
Abstract
Models of relapse have demonstrated that neuroadaptations in reward circuits following cocaine self-administration (SA) underlie reinstatement of drug-seeking. Dysregulation of the pathway from the prelimbic (PrL) cortex to the nucleus accumbens is implicated in reinstatement. A single BDNF infusion into the PrL cortex following a final cocaine SA session results in attenuation of reinstatement of cocaine-seeking. Inhibiting BDNF’s receptor, TrkB, ERK/MAP kinase activation, or NMDA receptors blocks this attenuating effect, indicating that the interaction between glutamate-mediated synaptic activity and TrkB signaling is imperative to BDNF’s suppressive effect on drug-seeking. Src family kinases (SFKs) are involved in both NMDA-mediated activation of TrkB- and TrkB-mediated tyrosine phosphorylation of NMDA receptors. We hypothesized that infusion of the SFK inhibitor, PP2, into the PrL cortex prior to a BDNF infusion, immediately after the end of the last cocaine SA session, would block BDNF’s ability to suppress reinstatement of cocaine-seeking in rats with a cocaine SA history. PP2, but not the negative control, PP3, blocked BDNF’s suppressive effect on context-induced relapse after 1 week of abstinence and cue-induced reinstatement after extinction. As previously reported, infusion of BDNF into the PrL cortex blocked cocaine SA-induced dephosphorylation of ERK, GluN2A, and GluN2B-containing receptors. Inhibition of SFKs using PP2 blocked BDNF-mediated phosphorylation of GluN2A, GluN2B, and ERK. These data indicate that SFK activity is necessary for BDNF-mediated suppression of cocaine-seeking and reversal of cocaine-induced dephosphorylation of key phosphoproteins in the prefrontal cortex related to synaptic plasticity.
Similar content being viewed by others

Introduction
Cocaine addiction is a prevalent neuropsychiatric disorder that imposes especially high societal costs owing to its chronic relapsing nature. For this reason, relapse prevention is a particularly relevant target for treatment. The dorsal medial prefrontal cortex, particularly the prelimbic (PrL) subregion, is the origin of the cortico-accumbens pathway to the nucleus accumbens core (NAcc) that has been implicated in reinstatement of drug-seeking in preclinical studies (Berglind et al, 2007; Go et al, 2016; McFarland and Kalivas, 2001; McFarland et al, 2003; Whitfield et al, 2011). Cocaine self-administration (SA) results in a transient period of dephosphorylation of a number of proteins associated with neuronal plasticity, including ERK, CREB, and NMDA receptor subunits, GluN2A and GluN2B. Reversing the dephosphorylation of these proteins with a single intra-PrL microinfusion of brain-derived neurotrophic factor (BDNF) immediately after a final session of cocaine SA results in a subsequent suppression of relapse to cocaine-seeking for up to 4 weeks post infusion (Berglind et al, 2007; Go et al, 2016; Whitfield et al, 2011) and normalizes cocaine-induced alterations in glutamate neurotransmitter levels in NAcc (Berglind et al, 2009). A single BDNF infusion had no effect on food-seeking in a similar paradigm, indicating the specificity of BDNF’s effects (Berglind et al, 2007). Intervention with a BDNF infusion immediately after cocaine SA is critical in preventing the cocaine-induced dephosphorylation of these proteins because their levels normalize within 24 h and a later infusion of BDNF does not suppress cocaine-seeking (Berglind et al, 2007; Whitfield et al, 2011).
The precise molecular events that underlie BDNF’s suppressive effects on relapse are only partially known. The high affinity BDNF receptor, tropomyosin kinase receptor B (TrkB), is expressed in rat cortex on the cell bodies and apical dendrites of pyramidal cells, as well as interneurons (Cellerino et al, 1996). Blocking activation of the TrkB receptor locally in the PrL cortex or its downstream target, extracellular signal-regulated kinase (ERK), prevents BDNF’s suppression of cocaine-seeking (Whitfield et al, 2011). Furthermore, NMDA receptor antagonists not only block the ability of BDNF to phosphorylate the NMDA receptor subunits, GluN2A and GluN2B, but also prevent BDNF’s suppression of cocaine-seeking (Go et al, 2016). We sought to understand how BDNF-induced TrkB activation and NMDA receptor subunit activation both underlie BDNF’s suppressive effect on cocaine-seeking.
A main link between these two receptors is mediated by src family kinases (SFKs). The SFKs, Fyn and Src, are highly expressed in neurons and have emerged as essential regulators of synaptic transmission specifically through tyrosine phosphorylation of the NMDA receptor subunits, GluN2B and GluN2A, respectively, to positively modulate synaptic transmission (Salter and Kalia, 2004; Ohnishi et al, 2011). Furthermore, TrkB-induced activation results in a feed forward activation of SFKs and a feedback enhancement of TrkB phosphorylation (Huang and McNamara, 2010). Thus, in this study, we investigated whether inhibition of SFKs using a non-selective SFK inhibitor, PP2 (Hanke et al, 1996), would block both the ability of BDNF to phosphorylate ERK, GluN2A, and GluN2B and BDNF’s suppression of cocaine-seeking. Our results indicate that SFK activation is critical for BDNF’s suppressive effects on cocaine-seeking, likely through SFK-mediated GluN2A, GluN2B, and ERK activation.
Materials and methods
See Supplementary Information for complete methodological details of previously published procedures.
Experimental Design
Figure 1a illustrates the behavioral paradigm used in Experiment 1 to determine the effect that SFK inhibition has on the ability of BDNF to suppress cocaine-seeking. Immediately following the last cocaine SA session, the PrL cortex of rats was infused bilaterally with the active SFK inhibitor, PP2, or its negative control, PP3, followed by an infusion of either BDNF or PBS. Rats were then returned to their home cages for 6 days of forced abstinence, following which they underwent a post-abstinence relapse test under extinction conditions. Animals then continued with extinction to criterion and underwent a cue-induced reinstatement test. Figure 1b displays the design used in Experiment 2 to determine the effects of SFK inhibition during early abstinence on the ability of an intra-PrL BDNF microinfusion to block cocaine SA-induced dephosphorylation of ERK, GluN2A, and GluN2B. All rats in Experiment 2, including yoked-saline control rats, were decapitated 2 h after the intracranial (i.c.) infusions at the end of cocaine SA and tissue punches from the PrL cortex were processed for phosphoprotein analysis.

(a) Experiment 1 design. Rats receive either the SFK inhibitor, PP2, or its negative control, PP3, 20 min prior to a BDNF or PBS infusion immediately after the final self-administration session. Rats then underwent abstinence, a post-abstinence (PA) test, extinction, and a cue induced reinstatement test. (b) Experiment 2 timeline for analysis of phosphoprotein expression. Rats were decapitated 2 h after intracranial infusions immediately after the last cocaine self-administration session. (c) Schematic depiction of cannula placements in the PrL cortex in Experiment 1 (closed circles). Animals with placements outside the area of interest were excluded. Open circles show the area included in tissue punches that were processed for phosphoprotein analysis in Experiment 2.
Subjects and Surgical Procedures
Male Sprague Dawley Rats (Charles River Laboratories, Wilmington, MA) weighing 275–325 g, were singly housed upon arrival in a temperature and humidity controlled vivarium in a room on a 12–12 reverse light/dark cycle. They had ad libitum access to standard rat chow (Harlan, Indianapolis, IN) and water. All experiments were conducted during the dark cycle and approved by the IACUC of the Medical University of South Carolina. Prior to surgery, rats were anesthetized, the left jugular veins were catheterized, and the PrL cortex was cannulated bilaterally as previously described (Berglind et al, 2007; Go et al, 2016). The antibiotic, cephazolin (10 mg/0.1 ml; 0.1 ml i.v.) and the anti-bacterial solution, taurolidine-citrate-saline (0.05 ml i.v.) were injected post surgery and for 5 days of recovery.
Cocaine SA
Rats were trained to self-administer cocaine on a FR1 schedule during 2 h sessions for 12–14 days as previously described (Go et al, 2016). In brief, following recovery, rats were food restricted to ⩽20 mg of chow one day prior to and during SA training. Rats were trained to press the active lever to self-administer cocaine hydrochloride (0.2 mg/infusion; NIDA, Research Triangle Park, NC) paired with a light and tone cue complex. Pressing the inactive lever had no programmed consequences. For rats in Experiment 2 (Figure 1b), a sender-receiver paradigm was used whereupon following receipt of a cocaine infusion in a sender animal, a corresponding receiver animal was infused with an equal volume of 0.9% saline.
Intracranial Drugs and Microinfusions in the PrL Cortex
Rats were habituated to an infusion room and their stylets manipulated after the last three SA sessions. On the day before the last SA session, a stylet (Plastics One, Roanoke, VA) that extended 1 mm beyond the implanted cannula was inserted. The PrL cortex of rats was infused bilaterally immediately following their final SA session. Ten mM stock solutions of the selective SFK inhibitor, 4-amino-5-(chlorophenyl)-7-(t-butyl)pyrazolo[3,4-D]pyrimidine (PP2) and its negative control, 4-amino-7-phenylpiryrazol[3,4-D]pyrimidine (PP3) (Tocris Bioscience-BioTechne) were prepared in 100% DMSO and diluted using double-filtered 1xPBS to 10 μM in a final concentration of 0.1% DMSO. Animals were infused with 10 μM of either PP2 (1.5 ng/0.5 μl per side, or PP3 (1.05 ng/0.5 μl per side) based on previous studies that demonstrated that intra-striatal infusion of 1.5 ng of PP2 bilaterally into the dorsal striatum was sufficient to suppress drug-seeking (Wang et al, 2007, 2010). Twenty min following the first i.c. infusion, rats were infused with either BDNF (0.75 μg/μl/side R&D Systems, Inc) or 10 mM PBS vehicle as previously described (Go et al, 2016).
Post-Abstinence Relapse Test, Extinction, and Cue-Induced Reinstatement
Following the final SA session and i.c. drug infusions, the rats in Experiment 1 were returned to the vivarium and underwent 6 days of abstinence in their home cages followed by a post-abstinence (PA) relapse test under extinction conditions as described (Go et al, 2016). After at least 6 days of additional extinction training to a criterion of less than 25 active lever presses for 2 consecutive days, rats underwent a cue-induced reinstatement test in which an active lever press resulted in presentation of the previously drug-paired cue complex, but no drug infusion.
Extractions and Histology
Brains from Experiment 1 were sectioned coronally at 40 μm in a cryostat and then Nissl-stained to verify cannula placements. In Experiment 2, bilateral, tissue punches were taken from the PrL cortex (approximately AP 2.5–4.5) in a cryostat using a 3 mm diameter biopsy punch (Braintree Scientific). Placements were verified and recorded during dissection and sections were taken anterior to and through the extent of the punch for Nissl staining and mapping. Punches were then stored at −80 °C until processed.
Tissue Processing
Punches from all experimental groups were processed simultaneously as previously described (Go et al, 2016; Sun et al, 2013).
Wes Immunoassay
The Wes capillary electrophoresis system (Protein Simple-BioTechne, San Jose, CA) was used for all protein quantitation. Sample aliquots were thawed and diluted to 0.8 μg/μl for all phosphoprotein targets and 0.4 μg/μl for all total protein targets using ¾ 0.1x Sample Buffer and ¼ 4 × Master Mix (1:1 mix of 40 mM DTT and 10 × Sample Buffer) according to supplier instructions. Samples were denatured at 95 °C for 5 min (ERK) or 70 °C for 10 min (GluN2A, GluN2B). Primary phosphoprotein antisera were loaded into 25-well plates as follows: pERK (1 : 12.5, Cell Signaling Technology), pGluN2A-Y1325 (1 : 12.5, Abcam), pGluN2B-Y1472 (1 : 12.5, Sigma-Aldrich). Calnexin (Enzo Life Sciences) was multiplexed with all phosphoprotein targets at a concentration of 1 : 2000. Total protein antisera were: ERK (1 : 100, Cell Signaling Technology) multiplexed with 1 : 1000 calnexin, GluN2A (1 : 50, Millipore) multiplexed with 1 : 2000 calnexin, and GluN2B (1 : 250, BD Biosciences) multiplexed with 1 : 250 calnexin. Supplier secondary antibodies were used as instructed (anti-rabbit, Protein Simple) for all protein targets with the exception of total GluN2B. In this case, supplier anti-mouse secondary was mixed 50 : 50 with anti-rabbit secondary to accommodate the two primary antisera. Each experimental group was equally represented in all plates. Analysis of the area under the curve of electropherogram peaks was performed with Compass software (Protein Simple) using the Dropped Lines analysis function (pGluN2A) or Gaussian distribution (all other proteins) of the luminol-peroxidase signal in the capillaries. Analysis was done by taking the normalized phosphoprotein (phosphor-protein target/corresponding multiplexed calnexin) to the total protein (also normalized to the corresponding multiplexed calnexin).
Statistical Analysis
All statistical analyses were performed using GraphPad PRISM 7 software (La Jolla, CA). For experiment one, two-way ANOVAs were performed followed by a priori planned Student-Neman-Keuls (SNK) pairwise comparison tests if significant main effects or interactions were observed. Grubb’s test was used to determine statistical outliers (n=1) that were removed from all analyses. Statistical significance was set at p<0.05. For experiment two, three-way ANOVA analyses were performed followed by a priori planned SNK tests. All data are graphically represented as the mean ±SEM.
Results
SFK Inhibition Blocks BDNF-Induced Suppression of Cocaine-Seeking
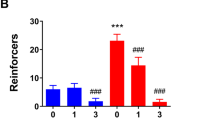
The timeline for Experiment 1 is depicted in Figure 1a. Cannula placements for Experiment 1 are shown in Figure 1c. Cocaine SA data during the final 12 days when all rats reached criterion are depicted in Supplementary Figure 1A. The starting number of rats was 48. Rats with placements outside the target region were excluded from mapping and analysis (n=6). Two rats were excluded due to lost head caps and four rats died due to surgical complications (final n=8–9/group). There were no significant differences in active lever presses among groups throughout cocaine SA (Supplementary Figure 1A) or during the last 3-days of cocaine SA, prior to i.c. infusions (F(1,29)=0.638, p=0.43, Figure 2a). Following cocaine SA for 12–14 days, rats received an i.c. infusion of either PP2 or PP3 followed by either BDNF or PBS. Following 6 days of home cage abstinence, a two-way ANOVA revealed a significant effect of i.c treatment #1 (PP2 or PP3) (F(1,29)=10.29, p=0.0033) and a significant effect of i.c. treatment #2 (PBS or BDNF) (F(1,29)=7.099, p=0.0125 Figure 2b) on non-reinforced active lever presses during the PA test. Planned SNK multiple comparison tests revealed that an i.c. infusion of BDNF significantly reduced active lever presses during the PA test when preceded by an infusion of PP3 (PP3-PBS vs PP3-BDNF). An infusion of PP2 prior to a BDNF infusion prevented the BDNF-induced reduction of active lever presses in comparison to PP3-infused rats (PP3-BDNF vs PP2-BDNF). In addition, rats that were infused with PP2 prior to BDNF showed no significant decrease in active lever presses in comparison to their PP2-PBS controls (PP2-PBS vs PP2-BDNF). Furthermore, there was no difference between the PP3-PBS and PP2-PBS groups, indicating that PP2 by itself did not alter cocaine- seeking in the PA test.

Self-administration, extinction, and relapse data. (a) There were no significant differences in active lever presses during SA among the rats that were subsequently assigned to one of four intracranial infusion groups. (b) Following 1 week of abstinence, rats that received PP3-BDNF pressed significantly less (**p<0.01) than rats that received PP3-PBS during a PA test. Rats that received PP2-BDNF pressed significantly more than animals that received PP3-BDNF (^^p<0.01). (c) Following the PA test (extinction day 1), rats that received PP3-BDNF pressed significantly less than other infusions groups on extinction day 2 only (^^vs. PP3-BDNF; ##vs. PP2-PBS; ***vs. PP3-PBS). (d) Following extinction to criteria, rats that received PP3-BDNF pressed significantly less (**p<0.01) than rats that received PP3-PBS. Rats that received PP2-BDNF pressed significantly more than animals that received PP3-BDNF (^^p<0.01).
There were no differences between groups during the last 2d of extinction as shown by a two-way ANOVA (F(1,29)=2.075, p=0.1604, Supplementary Figure S1C). Full extinction curves are shown in Figure 2c (active levers) and Supplementary Figure S1D (inactive levers). Following extinction, a two-way ANOVA revealed a significant interaction between i.c. treatment #1 (PP2 or PP3) and i.c. treatment #2 (PBS or BDNF) (F(1,29)=7.590. p=0.0100) in a cue-induced reinstatement test (Figure 2d). SNK multiple comparison tests revealed that there was a significant decrease in active lever pressing following BDNF treatment when preceded by PP3 (PP3-PBS vs PP3-BDNF). Infusion of PP2 prior to BDNF prevented the BDNF-induced reduction of active lever presses in the cue-induced reinstatement test (PP3-BDNF vs PP2-BDNF). There was no difference in the active lever presses between PP2-PBS vs PP2-BDNF and there was no difference between PP3-PBS and PP2-PBS, indicating that PP2 by itself when infused into PrL cortex immediately after cocaine SA had no effect on cue-induced reinstatement.
SFK Inhibition Prevents BDNF-Mediated Increases in Phosphoproteins in the PrL Cortex
In Experiment 2 (Figure 1b), animals were trained to self-administer cocaine as in Experiment 1 with added yoked-saline controls for all i.c. infusion groups (n=64). There were no differences between groups in lever pressing behavior during SA (Supplementary Figure S1B). Two rats were excluded due to failed catheters at the end of SA training, one rat was excluded because of a lost head cap, and eight rats were excluded owing to missed cannula placements. The experimental design estimated a higher surgical fail rate than was observed. Therefore, tissue from four rats (one per group) was not run for phosphoprotein analysis owing to a lack of space and ability to represent all eight experimental groups on one Wes plate. The final number per group was 6.
SFK inhibition blocks BDNF effects on pERK
A three-way, mixed factor ANOVA of pERK/tERK expression revealed no significant three-way interaction between saline vs cocaine, PP3 vsPP2, and PBS vsBDNF (F(1,40)=0.001, p=0.9762). There were main effects for all three variables tested (Saline vs Cocaine, PP3 vs PP2, PBS vs BDNF; p<0.0001). Because the simple main effect of i.v. drug was not complicated by higher level interactions, this main effect indicates that cocaine treatment significantly reduced ERK phosphorylation compared to yoked-saline controls (saline vs cocaine F(1,40)=32.36, p<0.0001). Pre-planned SNK multiple comparison tests (Figure 3) revealed that this main effect was driven by significant differences for all i.c. infusion groups PP3-PBS, PP2-PBS, PP3-BDNF by i.v. treatment (saline vs cocaine) except for PP2-BDNF. There was a significant interaction between PP3 vs PP2 and PBS vs BDNF (F(1,40)=32.36, p<0.0001) in addition to the main effects of each variable indicating that the change in BDNF’s effect compared with PBS depends on the pretreatment of PP3 or PP2. Pre-planned SNK multiple comparison tests (Figure 3) revealed that PP3 did not prevent BDNF-mediated induction of pERK (Saline-PP3-PBS vs Saline-PP3-BDNF and Cocaine-PP3-PBS vs Cocaine-PP3-BDNF). However, infusion of PP2 prior to BDNF blocked BDNF-mediated ERK phosphorylation in both the saline and the cocaine treatment groups (Saline-PP3-BDNF vs Saline-PP2-BDNF and Cocaine-PP3-BDNF vs Cocaine-PP2-BDNF). There were no significant BDNF-mediated increases in pERK in comparison with PBS controls when rats were pre-treated with PP2 (Saline-PP2-PBS vs Saline-PP2-BDNF and Cocaine-PP2-PBS vs Cocaine-PP2-BDNF). Finally, there was no effect of PP2 infusion alone on pERK expression, indicating that inhibition of SFKs alone is not sufficient to cause ERK dephosphorylation 2 h later (Saline-PP3-PBS vs Saline-PP2-PBS and Cocaine-PP3-PBS vs Cocaine-PP2-PBS). The ratio of total ERK to calnexin did not differ among groups (Supplementary Figure 2A).

Effect of PP2 infusion on BDNF’s ability to induce pERK in cocaine or yoked-saline groups. (a) Cocaine SA reduced pERK (PP3-PBS-cocaine vs PP3-PBS-saline, **p<0.01). In the presence of the SFK-inactive analogue, PP3, BDNF increased pERK in the yoked-saline group (PP3-PBS vs PP3-BDNF, ***p<0.001) and prevented the suppression of pERK in cocaine SA rats (PP3-PBS vs PP3-BDNF, ####p<0.0001). PP2 blocked BDNFs ability to increase pERK in yoked-saline rats (PP2-BDNF vs PP3-BDNF ^^^p<0.001) and in cocaine SA rats (PP2-BDNF vs PP3-BDNF $$p<0.01). pERK levels were significantly less in PP2-PBS rats treated with cocaine vs yoked-saline (PP2-PBS-Sal vs PP2-PBS-Coc ++p<0.01). (b) Representative electropherogram traces for each group are shown on the right of (a).
SFK inhibition blocks BDNF effects on pGluN2A
We examined the Y1325 phosphorylation site on the NMDA receptor subunit, GluN2A, because Y1325 phosphorylation increases NMDA channel open time probability (Cellerino et al, 1996; Taniguchi et al, 2009). Furthermore, we have previously observed that cocaine SA dephosphorylates Y1325-GluN2A (Go et al, 2016). A three-way ANOVA revealed no significant interaction between treatment #1 (Saline vs Cocaine) and treatment #2 (PP3 vs PP2) and treatment #3 (PBS vs BDNF) for pGluN2A (pGluN2a: F(1,40)=0.2828, p=0.5978). However, there were significant effects for all treatments. The significant main effect of i.v. drug (treatment #1) indicates that cocaine treatment significantly altered Y1325-GluN2A phosphorylation (treatment #1-Saline v Cocaine, F(1,40)=22.59, p<0.0001). Pre-planned SNK multiple comparison tests (Figure 4a) showed that an infusion of PP3 or PP2 when followed by PBS had no effect on cocaine-induced dephosphorylation of pGluN2A 2 h following the end of cocaine SA (Saline-PP3-PBS vs Cocaine-PP3-PBS and Saline-PP2-PBS vs Cocaine-PP2-PBS). However, there was no difference in BDNF-mediated GluN2A phosphorylation in the absence of PP2-mediated SFK inhibition between the i.v. drug groups (Saline-PP3-BDNF vs Cocaine-PP3-BDNF) which is consistent with previous analyses (Go et al, 2016). In the presence of PP2, there was no difference between i.v. drug groups infused with BDNF (Saline-PP2-BDNF vs Cocaine-PP2-BDNF). There was a significant interaction between treatment #2 (PP3 vs PP2) and #3 (PBS vs BDNF) (F(1,40)=17.15, p<0.0002). Pre-planned SNK multiple comparison tests showed that PP3 did not prevent BDNF-mediated induction of Y1325-pGluN2A in the yoked-saline (Saline-PP3-PBS vs Saline-PP3-BDNF) or cocaine self-administering (Cocaine-PP3-PBS vs Cocaine-PP3-BDNF) groups. However, infusion of PP2 blocked BDNF-mediated pGluN2A phosphorylation in both the yoked-saline and the cocaine treatment groups (Saline-PP3-BDNF vs Saline-PP2-BDNF and Cocaine-PP3-BDNF vs Cocaine-PP2-BDNF). There was no difference in pGluN2A levels when the rats were pre-treated with PP2 and BDNF vs PP2 and PBS in yoked-saline-treated rats (Saline-PP2-PBS vs Saline-PP2-BDNF) or in cocaine self-administering rats (Cocaine-PP2-PBS vs Cocaine-PP2-BDNF). Finally, there was no difference between rats infused with PP2 and PBS vs PP3 and PBS on pGluN2A expression in yoked-saline-treated rats (Saline-PP3-PBS vs Saline-PP2-PBS) or cocaine self-administering rats (Cocaine-PP3-PBS vs Cocaine-PP2-PBS), indicating that inhibition of SFKs immediately after cocaine SA does not cause pGluN2A dephosphorylation 2 h later. The ratio of total GluN2A to calnexin did not differ among groups (Supplementary Figure 2B).

Effect of PP2 infusion on BDNF’s ability to induce pGluN2A and pGluN2B in cocaine or yoked-saline groups. (a) Cocaine SA reduced pGluN2A (PP3-PBS-cocaine vs PP3-PBS-Saline, *p<0.05). In the presence of PP3, BDNF increased basal pGluN2A in the yoked-saline group (PP3-PBS vs PP3-BDNF, *p<0.05) and prevented the suppression of pGluN2A in cocaine SA rats (PP3-PBS vs PP3-BDNF, ####p<0.0001). PP2 blocked BDNFs ability to increase pGluN2A in yoked-saline rats (PP2-BDNF vs PP3-BDNF ^^p<0.01) and in cocaine SA rats (PP2-BDNF vs PP3-BDNF $$$$p<0.0001). pGluN2A levels were significantly less in PP2-PBS rats treated with cocaine vs yoked-saline (PP2-PBS-Sal vs PP2-PBS-Coc +p<0.05). (b) Representative electropherogram traces for each group are shown. (c) Cocaine SA reduced pGluN2B (PP3-PBS-cocaine vs PP3-PBS-saline, **p<0.01). In the presence of PP3, BDNF increased basal pGluN2B in the yoked-saline group (PP3-PBS vs PP3-BDNF, ***p<0.001) and prevented the suppression of pGluN2A in cocaine SA rats (PP3-PBS vs PP3-BDNF, ####p<0.0001). PP2 blocked BDNFs ability to increase pGluN2A in yoked-saline rats (PP2-BDNF vs PP3-BDNF ^^p<0.01) and in cocaine SA rats (PP2-BDNF vs PP3-BDNF $$$p<0.001). pGluN2B levels were significantly less in PP2-PBS rats treated with cocaine vs yoked-saline (PP2-PBS-Sal vs PP2-PBS-Coc +p<0.05). (d) Representative electropherogram traces for each group are shown.
SFK inhibition blocks BDNF effects on pGluN2B
The Y1472 phosphorylation site of the GluN2B subunit has been shown to suppress internalization of NMDA receptors and increase their surface expression (Carreño et al, 2011; Huang and McNamara, 2010). Also we have previously observed that this site is dysregulated 2 h after the end of cocaine SA (Go et al, 2016; Sun et al, 2013). A three-way ANOVA revealed no significant interaction between treatment #1 (saline vs cocaine), treatment #2 (PP3 vs PP2), and treatment #3 (PBS vs BDNF) for pGluN2B (pGluN2B: F(1,40)=0.3762, p=0.5431). However, there were significant main effects for all treatments. The significant main effect of saline vs cocaine indicates that cocaine treatment significantly altered GluN2B-Y1472 phosphorylation (treatment #1: F(1,1)=26.29, p<0.0001). A pre-planned SNK multiple comparison test (Figure 4c) showed that an infusion of PP3 or PP2 followed by a PBS infusion had no effect on cocaine-induced dephosphorylation of p-GluN2B in the PrL cortex 2 h after the end of cocaine SA (Saline-PP3-PBS vs Cocaine-PP3-PBS) or yoked-saline treatment (Saline-PP2-PBS vs Cocaine-PP2-PBS). There was no significant difference in pGluN2B levels between Saline-PP3-BDNF vs Cocaine-PP3-BDNF groups or the Saline-PP2-BDNF vs Cocaine-PP2-BDNF groups. There was a significant interaction between treatment #2 and #3 (PP3 vs PP2, PBS vs BDNF; F(1,40)=21.09, p<0.0001). A pre-planned SNK multiple comparison test (Figure 4c) showed PP3 did not prevent BDNF-mediated induction of pGluN2B in yoked-saline (Saline-PP3-PBS vs Saline-PP3-BDNF) or cocaine self-administering (Cocaine-PP3-PBS vs Cocaine-PP3-BDNF) groups. However, infusion of PP2 prior to BDNF blocked BDNF-mediated pGluN2B phosphorylation in both the saline and the cocaine treatment groups (Saline-PP3-BDNF vs Saline-PP2-BDNF and Cocaine-PP3-BDNF vs Cocaine-PP2-BDNF). There were no significant difference in pGluN2B levels when the rats were pre-treated with PP2 and BDNF vs PP2 and PBS in yoked-saline-treated rats (Saline-PP2-PBS vs Saline-PP2-BDNF) or in cocaine self-administering rats (Cocaine-PP2-PBS vs Cocaine-PP2-BDNF). Finally, there was no difference between rats infused with PP2 and PBS vs PP3 and PBS on pGluN2B levels in yoked-saline-treated rats (Saline-PP3-PBS vs Saline-PP2-PBS) or cocaine self-administering rats (Cocaine-PP3-PBS vs Cocaine-PP2-PBS), indicating that inhibition of SFKs immediately after cocaine SA does not cause pGluN2B dephosphorylation 2 h later. The ratio of total GluN2B to calnexin did not differ among groups (Supplementary Figure 2C).
Discussion
SFK Inhibition Prevents BDNF’s Suppressive Effects on Cocaine-Seeking
Intra-PrL infusion of a non-selective SFK inhibitor prior to BDNF blocked BDNF’s suppressive effects on cocaine-seeking. SFKs serve as points of convergence linking TrkB and NMDA receptors (Salter and Kalia, 2004). Binding of BDNF to TrkB activates SFKs, leading to tyrosine phosphorylation of GluN2A and GluN2B (Iwasaki et al, 1998; Tezuka et al, 1999; Goebel-Goody et al, 2009; Huang and McNamara, 2010). In addition, although the mechanism is unknown, SFK activation has a positive feedback on TrkB, fully activating it. Conversely, SFK inhibition reduces TrkB phosphorylation and glutamate release from cortical neurons (Huang and McNamara, 2010; Zhang et al, 2012). TrkB-activation of GluN2A and GluN2B in the PrL cortex is essential to BDNF’s suppressive effects on cocaine-seeking. The critical nature of this interaction was demonstrated after an intra-PrL infusion of a selective GluN2A or a GluN2B antagonist before a BDNF infusion blocks the suppressive effect of BDNF on subsequent cocaine-seeking. These data indicate that activation of GluN subunits is essential for BDNF-mediated behavioral effects, with SFKs as the likely link between the TrkB/NMDA receptor systems (Go et al, 2016). TrkB/SFK interactions also underlie acquisition of spatial memory in tasks mediated by NMDA receptors in the hippocampus (Mizuno et al, 2003). PP2-induced inhibition of SFKs in the dorsal hippocampus suppresses context-induced reinstatement of cocaine-seeking (Xie et al, 2013) and contributes to reconsolidation of cocaine-related memories (Wells et al, 2015), likely through mediation of GluN2B and GluN2A subunit phosphorylation, respectively. Similarly, inhibition of SFKs by PP2 infusion into the dorsal striatum (but not the ventral striatum) disrupted maladaptive alcohol-mediated GluN2B phosphorylation and reinstatement of alcohol-seeking (Wang et al, 2010). These studies indicate that SFKs contribute to drug-seeking in specific brain regions at the time of reinstatement. In contrast, our data indicate the importance of SFK activation in the PrL cortex during early withdrawal from cocaine SA on the ability of BDNF to suppress subsequent cocaine-seeking 1–3 weeks later. PP2 by itself had no effect on reinstatment when administered immediately after cocaine SA, likely because the GluN2A and GluN2B-containing NMDA receptors in the PrL cortex are dephosphorylated and inactivated by cocaine at that time (Go et al, 2016).
SFK Inhibition Blocks BDNF’s Ability to Prevent Cocaine-Induced ERK Dephosphorylation
BDNF’s suppressive effects on cocaine-seeking depend on both TrkB and ERK activation. Blocking ERK activation with a MEK inhibitor prevents BDNF’s attenuation of subsequent cocaine-seeking, indicating the importance of preventing cocaine-induced deactivation of ERK during early withdrawal (Whitfield et al, 2011). Our data indicate that BDNF-mediated phosphorylation of ERK depends on SFK activation. BDNF-induced TrkB activation results in dimerization and autophosphorylation of intracellular tyrosine residues in the receptor that results in activation of SFKs. PP2 also prevents glutamate-induced ERK phosphorylation in neuronal cultures; however, this inhibition was mediated indirectly by PKA (Poddar et al, 2016). We hypothesized that SFKs would be essential for BDNF-mediated suppression of cocaine-seeking primarily because early withdrawal from cocaine SA results in activation of STEP, a key regulator of SFK family members, SFK substrates, and ERK (Baum et al, 2010; Sun et al, 2013). STEP dephosphorylates Fyn, GluN2A-Y1325, and GluN2B-Y1472 (Tian et al, 2016). Further, inhibition of STEP activity with a relatively selective inhibitor, TC-2153, immediately after cocaine SA suppresses cocaine-seeking by activating ERK (Siemsen et al, 2017). These data indicate that BDNF-mediated ERK phosphorylation during early withdrawal is crucial for suppression of cocaine-seeking.
SFK Inhibition Blocks BDNF’s Ability to Prevent Cocaine-Induced GluN Dephosphorylation
By augmenting SFK-mediated tyrosine phosphorylation of GluN2A/B, BDNF-induced TrkB stimulation enhances NMDA receptor function (Iwasaki et al, 1998; Levine et al, 1998; Lin et al, 1999). The SFK, Fyn, phosphorylates GluN2B at Y1472, suppressing internalization and enhancing synaptic activity and LTP (Carreño et al, 2011; Huang and McNamara, 2010). In turn, GluN2B activation directly regulates the ERK MAP kinase pathway (Krapivinsky et al, 2003). During early withdrawal from cocaine SA, the GluN2B-Y1472 residue in the PrL cortex is dephosphorylated in comparison to yoked-saline controls. Preventing phosphorylation of this residue prior to a BDNF infusion blocks BDNF’s suppressive effects (Go et al, 2016). In addition, the SFK, Src, phosphorylates GluN2A at Y1325, potentiating NMDA receptor currents and increasing open channel probability (Taniguchi et al, 2009). Phosphorylation of this residue is also downregulated during early withdrawal from cocaine and blocking BDNF-induced phosphorylation of this receptor prevents BDNF’s suppressive effects (Go et al, 2016). Our data indicate that by nonselectively inhibiting SFKs, BDNF’s ability to induce phosphorylation of GluN subunits is inhibited. SFK activity is crucial because NMDA receptor subunit phosphorylation contributes to sustained ERK activation following TrKB activation (Matsumoto et al, 2008). One possible mechanism for this effect is release of Zn2+ from glutamatergic terminals that can induce ERK phosphorylation via NMDA receptors. This Zn2+ induced ERK activation is accompanied by a corresponding deactivation of STEP (Poddar et al, 2016). In addition, Zn2+ can stimulate the residues in TrkB that cause autophosphorylation resulting in activation of downstream pathways including ERK, an effect that is SFK-dependent (Huang and McNamara, 2010).
Taken together these data indicate that SFKs mediate mutual activation loops between NMDA receptors and TrkB receptors (Figure 5). BDNF-induced stimulation of TrkB activates SFKs. SFKs further activate TrKB and phosphorylate their associated substrates including NMDA receptors. NMDA receptor activation results in increased extracellular Zn2+ entry into the cell, causing further Zn2+ induced transactivation of TrkB and ERK phosphorylation, also resulting in deactivation of STEP. The deactivation of STEP, which is hyperactivated during early withdrawal from cocaine SA, results in less dephosphorylation of all of these key residues, specifically GluN2A, GluN2B, ERK, and Fyn. Which SFK family members are deregulated during early cocaine abstinence is a subject of future studies.

Proposed model of the crosstalk between TrkB and NMDARs in pyramidal neurons of the PrL cortex after cocaine self-administration in rats infused with PBS (a) or BDNF (b). (a) During early withdrawal from cocaine, the tyrosine phosphatase, STEP, is activated by dephosphorylation. STEP dephosphorylates and inactivates ERK and GluN2A/B. (b) BDNF binding to TrkB induces autophosphorylation of TrkB at Y705/Y706 and Y515, stimulating the ERK MAP kinase cascade. Autophosphorylated and activated TrkB phosphorylates and activates SFK and active SFK promotes further phosphorylation of TrkB. Activated SFKs phosphorylate GluN2A and GluN2B via binding to a scaffolding protein (ND 2.1 in the case of Src) that results in the upregulation of GluN2A- and GluN2B-containing NMDAR function at the synapse.
Funding and disclosure
This research was supported by P50 DA015369, RO1 DA033479, T32 DA007288, and F31 DA039709 (SMB). The authors declare no conflict of interest.
References
Baum ML, Kurup P, Xu J, Lombroso PJ (2010). A STEP forward in neural function and degeneration. Commun Integr Biol 3: 419–422.
Berglind WJ, See RE, Fuchs RA, Ghee SM, Whitfield TW Jr, Miller SW et al (2007). A BDNF infusion into the medial prefrontal cortex suppresses cocaine seeking in rats. Eur J Neurosci 26: 757–766.
Berglind WJ, Whitfield TW, LaLumiere RT, Kalivas PW, McGinty JF (2009). A Single intra-PFC infusion of BDNF prevents cocaine-induced alterations in extracellular glutamate within the nucleus accumbens. J Neurosci 29: 3715–3719.
Carreño FR, Walch JD, Dutta M, Nedungadi TP, Cunningham JT (2011). Brain-derived neurotrophic factor-tyrosine kinase B pathway mediates NMDA receptor NR2B subunit phosphorylation in the supraoptic nuclei following progressive dehydration. J Neuroendocrinol 23: 894–905.
Cellerino A, Maffei L, Domenici L (1996). The distribution of brain-derived neurotrophic factor and its receptor trkB in parvalbumin-containing neurons of the rat visual cortex. Eur J Neurosci 8: 1190–1197.
Go BS, Barry SM, McGinty JF (2016). Glutamatergic neurotransmissionin the prefrontal cortex mediatesthe suppressive effect of intra-prelimbic cortical infusion of BDNF on cocaine-seeking. Eur Neuropsychopharmacol 26: 1989–1999.
Goebel-Goody SM, Davies KD, Alvestad Linger RM, Freund RK, Browning MD (2009). Phospho-regulation of synaptics and extrasynaptic N-methyl-d-aspartate receptors in adult hippocampal slices. Neuroscience 158: 1446–1459.
Hanke JH, Gardner JP, Dow RL, Changelian PS, Brissette WH, Weringer EJ et al (1996). Discovery of a novel, potent, and Src family-selective tyrosine kinase inhibitor: Study of Lck- and FynT-dependent T cell activation. J Biol Chem 271: 695–701.
Huang YZ, McNamara JO (2010). Mutual regulation of Src family kinases and the neurotrophin receptor TrkB. J Biol Chem 285: 8207–8217.
Iwasaki Y, Gay B, Wada K, Koizumi S (1998). Association of the Src family tyrosine kinase Fyn with TrkB. J Neurochem 71: 106–111.
Krapivinsky G, Krapivinsky L, Manasian Y, Ivanov A, Tyzio R, Pellegrino C et al (2003). The NMDA receptor is coupled to the ERK pathway by a direct interaction between NR2B and RasGRF1. Neuron 40: 775–784.
Levine ES, Crozier RA, Black IB, Plummer MR (1998). Brain-derived neurotrophic factor modulates hippocampal synaptic transmission by increasing N-methyl-D-aspartic acid receptor activity. Proc Natl Acad Sci USA 95: 10235–10239.
Lin SY, Wu K, Len GW, Xu JL, Levine ES, Suen PC et al (1999). Brain-derived neurotrophic factor enhances association of protein tyrosine phosphatase PTP1D with the NMDA receptor subunit NR2B in the cortical postsynaptic density. Brain Res Mol Brain Res 70: 18–25.
Matsumoto T, Rauskolb S, Polack M, Klose J, Kolbeck R, Korte M et al (2008). Biosynthesis and processing of endogenous BDNF: CNS neurons store and secrete BDNF, not pro-BDNF. Nat Neurosci 11: 131–133.
McFarland K, Kalivas PW (2001). The circuitry mediating cocaine-induced reinstatement of drug-seeking behavior. J Neurosci 21: 8655–8663.
McFarland K, Lapish CC, Kalivas PW (2003). Prefrontal glutamate release into the core of the nucleus accumbens mediates cocaine-induced reinstatement of drug-seeking behavior. J Neurosci 23: 3531–3537.
Mizuno M, Yamada K, He J, Nakajima A, Nabeshima T (2003). Involvement of BDNF receptor TrkB in spatial memory formation. Learn Mem 10: 108–115.
Ohnishi H, Murata Y, Okazawa H, Matozaki T (2011). Src family kinases: modulators of neurotransmitter receptor function and behavior. Trends Neurosci 34: 629–637.
Poddar R, Rajagopal S, Shuttleworth CW, Paul S (2016). Zn2+-dependent activation of the Trk signaling pathway induces phosphorylation of the brain-enriched tyrosine phosphataseSTEP. J Biol Chem 291: 813–825.
Salter MW, Kalia LV (2004). Src kinases: a hub for NMDA receptorregulation. Nat Rev Neurosci 5: 317–328.
Siemson BM, Lombroso PJ, McGinty JF (2017). Intra-prelimbic cortical inhibition of striatal-enriched protein tyrosine phosphatase suppresses cocaine-seeking in rats. Addict Biol (doi:10.1111/adb.12504).
Sun WL, Zelek-Molik A, McGinty JF (2013). Short and long access to cocaine self-administration activates tyrosine phosphatase STEP and attenuates GluN expression but differentially regulates GluA expression in the prefrontal cortex. Psychopharmacology 229: 603–613.
Taniguchi S, Nakazawa T, Tanimura A, Kiyama Y, Tezuka T, Watabe AM et al (2009). Involvement of NMDAR2A tyrosine phosphorylation in depression-related behaviour. EMBO J 28: 3717–3729.
Tezuka T, Umemori H, Akiyama T, Nakanishi S, Yamamoto T (1999). PSD-95 promotes Fyn-mediated tyrosine phosphorylation of the N-methyl-D-aspartate receptor subunit NR2A. Proc Natl Acad Sci 96: 435–440.
Tian M, Xu J, Lei G, Lombroso PJ, Jackson MF, MacDonald JF (2016). STEP activation by Gaq coupled GPCRs opposes Src regulation of NMDA receptors containing the GluN2A subunit. Sci Rep 6: 36684.
Wang J, Carnicella S, Phamluong K, Jeanblanc J, Ronesi JA, Chaudhri N et al (2007). Ethanol induces long-term facilitation of NR2B-NMDA receptor activity in the dorsal striatum: implications for alcohol drinking behavior. J Neurosci 27: 3593–3602.
Wang J, Lanfranco MF, Gibb SL, Yowell QV, Carnicella S, Ron D (2010). Long-lasting adaptations of the NR2B-containing NMDA receptors in the dorsomedial striatum play a crucial role in alcohol consumption and relapse. J Neurosci 30: 10187–10198.
Wells AM, Xie X, Higginbotham JA, Arguello AA, Healey KL, Blanton M et al (2015). Contribution of an SFK-mediated signaling pathway in the dorsal hippocampus to cocaine-memory reconsolidation in rats. Neuropsychopharmacology 1–11.
Whitfield TW, Shi X, Sun WL, McGinty JF (2011). The suppressive effect of an intra-prefrontal cortical infusion of BDNF on cocaine-seeking is Trk receptor and extracellular signal-regulated protein kinase mitogen-activated protein kinase dependent. J Neurosci 31: 834–842.
Xie X, Arguello AA, Wells AM, Reittinger AM, Fuchs RA (2013).Role of a Hippocampal Src-family kinase-mediated glutamatergic mechanism in drug context-induced cocaine seeking. Neuropsychopharmacology 38: 2657–2665.
Zhang Z, Fan J, Ren Y, Zhou W, Yin G (2012). The release of glutamate from cortical neurons regulated by BDNF via the TrkB/Src/PLC-γ1 pathway. J Cell Biochem 114: 144–151.
Acknowledgements
We would like to thank Erica Herzig, Ben Siemsen, Stephen Saunier, and Kailey Ray for excellent technical work.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on the Neuropsychopharmacology website
Rights and permissions
About this article

Cite this article
Barry, S., McGinty, J. Role of Src Family Kinases in BDNF-Mediated Suppression of Cocaine-Seeking and Prevention of Cocaine-Induced ERK, GluN2A, and GluN2B Dephosphorylation in the Prelimbic Cortex. Neuropsychopharmacol 42, 1972–1980 (2017). https://doi.org/10.1038/npp.2017.114
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/npp.2017.114
This article is cited by
-
Cellular mechanisms of contextual fear memory reconsolidation: Role of hippocampal SFKs, TrkB receptors and GluN2B-containing NMDA receptors
Psychopharmacology (2024)
-
Inhibition of CSF1R, a receptor involved in microglia viability, alters behavioral and molecular changes induced by cocaine
Scientific Reports (2021)
-
The NMDA Receptor Subunit (GluN1 and GluN2A) Modulation Following Different Conditions of Cocaine Abstinence in Rat Brain Structures
Neurotoxicity Research (2021)
-
Biphasic effect of abstinence duration following cocaine self-administration on spine morphology and plasticity-related proteins in prelimbic cortical neurons projecting to the nucleus accumbens core
Brain Structure and Function (2019)
-
Mechanisms underlying the efficacy of exercise as an intervention for cocaine relapse: a focus on mGlu5 in the dorsal medial prefrontal cortex
Psychopharmacology (2019)

{kind=link}
{kind=link}