstrate that increasing basal levels of IκK activity is sufficient to induce anxiety and depression-like behavior in stress-naive mice, and increase susceptibility to acute stressors. We also found that following acute social defeat stress, which normally is insufficient for induction of depression-like behavior, overexpression of IκKca is sufficient to drive synaptic structural adaptations and promote social avoidance behavior. These findings highlight the critical regulatory role of IκK in controlling NAc MSN dendritic spine structural plasticity to promote anxiety and depression-like behavior. Previously, we demonstrated the capability of IκK inhibition to reverse chronic stress-induced neuroadaptations and behavior, yet our current findings suggest inhibition of IκK activity via IκKdn does not affect baseline behavior or response to a minor acute stressor. These results also show that under both stress naive and acute stress conditions, IκKdn has minimal effects on spine morphology, complementing our current behavioral findings (Christoffel et al, 2011a and current data set). Though further studies are needed, perhaps this indicates low basal levels of IκK activity in MSNs, contrasting what is found in pyramidal neurons. Collectively, our results do suggest that stress promotes depression and anxiety-like behavior through an activity-dependent intracellular mechanism requiring IκK activation leading to synaptic remodeling.
The presented results also support the hypothesis that hyperactivity of the IκK/NFκB pathway may serve as a risk factor for mood and anxiety disorders, and detection of increased basal activity could serve as a biomarker for diagnosis. Indeed, it has been shown that patients with major depressive disorder have increased blood levels of cytokines, an established upstream regulator of this pathway (Liu et al, 2011). Further evidence of the important role of cytokine signaling is the ability of celecoxib, a cyclooxygenase-2 inhibitor, to reduce serum levels of IL-6 in patients with major depressive disorder and concurrently reduce depressive symptoms (Abbasi et al, 2012). From a more basic perspective, transgenic mice lacking the p50 subunit of the NFκB transcriptional complex exhibit increased basal levels of anxiety as measured by both open field and the light/dark box test (Lehmann et al, 2010). The authors suggest that p50 in neurons serves to dampen the intracellular activity of NFκB and thus in the knockout, the activity of this pathway is heightened. Together with the current findings, this demonstrates that over-activity of this pathway, either throughout development or only discrete periods during adulthood, produces exaggerated anxiety behaviors.
The observed increase in spine density suggests that restructuring of synaptic connectivity is crucial for the observed maladaptive behavior. Similar results have been observed in the amygdala, where prolonged restraint stress leads to increased dendritic spine density accompanied by an increase in anxious behavior (Vyas et al, 2006). Similarly, we recently reported that chronic social defeat stress results in an increase in immature dendritic spines that have smaller postsynaptic densities, which strongly correlated with the social interaction scores of both defeated and control mice. Additionally, overexpression of IκKdn appears only to prevent the maintenance of the stress-induced increases in stubby spines, while not significantly changing the type or density otherwise. These results are consistent with the idea that under control conditions inhibition of IκK does not lead to significant synaptic remodeling (Christoffel et al, 2011a). Conversely, increased IκK activity is sufficient to induce thin spine formation and promote susceptibility to maladaptive behavioral responses following an acute social defeat stress. Thin dendritic spines are immature synaptic structures that are highly plastic in nature, and possess smaller spine heads, consistent with our finding of an overall decrease in the average spine head volume. These spines readily stabilize or retract in response to increased or weakened synaptic input (Bourne and Harris, 2007), making them prime candidates for synaptic reorganization. Interestingly, immature synaptic structures are known to predominately express the GluN2B receptor (Sheng et al, 1994), and ketamine, a potent NMDA receptor antagonist, along with GluN2B-specific antagonists have rapid antidepressant effects in multiple models of depression (Autry et al, 2011; Li et al, 2010). Collectively, this suggests that the formation of new immature glutamatergic synaptic structures on NAc MSNs is a critical step in the development and expression of maladaptive behavioral stress responses.
IκK exerts many of its downstream effects through NFκB, and expression of the IκK mutant constructs alters levels of phospho-p65. Yet, there are other effectors that may be involved in regulating synaptic plasticity mechanisms. Recent evidence suggests that IκK acts directly on other pathways, such as insulin and neurotrophic signaling cascades, to alter dendritic spines and neuronal function (Lee et al, 2007; Nakamori et al, 2006; Schmeisser et al, 2012). Schmeisser et al (2012) demonstrate that inhibition of IκK, via conditional expression of an IκKdn allele or knockout of IκK specifically in excitatory neurons, prevents synapse formation and maintenance in a manner that is dependent upon Igf2 signaling. Along similar lines, phosphorylation of TSC1 by IκK activates the mTOR pathway, which is known to be involved in spine plasticity, and mediates the rapid antidepressants effects of ketamine. Taken as a whole, these findings implicate IκK as a broadly acting kinase, capable of modulating multiple signaling cascades to exert its effects on neuronal function.
The IκK-dependent synaptic adaptations mentioned above suggest that IκK affects not only anxiety and depressive behaviors, but more broadly can serve as a molecular mechanism for experience-dependent synaptic plasticity. For example, the IκK signaling pathway is recruited following cocaine administration, and is necessary for psychostimulant-induced structural plasticity in NAc MSNs (For review, see Russo et al, 2010). Similar to our findings, overexpression of the NFκB transcriptional complex subunit p65 in cultured hippocampal pyramidal neurons results in increased spine density. Conversely, knockout of the p65 gene in a transgenic line leads to decreased spine density in vivo (Boersma et al, 2011). IκK activity in the hippocampus has also been found to affect histone acteylation, and this action was shown to be a critical regulator of reconsolidation of conditioned fear memories (Lubin and Sweatt, 2007). Interestingly, activation of NFκB in the hippocampus by IL-1β has been shown to inhibit neurogenesis and is important in the pro-depressant effects of chronic unpredictable stress (Koo and Duman, 2008; Koo et al, 2010). Complementary to our findings, Koo et al (2010) found that inhibition of NFκB in hippocampus blocked the anhedonic effects of CUS as measured by sucrose consumption. However, stress-induced IκK or NFκB activity has not been observed in other brain regions that show decreased spine density in response to stress, such as in prefrontal cortical neurons (Radley et al, 2008). It is tempting to speculate whether IκK activity is increased or decreased in these regions and correlates with the observed spine adaptations. Future studies characterizing brain-wide alterations in IκK activity in response to stress will provide important information regarding the global regulation of IκK signaling in psychiatric disorders.
In addition to brain region–specific regulation of IκK activity, there is also likely to be cell-type specificity. Studies of cocaine's effects in the NAc have shown dopamine receptor 1 (D1R)-expressing cells to be the primary long-term locus of spine change (Lee et al, 2006; Lobo et al, 2010), and as mentioned above, IκK is a potent regulator of spine density in this region. IκK activity in response to stress or stimulant administration may similarly be regulated in a cell-type–specific manner. In fact, a transgenic line expressing the super-repressor, IκBα-SR, predominately in interneurons, leads to a hyperexcitable state characterized by increased long-term potentiation, spatial learning, and seizures (O’Mahony et al, 2006). Perhaps not surprising, a transgenic line expressing the Tet-O inducible super-repressor under the CamKII-α promoter, shows opposite behavioral changes, namely deficits in spatial learning (Kaltschmidt et al, 2006). These behavioral deficits were accompanied by impaired long-term potentiation and reduced forskolin-induced CREB phosphorylation. Similarly, transgenic mice lacking the p65 subunit are impaired in spatial learning tasks (Meffert et al, 2003). Interestingly, in the p50 knockout, there is a paradoxical increase in NFκB activity, and better performance in the Morris water maze, but not in the less anxiety-provoking Barnes maze. This suggests that developmental effects of p50 knockout may lead to a compensatory increase in NFκB activity and subsequent anxiety profile consistent with our results. We are able to avoid the difficulties of these subunit specific developmental effects of IκK on behavior by controlling the activity of the pathway at a higher regulatory level. Using viral-mediated gene transfer to provide greater spatiotemporal precision (Carlezon and Neve, 2003, we manipulated IκK specifically within the adult NAc. Together, our findings provide strong evidence for a critical role of IκK in the NAc in synaptic plasticity and behavior.
Ultimately, it appears that elevation of the activity of the IκK pathway regulates biochemical or transcriptional events to induce a highly plastic state. This permissive state is essential to the formation of novel behavioral responses, whether in response to normal experience or noxious stimuli, such as stress or drugs of abuse. Repeated induction of this state by either type of stimuli appears to enhance the behavioral response through restructuring of synaptic contacts. Increased IκK activity and immature spine formation occur in response to chronic social defeat in susceptible mice, and increasing IκK activity during an acute social stress is sufficient to promote immature thin spines and social avoidance behavior. Although only speculative at this point, stabilization of these new contacts is potentially the root problem in reversing maladaptive behaviors. There has been much discussion in the literature concerning the slow onset of efficacy of traditional antidepressants, and whether this is due to a slow onset of plasticity mechanisms has yet to be shown definitively. The rapid relief of depressive symptoms via ketamine, acting on glutamate transmission and inducing plasticity of spines, suggests that dysregulation of plasticity mechanisms is a primary cause of depression-like behaviors.
In conclusion, we found that IκK activity affects emotional behaviors and regulates vulnerability to acute stress, likely through modulation of synaptic plasticity mechanisms. These findings point to the induction of immature synaptic structures in the NAc as a key neuroadaptation-regulating vulnerability to stress. Furthermore, the common effect of multiple molecules on depressive behaviors, suggest many signaling cascades might interact to alter the state of plasticity in the brain. Gaining a further understanding of these interactions will further elucidate the most effective means to modulate neuronal function in psychiatric disorders.
References
Abbasi SH, Hosseini F, Modabbernia A, Ashrafi M, Akhondzadeh S (2012). Effect of celecoxib add-on treatment on symptoms and serum IL-6 concentrations in patients with major depressive disorder: randomized double-blind placebo-controlled study. J Affect Disord; e-pud ahead of print 18 April 2012.
Ahn HJ, Hernandez CM, Levenson JM, Lubin FD, Liou HC, Sweatt JD (2008). c-Rel, an NF-kappaB family transcription factor, is required for hippocampal long-term synaptic plasticity and memory formation. Learn Mem 15: 539–549.
Autry AE, Adachi M, Nosyreva E, Na ES, Los MF, Cheng PF et al (2011). NMDA receptor blockade at rest triggers rapid behavioural antidepressant responses. Nature 475: 91–95.
Berton O, McClung CA, Dileone RJ, Krishnan V, Renthal W, Russo SJ et al (2006). Essential role of BDNF in the mesolimbic dopamine pathway in social defeat stress. Science 311: 864–868.
Bewernick BH, Hurlemann R, Matusch A, Kayser S, Grubert C, Hadrysiewicz B et al (2010). Nucleus accumbens deep brain stimulation decreases ratings of depression and anxiety in treatment-resistant depression. Biol Psychiatry 67: 110–116.
Boersma MC, Dresselhaus EC, De Biase LM, Mihalas AB, Bergles DE, Meffert MK (2011). A requirement for nuclear factor-kappaB in developmental and plasticity-associated synaptogenesis. J Neurosci 31: 5414–5425.
Bourne J, Harris KM (2007). Do thin spines learn to be mushroom spines that remember? Curr Opin Neurobiol 17: 381–386.
Bunting K, Rao S, Hardy K, Woltring D, Denyer GS, Wang J et al (2007). Genome-wide analysis of gene expression in T cells to identify targets of the NF-kappa B transcription factor c-Rel. J Immunol 178: 7097–7109.
Carlezon Jr WA, Neve RL (2003). Viral-mediated gene transfer to study the behavioral correlates of CREB function in the nucleus accumbens of rats. Methods Mol Med 79: 331–350.
Chen LF, Greene WC (2004). Shaping the nuclear action of NF-kappaB. Nat Rev 5: 392–401.
Chen YL, Law PY, Loh HH (2006). Nuclear factor kappaB signaling in opioid functions and receptor gene expression. J Neuroimmune Pharmacol 1: 270–279.
Christoffel DJ, Golden SA, Dumitriu D, Robison AJ, Janssen WG, Ahn HF et al (2011a). IkappaB kinase regulates social defeat stress-induced synaptic and behavioral plasticity. J Neurosci 31: 314–321.
Christoffel DJ, Golden SA, Russo SJ (2011b). Structural and synaptic plasticity in stress-related disorders. Rev Neurosci 22: 535–549.
Covington 3rd HE, Maze I, LaPlant QC, Vialou VF, Ohnishi YN, Berton O et al (2009). Antidepressant actions of histone deacetylase inhibitors. J Neurosci 29: 11451–11460.
Frensing T, Kaltschmidt C, Schmitt-John T (2008). Characterization of a neuregulin-1 gene promoter: positive regulation of type I isoforms by NF-kappaB. Biochim Biophys Acta 1779: 139–144.
Golden SA, Covington 3rd HE, Berton O, Russo SJ (2011). A standardized protocol for repeated social defeat stress in mice. Nat Protoc 6: 1183–1191.
Häcker H, Karin M (2006). Regulation and function of IKK and IKK-related kinases. Sci STKE 2006: re13.
Israel A (2010). The IKK complex, a central regulator of NF-kappaB activation. Cold Spring Harb Perspect Biol 2: a000158.
Kaltschmidt B, Ndiaye D, Korte M, Pothion S, Arbibe L, Prullage M et al (2006). NF-kappaB regulates spatial memory formation and synaptic plasticity through protein kinase A/CREB signaling. Mol Cell Biol 26: 2936–2946.
Kaltschmidt C, Kaltschmidt B, Baeuerle P (1993). Brain synapses contain inducible forms of the transcription factor NF-κB. Mech Dev 43: 135–147.
Koo JW, Duman RS (2008). IL-1beta is an essential mediator of the antineurogenic and anhedonic effects of stress. Proc Natl Acad Sci USA 105: 751–756.
Koo JW, Russo SJ, Ferguson D, Nestler EJ, Duman RS (2010). Nuclear factor-kappaB is a critical mediator of stress-impaired neurogenesis and depressive behavior. Proc Natl Acad Sci USA 107: 2669–2674.
Krishnan V, Han MH, Graham DL, Berton O, Renthal W, Russo SJ et al (2007). Molecular adaptations underlying susceptibility and resistance to social defeat in brain reward regions. Cell 131: 391–404.
LaPlant Q, Chakravarty S, Vialou V, Mukherjee S, Koo JW, Kalahasti G et al (2009). Role of nuclear factor kappaB in ovarian hormone-mediated stress hypersensitivity in female mice. Biol Psychiatry 65: 874–880.
Lee DF, Kuo HP, Chen CT, Hsu JM, Chou CK, Wei Y et al (2007). IKK beta suppression of TSC1 links inflammation and tumor angiogenesis via the mTOR pathway. Cell 130: 440–455.
Lee KW, Kim Y, Kim AM, Helmin K, Nairn AC, Greengard P (2006). Cocaine-induced dendritic spine formation in D1 and D2 dopamine receptor-containing medium spiny neurons in nucleus accumbens. Proc Natl Acad Sci USA 103: 3399–3404.
Lehmann ML, Brachman RA, Listwak SJ, Herkenham M (2010). NF-kappaB activity affects learning in aversive tasks: possible actions via modulation of the stress axis. Brain Behav Immun 24: 1008–1017.
Li N, Lee B, Liu RJ, Banasr M, Dwyer JM, Iwata M et al (2010). mTOR-dependent synapse formation underlies the rapid antidepressant effects of NMDA antagonists. Science 329: 959–964.
Liu Y, Ho RC, Mak A (2011). Interleukin (IL)-6, tumour necrosis factor alpha (TNF-alpha) and soluble interleukin-2 receptors (sIL-2R) are elevated in patients with major depressive disorder: a meta-analysis and meta-regression. J Affect Disord 139: 230–239.
Lobo MK, Covington 3rd HE, Chaudhury D, Friedman AK, Sun H, Damez-Werno D et al (2010). Cell type-specific loss of BDNF signaling mimics optogenetic control of cocaine reward. Science 330: 385–390.
Lubin FD, Sweatt JD (2007). The IkappaB kinase regulates chromatin structure during reconsolidation of conditioned fear memories. Neuron 55: 942–957.
Meffert MK, Chang JM, Wiltgen BJ, Fanselow MS, Baltimore D (2003). NF-kappa B functions in synaptic signaling and behavior. Nat Neurosci 6: 1072–1078.
Mercurio F, Murray BW, Shevchenko A, Bennett BL, Young DB, Li JW et al (1999). IkappaB kinase (IKK)-associated protein 1, a common component of the heterogeneous IKK complex. Mol Cell Biol 19: 1526–1538.
Mercurio F, Zhu H, Murray BW, Shevchenko A, Bennett BL, Li J et al (1997). IKK-1 and IKK-2: cytokine-activated IkappaB kinases essential for NF-kappaB activation. Science 278: 860–866.
Mogenson GJ, Jones DL, Yim CY (1980). From motivation to action: functional interface between the limbic system and the motor system. Prog Neurobiol 14: 69–97.
Monteggia LM, Luikart B, Barrot M, Theobold D, Malkovska I, Nef S et al (2007). Brain-derived neurotrophic factor conditional knockouts show gender differences in depression-related behaviors. Biol Psychiatry 61: 187–197.
Nakamori Y, Emoto M, Fukuda N, Taguchi A, Okuya S, Tajiri M et al (2006). Myosin motor Myo1c and its receptor NEMO/IKK-gamma promote TNF-alpha-induced serine307 phosphorylation of IRS-1. J Cell Biol 173: 665–671.
O’Mahony A, Raber J, Montano M, Foehr E, Han V, Lu SM et al (2006). NF-kappaB/Rel regulates inhibitory and excitatory neuronal function and synaptic plasticity. Mol Cell Biol 26: 7283–7298.
O’Neill LA, Kaltschmidt C (1997). NF-kappa B: a crucial transcription factor for glial and neuronal cell function. Trends Neurosci 20: 252–258.
Papp M, Willner P, Muscat R (1991). An animal model of anhedonia: attenuation of sucrose consumption and place preference conditioning by chronic unpredictable mild stress. Psychopharmacology 104: 255–259.
Porsolt RD, Anton G, Blavet N, Jalfre M (1978). Behavioural despair in rats: a new model sensitive to antidepressant treatments. Eur J Pharmacol 47: 379–391.
Radley JJ, Rocher AB, Miller M, Janssen WG, Liston C, Hof PR et al (2006). Repeated stress induces dendritic spine loss in the rat medial prefrontal cortex. Cereb Cortex 16: 313–320.
Radley JJ, Rocher AB, Rodriguez A, Ehlenberger DB, Dammann M, McEwen BS et al (2008). Repeated stress alters dendritic spine morphology in the rat medial prefrontal cortex. J Comp Neurol 507: 1141–1150.
Richter M, Suau P, Ponte I (2002). Sequence and analysis of the 5′ flanking and 5′ untranslated regions of the rat N-methyl-D-aspartate receptor 2A gene. Gene 295: 135–142.
Rodriguez A, Ehlenberger DB, Dickstein DL, Hof PR, Wearne SL (2008). Automated three-dimensional detection and shape classification of dendritic spines from fluorescence microscopy images. PLoS One 3: e1997.
Russo SJ, Dietz DM, Dumitriu D, Morrison JH, Malenka RC, Nestler EJ (2010). The addicted synapse: mechanisms of synaptic and structural plasticity in nucleus accumbens. Trends Neurosci 2: 1–10.
Russo SJ, Wilkinson MB, Mazei-Robison MS, Dietz DM, Maze I, Krishnan V et al (2009). Nuclear factor kappa B signaling regulates neuronal morphology and cocaine reward. J Neurosci 29: 3529–3537.
Saha RN, Liu X, Pahan K (2006). Up-regulation of BDNF in astrocytes by TNF-alpha: a case for the neuroprotective role of cytokine. J Neuroimmune Pharmacol 1: 212–222.
Schmeisser MJ, Baumann B, Johannsen S, Vindedal GF, Jensen V, Hvalby OC et al (2012). IkappaB kinase/nuclear factor kappaB-dependent insulin-like growth factor 2 (Igf2) expression regulates synapse formation and spine maturation via Igf2 receptor signaling. J Neurosci 32: 5688–5703.
Schölzke MN, Potrovita I, Subramaniam S, Prinz S, Schwaninger M (2003). Glutamate activates NF-kappaB through calpain in neurons. Eur J Neurosci 18: 3305–3310.
Sheng M, Cummings J, Roldan LA, Jan YN, Jan LY (1994). Changing subunit composition of heteromeric NMDA receptors during development of rat cortex. Nature 368: 144–147.
Vialou V, Robison AJ, Laplant QC, Covington 3rd HE, Dietz DM, Ohnishi YN et al (2010). DeltaFosB in brain reward circuits mediates resilience to stress and antidepressant responses. Nat Neurosci 13: 745–752.
Vyas A, Jadhav S, Chattarji S (2006). Prolonged behavioral stress enhances synaptic connectivity in the basolateral amygdala. Neuroscience 143: 387–393.
Acknowledgements
We thank Kevin Guise for his assistance in performing the cumulative frequency plots of average spine head volume. This work was supported by funding from the US National Institute of Mental Health (R01MH090264-01) and the National Alliance for Research on Schizophrenia and Depression (SJR).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Christoffel, D., Golden, S., Heshmati, M. et al. Effects of Inhibitor of κB Kinase Activity in the Nucleus Accumbens on Emotional Behavior. Neuropsychopharmacol 37, 2615–2623 (2012). https://doi.org/10.1038/npp.2012.121
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/npp.2012.121
Keywords
This article is cited by
-
Prokineticin-2 Participates in Chronic Constriction Injury-Triggered Neuropathic Pain and Anxiety via Regulated by NF-κB in Nucleus Accumbens Shell in Rats
Molecular Neurobiology (2023)
-
Dendritic remodeling of D1 neurons by RhoA/Rho-kinase mediates depression-like behavior
Molecular Psychiatry (2020)
-
Dendritic spine density is increased on nucleus accumbens D2 neurons after chronic social defeat
Scientific Reports (2020)
-
The molecular and cellular mechanisms of depression: a focus on reward circuitry
Molecular Psychiatry (2019)
-
A diet enriched with curcumin promotes resilience to chronic social defeat stress
Neuropsychopharmacology (2019)
