
Key Points
-
The recent failure of several high-profile clinical trials in sepsis has led to a rethinking of the fundamental pathophysiological basis for the disorder. Most patients with sepsis survive the initial few days of the disorder and progress to a more indolent and protracted disease.
-
Patients with unresolved, prolonged sepsis enter a phase of immunosuppression that is characterized by a failure to eradicate the primary infection, the acquisition of lethal secondary infections and/or the reactivation of numerous latent viruses.
-
The immunosuppressive phase of sepsis is multifactorial and features defects in both innate and adaptive immunity, including apoptotic depletion of immune effector cells, increased numbers of regulatory T cells, decreased expression of positive co-stimulatory molecules, increased expression of negative co-stimulatory molecules, increased numbers of myeloid-derived suppressor cells and T cell exhaustion.
-
Surprisingly, cancer and sepsis share many immune defects, and the remarkable success of new immunotherapeutics in cancer has highlighted the potential for this new approach in sepsis. In this regard, a programmed cell death 1 (PD1)-specific antibody has been highly effective in clinically relevant animal models of sepsis and has reversed sepsis-induced defects in patient blood cells in in vitro studies.
-
Effective therapeutic approaches to sepsis are likely to be individualized and determined by whether the patient is in the early hyperinflammatory phase of the disorder or has entered the more protracted immunosuppressive phase.
-
In patients with sepsis, the appropriate immune-boosting therapy is likely to be chosen on the basis of a number of clinical and laboratory parameters. Immunological phenotype analyses, functional assays (such as assays to determine tumour necrosis factor production by stimulated whole blood cells) and biomarkers will probably be useful in guiding new immunotherapies for sepsis.
Abstract
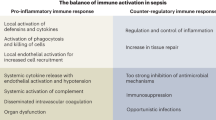
Sepsis — which is a severe life-threatening infection with organ dysfunction — initiates a complex interplay of host pro-inflammatory and anti-inflammatory processes. Sepsis can be considered a race to the death between the pathogens and the host immune system, and it is the proper balance between the often competing pro- and anti-inflammatory pathways that determines the fate of the individual. Although the field of sepsis research has witnessed the failure of many highly touted clinical trials, a better understanding of the pathophysiological basis of the disorder and the mechanisms responsible for the associated pro- and anti-inflammatory responses provides a novel approach for treating this highly lethal condition. Biomarker-guided immunotherapy that is administered to patients at the proper immune phase of sepsis is potentially a major advance in the treatment of sepsis and in the field of infectious disease.
Similar content being viewed by others
Main
Sepsis is defined as the host inflammatory response to severe, life-threatening infection with the presence of organ dysfunction1. Sepsis is the most frequent cause of mortality in most intensive care units (ICUs) and is responsible for more than 250,000 deaths in the United States annually2. The incidence of sepsis is increasing owing to the ageing population, who have impaired immunity as a result of immunosenescence2. Despite the litany of failed clinical trials in sepsis, a better understanding of different immunological phases of the disorder and encouraging results from several Phase II clinical trials of immunotherapies in sepsis have brought cautious optimism to the field3,4,5,6,7.
Until recently, most research on sepsis was focused on blocking the initial hyperinflammatory, cytokine-mediated phase of the disorder. Improved treatment protocols have resulted in most patients surviving this initial hyperinflammatory phase and entering a protracted immunosuppressive phase8,9,10,11,12,13. Deaths in this immunosuppressive phase are typically due to failure to control the primary infection or the acquisition of secondary hospital-acquired infections, often with opportunistic pathogens14,15. The recent remarkable success of cytotoxic T lymphocyte antigen 4 (CTLA4)- and programmed cell death 1 (PD1)-specific antibodies as immunotherapies to improve host immunity and to increase survival in cancer patients16,17 is highly encouraging because of the many similarities between the immune defects observed in cancer and those observed in sepsis and because both agents result in improved survival in animal models of sepsis7,10. In this Review, we discuss the panoply of sepsis-induced defects in innate and adaptive immune cells and highlight several promising immunotherapies for the treatment of sepsis.
Controversies over host immunity in sepsis
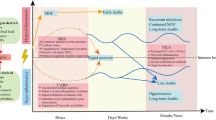
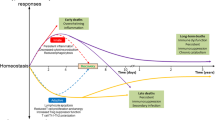
The current paradigm regarding the host immune response to sepsis is debated2,3,4,5,6,7,18,19. Traditionally, the host immune response to sepsis was considered to be characterized by an initial hyperinflammatory phase that evolved over several days into a more protracted immunosuppressive phase7,8,9 (theory 1). However, recent studies have shown that both pro-inflammatory and anti-inflammatory responses occur early and simultaneously in sepsis18,19,20 (Fig. 1a), although the net initial effect of these competing processes is typically manifested by an early, dominant, hyperinflammatory phase characterized by shock, fever and hypermetabolism. The robustness of the hyperinflammatory phase depends on numerous factors, including pre-existing co-morbidities, nutritional status, microorganism load and virulence factors8,9.

a | Theory 1: recent studies show that activation of both pro- and anti-inflammatory immune responses occurs promptly after sepsis onset. Cells of the innate immune system, including monocytes and neutrophils, release high levels of pro-inflammatory cytokines that drive inflammation (blue line; days 1–3). The intensity of the initial inflammatory response varies in individual patients depending on multiple factors, including pathogen load and virulence, patient co-morbidities and host genetic factors. Early deaths in sepsis (top red line; day 3) are typically due to a hyperinflammatory 'cytokine storm' response with fever, refractory shock, acidosis and hypercatabolism. An example of this scenario would be a young patient dying of toxic shock syndrome or meningococcaemia. Most patients have a restoration of innate and adaptive immunity and survive the infection (recovery; blue and green lines; day 6). If sepsis persists, the failure of crucial elements of both the innate and the adaptive immune systems occurs such that patients enter a marked immunosuppressive state (blue and red lines; after day 6). Deaths are due to an inability of the patient to clear primary infections and the development of secondary infections. b | Theory 2: a competing theory of sepsis agrees that there is an early activation of innate immunity and a suppression of adaptive immunity; however, this theory holds that deaths in sepsis are due to the persistent activation of innate immunity that results in intractable inflammation and organ injury. According to this theory, late deaths in sepsis are due to persistent, underlying innate immune-driven inflammation.
Investigators recently reported a new paradigm (Fig. 1b) to describe the host immune response in trauma and sepsis (theory 2). Circulating leukocyte gene expression data from people suffering from trauma and burns showed the rapid and sustained upregulation of genes that regulate the innate immune response and the simultaneous downregulation of genes that regulate the adaptive immune response19. These investigators hypothesized that the best model to describe the host immune response in trauma and sepsis is one of protracted, unabated inflammation driven by the innate immune system with resultant organ dysfunction and failure. Although these investigators agree that the adaptive immune system is impaired, they theorize that patients who die of sepsis have a longer duration and a greater degree of organ injury caused by unabated innate immune-driven inflammation than those who survive. They postulate that this inflammation persists despite the downregulation of the expression of genes that regulate the adaptive immune response and that it is ultimately responsible for patient morbidity and mortality19.
Although we agree with the provocative findings of this group, we believe that this new model — which proposes that morbidity and mortality in sepsis are due to unremitting innate immune-driven inflammation — is unlikely to reflect the actual clinical scenario in most patients. We have shown that patients who die of sepsis have marked immunosuppression12. Immune cells that were harvested from the spleen or lungs of patients with sepsis within 30 to 180 minutes of death showed markedly decreased production of both pro- and anti-inflammatory cytokines, upregulated expression of inhibitory receptors (including PD1), expansion of regulatory T (TReg) cell and myeloid-derived suppressor cell (MDSC) populations, and downregulation of CD28 and HLA-DR-mediated activation pathways12. Collectively, these results show that sepsis induces numerous overlapping mechanisms of immunosuppression involving both the innate and adaptive immune systems.
There are several potential explanations for the different findings in the two studies. First, the results of the gene expression study involved patients with trauma and burns19, and although many of these patients became septic, these patients differ from those who developed sepsis as a primary phenomenon. The mean age of the patients with trauma in the transcriptome study was 33 years, whereas the mean age of patients with sepsis in most developed countries is at least twice this age (65–68 years). This difference in age in the two studies is important because the older patients have underlying defects in immunity due to immunosenescence21. Second, these investigators based their conclusions on gene expression data that, because of post-transcriptional regulation, might not reflect protein levels. Conversely, the post-mortem study of patients with sepsis quantitated actual cytokine levels. Other possible reasons for differences between these two studies are the duration and severity of illness of the patients who were analysed. Many patients with sepsis had protracted disease with a high severity of illness and increased mortality.
We believe that immunosuppression rather than low-grade inflammation is the predominant driving force for morbidity and mortality in sepsis. First, our post-mortem findings are consistent with numerous studies that have examined cytokine production in peripheral-blood mononuclear cells and whole blood from patients with sepsis and have shown severely decreased pro-inflammatory cytokine production22,23,24,25. Second, recent post-mortem studies have reported that a high number of patients who die of sepsis have unresolved opportunistic infections14,15, which is consistent with defective host immunity as a dominant cause of death. Third, the failure of more than 30 clinical trials of different anti-inflammatory agents is inconsistent with the hypothesis that inflammation is a key driving mechanism. Undoubtedly, focal regions of inflammation occur in infected tissue in patients with sepsis. Areas of ischaemic and necrotic tissue are likely to contribute to local inflammation. It is possible that this focal inflammation contributes to organ dysfunction, morbidity and mortality in a subset of patients with sepsis. Nevertheless, we hypothesize that protracted sepsis is predominantly characterized by systemic immunosuppression leading to a failure to eradicate primary infections and to the acquisition of lethal secondary infections. Intriguingly, the gene expression data from children who develop sepsis show similarities to the data from adult patients with trauma-related sepsis: for example, the expression of genes that modulate innate immunity is upregulated, whereas the expression of genes that modulate adaptive immunity is downregulated26. Although these gene expression data would seem to favour theory 2, post-mortem studies of paediatric patients who die of sepsis have shown immunosuppressive features that are characteristic of those observed in adult patients with sepsis, who show a remarkable depletion of immune effector cells (see below). Hopefully, future studies will determine which of these two conflicting hypotheses is correct, as the resolution of this controversy has important therapeutic implications.
Apoptosis and immunosuppression
A fundamental discovery in the field of sepsis occurred when investigators showed that apoptosis causes the marked depletion of immune cells, including CD4+ and CD8+ T cells, B cells, follicular dendritic cells (follicular DCs) and interdigitating DCs, in various organs of patients dying of sepsis, leading to immunosuppression27,28,29,30,31 (Fig. 2). Sepsis-induced immune cell apoptosis has now been confirmed in several post-mortem studies; it affects all age groups (neonatal, paediatric and adult populations)27,30,31 and occurs in response to various microorganisms. Apoptosis of immune cells occurs in lymphoid tissues (spleen, thymus and lymph nodes) and gut-associated lymphoid tissues (GALTs)32. The loss in intestinal intraepithelial and lamina propria lymphocytes might facilitate bacterial translocation into the systemic circulation, thereby perpetuating the systemic inflammatory response and predisposing to secondary infections. Sepsis-induced apoptosis occurs through both death receptor- and mitochondrial-mediated pathways, suggesting that multiple cell death stimuli are activated during sepsis33. Therefore, it is unlikely that blockade of a single apoptotic trigger will prevent lymphocyte cell death during the disorder.

Spleens from patients with sepsis or patients who required a splenectomy for traumatic injury were obtained and underwent immunostaining for CD4+ T cells, CD8+ T cells or HLA-DR. Sepsis induces the apoptotic death of splenocytes, including CD4+ and CD8+ T cells, leading to marked depletion of these crucial immune effector cells. Note the decrease in numbers of periarteriolar CD4+ and CD8+ T cells (stained brown) in spleens from patients with sepsis compared with patients with trauma (original magnification ×200). The decrease in numbers of CD4+ T cells in patients with sepsis is frequently as severe as occurs in patients with AIDS. The number of HLA-DR+ cells (predominantly B cells) and the levels of HLA-DR expression (as determined by the intensity of the staining) are both decreased in patients with sepsis compared with patients with trauma (HLA-DR+ cells are stained brown). Tissue samples were stained for CD4, CD8 or HLA-DR. 3,3′-diaminobenzidine 4-HCl was used as a chromogen to stain the cells of interest (brown), and a hematoxylin counterstain (blue) was used for background staining. Images courtesy of P. Swanson, University of Washington School of Medicine in Seattle, Washington, USA.
A key question is whether the extensive sepsis-induced apoptosis of immune cells is an epiphenomenon or a major pathophysiological mechanism. Multiple independent laboratories have shown — through the use of various strategies, including transgenic and knockout mice, anti-apoptotic cytokines, caspase inhibitors and death receptor antagonists — that preventing lymphocyte apoptosis improves survival in sepsis34,35,36,37. The detrimental effects of apoptosis are not only related to the severe loss of immune cells but also the impact that apoptotic cell uptake has on the surviving immune cells38. Uptake of apoptotic cells by monocytes, macrophages and DCs results in immune tolerance by inducing anergy or a T helper 2 (TH2) cell-associated immune phenotype with increased interleukin-10 (IL-10) production39. The net result of this effect is that the surviving phagocytic cells cannot combat the remaining pathogenic organisms. Thus, sepsis-induced apoptosis has multiple important effects that impair host defences.
Impact of sepsis on immune cells
Sepsis directly or indirectly impairs the function of almost all types of immune cells. The following section provides an overview of these various immunosuppressive effects on the different cells of the innate and adaptive immune systems.
Neutrophils and MDSCs. Neutrophils are essential for the early control of invading pathogens40. Investigations on neutrophils obtained during the first hours of sepsis uncovered numerous abnormalities (Fig. 3). Most neutrophils normally undergo apoptosis within 24 hours after release from the bone marrow40. Surprisingly, and in contrast to lymphocytes, which undergo accelerated apoptosis, neutrophil apoptosis is delayed during sepsis40. Because of an increase in the release of immature neutrophils and the delayed apoptosis of circulating neutrophils, patients with sepsis typically have markedly increased numbers of circulating neutrophils of various degrees of maturation41. Animal models of sepsis and experimental studies in patients revealed disrupted neutrophil functions, including impaired clearance of bacteria, reduced production of reactive oxygen species (ROS) and decreased recruitment to infected tissues42,43. Loss of chemotactic activity is probably the most frequently documented dysfunction of circulating neutrophils during sepsis43,44. Possible explanations for this include nitric oxide-mediated suppression and reduced expression of CXC-chemokine receptor 2 (CXCR2)44. A current theory is that these alterations in neutrophil function are due to abnormalities in Toll-like receptor (TLR) signalling, which is analogous to the phenomenon that occurs in monocytes during endotoxin tolerance43 (see below).

a | Sepsis has diverse and important effects on all cellular elements that comprise the innate immune system. Sepsis rapidly triggers extensive apoptosis in follicular dendritic cells, dendritic cells, immature macrophages, natural killer (NK) cells and myeloid-derived suppressor cells (MDSCs). Conversely, sepsis delays neutrophil apoptosis — a result that is thought to be secondary to the mechanisms of neutrophil activation. After initial mobilization and activation of neutrophils, subsequent neutrophils that are released from the bone marrow have lower bactericidal functions and decreased cytokine production. Recent data show that a subset of neutrophils release large amounts of the immunosuppressive cytokine interleukin-10 (IL-10). Decreased HLA-DR expression on antigen-presenting cells, including monocytes, macrophages and dendritic cells, is a hallmark of sepsis, which may impair the optimal presentation of microbial antigens to T cells. b | Sepsis causes a marked loss of CD4+ and CD8+ T cells, as well as B cells. Regulatory T (TReg) cells are more resistant to sepsis-induced apoptosis and, consequently, there is an increased percentage of TReg cells in the circulation of patients with sepsis relative to the other lymphocyte subsets. This contributes to the formation of a more immunosuppressive phenotype. Surviving CD4+ and CD8+ T cells either shift from a pro-inflammatory T helper 1 (TH1) cell phenotype to an anti-inflammatory TH2 cell phenotype or develop an 'exhaustive' phenotype that is characterized by increased programmed cell death 1 expression and reduced cytokine secretion. CD4+ T cells have decreased expression of CD28 and reduced T cell receptor (TCR) diversity, which are both likely to contribute to the impaired antimicrobial response to invading pathogens.
Several studies have reported that impaired neutrophil function precedes the development of nosocomial infections. Patients with the most severely reduced neutrophil functions have the highest risk of acquiring nosocomial infections45. Reduced neutrophil function has also been reported in a mouse model of polymicrobial sepsis, in which the mice had an increased susceptibility to secondary infection with Pseudomonas aeruginosa and the subsequent development of pneumonia46. Furthermore, ten patients with septic shock who underwent extracorporeal cell therapy with donor granulocytes showed improvement in various biomarkers of sepsis, as well as decreased sepsis severity47.
Neutrophils show great plasticity in response to a wide range of physiological or pathological conditions43. Sepsis can induce the formation of a subset of neutrophils with suppressive properties. Although neutrophils are often not considered to produce large amounts of cytokines, it has been shown that they produce large amounts of the immunosuppressive cytokine IL-10 during sepsis48. Recently, a subset of mature CD16hiCD62L (also known as L-selectin)low neutrophils that suppressed T cell proliferation was identified in a human model of endotoxaemia in which healthy volunteers were administered with a low dose of endotoxin49. Immunosuppressive neutrophils were also detected in five patients suffering from severe trauma49.
The recently investigated heterogeneity in neutrophils, including immunosuppressive subsets of myeloid cells, offers parallels with MDSCs. In experimental models of sepsis, these cells have been shown to block specific T cell functions, including proliferation and production of interferon-γ (IFNγ) and IL-2 (Refs 50, 51). However, the actual effect of MDSCs might evolve during sepsis, as studies show that MDSCs can either enhance or attenuate the sepsis-associated inflammatory response, depending on the stage of the disorder50,51,52,53. Our group recently showed that there was an increase in the number of cells that were consistent with an MDSC phenotype in the lungs of patients who died of sepsis12. Currently, few clinical studies have investigated MDSCs in patients with sepsis, possibly owing to the complexity of immunophenotyping these cells and the absence of a universally accepted phenotypic definition for these cells in humans54.
DCs. DCs are particularly vulnerable to sepsis-induced apoptosis29. In a post-mortem study of patients with trauma or with sepsis, researchers found a marked reduction in the number of splenic DCs and the percentage area of the spleen that was occupied by DCs in the patients with sepsis compared with patients with trauma29. Similarly, reduced numbers of circulating DCs have been recorded in patients suffering from burns and subsequent sepsis and in patients with severe sepsis55,56,57. Both plasmacytoid and myeloid DCs are effected by sepsis-induced apoptosis55,58 (Fig. 3). Interestingly, DC loss was more apparent in patients with sepsis who died than in survivors55, and it was also more marked in patients who subsequently developed nosocomial infections than in those patients who did not59.
Not only are DC numbers decreased in patients with sepsis, but the surviving DCs also have lower expression of HLA-DR and produce increased levels of IL-10 (Refs 57, 60). In addition, monocyte-derived DCs from patients with sepsis were unable to induce a robust effector T cell response but instead induced either T cell anergy or TReg cell proliferation61. In mouse models of burn injury, DCs possess immunosuppressive properties that impair defences against a subsequent bacterial challenge. Several investigators have shown that preventing sepsis-induced DC apoptosis or improving DC function during the disorder results in enhanced survival62,63,64. Indeed, mice that had selective overexpression of the anti-apoptotic factor B cell lymphoma 2 (BCL-2) in DCs were reported to have improved survival in a lethal model of endotoxic shock62. Importantly, these BCL-2-overexpressing DCs were resistant to sepsis-induced apoptosis, which suggests that DC death is an important determinant of sepsis-induced immunosuppression and mortality.
FMS-related tyrosine kinase 3 ligand (FLT3L) is a DC growth factor that induces a rapid increase in DC numbers. Treatment of burn-injured animals with FLT3L restored DC function and enhanced survival when mice were challenged with P. aeruginosa, which is a pathogen that commonly infects patients with burns63,64,65. The protective effect of FLT3L was also observed following the adoptive transfer of FLT3L-treated DCs. Additional studies documented that treatment with FLT3L increased the secretion of IL-12, IL-15 and IFNγ and had broad effects on the function of CD4+ T cells, natural killer (NK) cells and neutrophils in models of burn infection63,64,65. Intrapulmonary transfer of bone marrow-derived DCs restored DC function and prevented fatal Aspergillus fumigatus infection in mice that had recovered from primary peritonitis66. TLR agonists have also been shown to improve survival from pneumonia occurring in mice after haemorrhagic shock67,68. Possible mechanisms for the beneficial effect of TLR agonists include the increased expression of MHC class II molecules and of the co-stimulatory receptors CD80 and CD86 on DCs67,68. Given these encouraging results, some authors have argued that preserving and/or restoring DC function should be a primary target of investigations into sepsis63,64,65,66,67,68,69.
Monocytes and macrophages. A hallmark of sepsis is the diminished capacity of monocytes to release pro-inflammatory cytokines in response to endotoxin (lipopolysaccharide (LPS)), other TLR agonists and various other bacterial compounds70,71 (Fig. 3). This finding is consistent with the phenomenon of endotoxin tolerance. Monocytes from patients with sepsis typically show a diminished ability to release pro-inflammatory cytokines such as tumour necrosis factor (TNF), IL-1α, IL-6 and IL-12, whereas their ability to release anti-inflammatory mediators, such as IL-1 receptor antagonist (IL-1RA) and IL-10, is either unimpaired or enhanced70,71. These findings show that LPS can still activate monocytes but that their intracellular signalling has shifted towards the production of anti-inflammatory molecules, thereby supporting the concept of monocyte reprogramming72. In clinical studies, the magnitude and the persistent nature of this refractory state is associated with increased mortality and nosocomial infections73.
The mechanisms responsible for endotoxin tolerance are not fully understood70,72. Analysis of mRNA expression levels has shown increased expression of genes encoding inhibitory signalling molecules and inhibitory cytokines and reduced expression of pro-inflammatory and chemokine receptor genes71,72,73,74. Although they have mainly been reported as useful biomarkers for predicting sepsis onset or prognosis, microRNAs are also thought to participate in endotoxin tolerance71. Similarly, recent work highlights a prominent role for epigenetic regulation at different levels, such as histone modification and chromatin remodelling75,76. These epigenetic modifications are restored in vitro after IFNγ stimulation and are associated with the recovery of monocyte cytokine release76,77. Thus, IFNγ might abrogate endotoxin tolerance by facilitating TLR-induced chromatin remodelling78.
Two major consequences of endotoxin tolerance on monocytes and macrophages are an increase in the release of immunosuppressive mediators (mainly IL-10) and a decrease in antigen presentation as a result of reduced expression of HLA-DR; both are associated with a worse outcome in sepsis22,79,80,81. Continued release of IL-10 might contribute to or amplify sepsis-induced immunosuppression and thus might augment susceptibility to secondary microbial infections82,83,84. Indeed, blocking IL-10 reverses endotoxin tolerance ex vivo83,85 and can reverse sepsis-induced immunosuppression and improve survival in a clinically relevant animal model of sepsis86.
Low levels of monocyte HLA-DR expression function as a surrogate marker of monocyte unresponsiveness79,87. Several studies showed an association of reduced monocyte HLA-DR expression with impaired monocyte function (for example, lower levels of TNF and IL-1β release from monocytes in response to bacterial challenges88 and decreased lymphocyte proliferation in response to tetanus toxin, presumably due to impaired antigen presentation89). Most importantly, decreased monocyte HLA-DR expression is associated with increased risk of nosocomial infections and death90,91. It is noteworthy that after adjustment for usual clinical confounders through multivariate analysis, decreased monocyte HLA-DR expression remains an independent predictor of nosocomial infection occurrence and mortality after sepsis73,92. This finding shows that monocyte unresponsiveness and immunosuppression independently contribute to the increased risk of adverse events in sepsis.
NK cells. The preferential location of NK cells in tissues along with their low numbers in peripheral blood render the study of this cell subset difficult, especially in the context of human sepsis. These facts explain why NK cells have not been heavily investigated in sepsis93,94. Studies indicate that sepsis affects both of the main human NK cell subsets (that is the CD56hi and CD56low NK cell subsets); in patients with sepsis the number of circulating NK cells is markedly decreased95,96, often for weeks97, and low numbers of NK cells are associated with increased mortality98. Furthermore, the absolute number of both NK cell subsets is decreased in sepsis99. Reduced NK cell cytotoxic function and cytokine secretion occur during sepsis and following burns and traumatic injuries in animal models and in patients96,100,101,102,103.
Cytokine production by NK cells in response to TLR agonists was impaired after polymicrobial sepsis104, which suggests that NK cells become tolerant to TLR agonists, and have features similar to those of endotoxin tolerance in monocytes105. Impaired IFNγ production in response to TLR agonists such as LPS and CpG has also been reported in NK cells from patients with sepsis99. As NK cells have a central role in antiviral defence, it is possible that impaired NK cell function could lead to the reactivation of latent viruses; this phenomenon has been frequently described in patients in ICUs106,107,108,109. Decreased IFNγ production by NK cells occurs in patients with sepsis and frequently precedes cytomegalovirus reactivation105 in patients with protracted sepsis96. In addition, defective NK cell production of IFNγ could be a factor in the increased incidence of secondary infections and the decreased monocyte HLA-DR expression that occur in septic patients110.
γδ T cells. γδ T cells are a distinct subset of lymphocytes that reside in large numbers in the intestinal mucosa and that possess qualities common to both innate and adaptive immune cells. Although the breadth of antigens to which γδ T cells respond is not completely known, it is clear that they recognize lipid antigens that are present on various invading pathogens111. γδ T cells reside in the intestines and on other mucosal surfaces, where they recognize invading pathogens and mount a prompt, innate-like immune response by releasing IFNγ, IL-17 and various chemokines. As such, γδ T cells can be considered to represent a first line of defence against particular pathogens112. The number of circulating γδ T cells is significantly decreased in patients with sepsis, and more severe depletion occurs in patients who have the greatest severity of illness and mortality112. The loss of γδ T cells in the intestinal mucosa might be particularly detrimental to the host because it might lead to the invasion of intestinal pathogens into the circulation or the peritoneal cavity, thereby causing secondary infections.
CD4+ TH cell subsets. Numerous studies have reported marked effects of sepsis on circulating and tissue T cells, with some of the most considerable effects occurring in CD4+ T cells113,114,115,116,117,118. Mature CD4+ TH cells have been characterized into TH1, TH2 and TH17 cell subsets on the basis of the type of cytokines that they produce on stimulation. Early studies suggested that both TH1 and TH2 cell-associated cytokine production are decreased during the initial immune response to sepsis and trauma119,120,121,122 (Fig. 3). Additional work showed marked reductions in the expression of T-bet and GATA-binding protein 3 (GATA3), which are transcription factors that modulate the TH1 and TH2 cell response, respectively, and this has reinforced the idea that both the TH1 and TH2 cell lineages are suppressed after trauma and sepsis123. These studies also showed that expression of the TReg cell-associated transcription factor forkhead box P3 (FOXP3) is not altered during sepsis, which supports the idea that regulatory functions are maintained or increased during sepsis, whereas effector T cell responses are downregulated (see below)95,122,124. Histone methylation and chromatin remodelling are thought to contribute to the suppression of TH1 and TH2 cell functions by acting at promoter regions of the IFNG and GATA3 genes125.
Although the TH17 cell subset has been less well studied than the TH1 and TH2 cell subsets, there is now general agreement that TH17 cells protect against extracellular bacterial and fungal infections by producing IL-17 and IL-22 (Refs 126, 127, 128, 129). The TH17 cell response is reduced in sepsis, possibly as a result of decreased expression of retinoic acid receptor-related orphan receptor-γt (RORγt), which is the transcription factor that is specific for TH17 cells95,123. This defect in the TH17 cell phenotype in sepsis is likely to be a contributing factor to the increased susceptibility of these patients to secondary fungal infections129. Indeed, mice that survived an initial polymicrobial bacterial infection (but not control mice) become highly susceptible to and rapidly succumb to pulmonary A. fumigatus infection66. Recently, IL-7 therapy has been shown to increase the TH17 cell response and to decrease the number of deaths from secondary infection with Candida albicans130,131.
T cell exhaustion. T cell exhaustion was first described in mice suffering from chronic viral infections and is typified by T cells that have severely impaired effector functions132. Subsequently, T cell exhaustion has been shown in patients with bacterial and parasitic infections, HIV and cancer133. The prolonged duration of sepsis is characterized by high antigen load and increased levels of pro- and anti-inflammatory cytokines, which is an ideal setting for the development of T cell exhaustion. A recent study12 in which spleens were obtained rapidly after the death of patients with sepsis showed evidence that is highly consistent with the occurrence of T cell exhaustion, including: profound suppression of the production of IFNγ and TNF by stimulated T cells; increased expression of PD1 on CD4+ T cells and of programmed cell death 1 ligand 1 (PDL1) on macrophages; and decreased T cell expression of CD127 (the IL-7R α-chain), which is another phenotypic feature of exhausted T cells. Furthermore, this study showed that capillary endothelial cells and bronchial epithelial cells in patients with sepsis had increased PDL1 expression, which in this setting can impair the function of T cells that have migrated to the local area of infection, thereby seriously compromising the ability of the host to eradicate the organisms. A potential causal link between T cell exhaustion and morbidity and mortality in sepsis was provided by studies showing that increased expression of PD1 on circulating T cells from patients with sepsis correlated with decreased T cell proliferative capacity, increased nosocomial infections, and mortality134. Finally, animal studies show that inhibition of the PD1–PDL1 interaction improves survival in several clinically relevant models of sepsis, which is consistent with a key role for T cell exhaustion in the pathogenesis of sepsis (see below)135,136,137.
TReg cells. An increased percentage of circulating TReg cells has been described in patients suffering from septic shock. This increase was observed immediately after the onset of sepsis but persisted only in those patients who subsequently died138. These results were further extended by showing that this relative increase was due to a decrease in effector T cell numbers rather than changes in the absolute numbers of TReg cells119. This finding suggests that TReg cells are more resistant to sepsis-induced apoptosis, possibly because of increased expression of the anti-apoptotic protein BCL-2. Alarmins, including heat shock proteins and histones, are increased in sepsis, and as strong inducers of TReg cells they are also likely to contribute to increased TReg cell numbers in sepsis139. Since this initial work, many groups have confirmed an increase in TReg cell numbers in the blood or the spleen in various animal models of and in patients suffering from trauma and sepsis114,140,141.
Initial studies investigating the role of TReg cells in sepsis-induced immune dysfunction in mice were inconclusive, possibly owing to the use of CD25-specific antibodies, which lack specificity in inhibiting TReg cells142. Recent studies indicate that increased TReg cells are deleterious in sepsis and are associated with decreased effector T cell proliferation and function141. Importantly, these suppressive effects were totally abrogated by using small interfering RNA (siRNA) specific for FOXP3 to block the differentiation of TReg cells141. Other investigators used glucocorticoid-induced TNF-receptor-related protein (GITR; also known as TNFRSF18)-specific antibodies to block TReg cells, which led to improved immune function in sepsis and enhanced microbial killing143. In addition, sepsis-induced immunosuppression was shown to facilitate rapid growth of solid tumours via a TReg cell-mediated effect144.
TReg cells can also suppress innate immune cells. In the context of LPS stimulation, TReg cells inhibit both monocyte and neutrophil function145. Furthermore, TReg cells inhibit IFNγ production by γδ T cells in response to Mycobacterium tuberculosis and precipitate an NK cell-dependent endotoxin tolerance-like phenomenon that is characterized by decreased production of IFNγ and granulocyte–macrophage colony-stimulating factor (GM-CSF)113,146,147. In summary, there is extensive evidence that patients with sepsis and trauma have increased numbers of TReg cells, which, by acting both on innate and adaptive immune cells, impair immunity and contribute to nosocomial infections and mortality.
Immunotherapies in sepsis
Recombinant human IL-7. Considering the severe quantitative and qualitative alterations in T cells that are induced by sepsis, IL-7 has recently emerged as a promising therapeutic agent for septic patients. IL-7 is essential for T cell development and function148. Its effects are mediated via the heterodimeric IL-7R, which is composed of the IL-7R α-chain (CD127) and the common cytokine receptor γ-chain (γc; CD132). This receptor is expressed by most resting human T cells: naive and central memory T cells express the highest levels and TReg cells express the lowest levels. IL-7 upregulates expression of the anti-apoptotic molecule BCL-2, induces the proliferation of peripheral T cells and sustains increased numbers of circulating blood CD4+ and CD8+ T cells. In addition, IL-7 increases the diversity of the T cell receptor (TCR) repertoire, is associated with a reduction in the proportion of TReg cells in the circulation, rejuvenates exhausted T cells by decreasing PD1 expression and increases the expression of cell adhesion molecules, thereby facilitating the trafficking of T cells to sites of infection149,150 (Fig. 4). Recently, a massive loss in diversity of the TCR repertoire was observed in patients with sepsis151. Importantly, this low TCR diversity was also associated with increased risk of death and nosocomial infections. An optimal diversity of the TCR repertoire greatly facilitates an effective immune response against invading pathogens, and this might be one of the most important benefits of IL-7. Thus, beyond supporting the replenishment of lymphocytes, which are markedly depleted during sepsis, IL-7 improves multiple functional aspects of T cells that are considerably altered in sepsis.

There are several immunotherapeutic agents that have shown promise in reversing the immunosuppressive phase of sepsis, including recombinant interleukin-7 (IL-7), programmed cell death 1 (PD1)- or programmed cell death 1 ligand 1 (PDL1)-specific antibodies, recombinant interferon-γ (IFNγ) and recombinant granulocyte–macrophage colony-stimulating factor (GM-CSF). GM-CSF and IFNγ primarily affect monocytes and macrophages to increase HLA-DR expression and to induce activation. IL-7 and PD1-specific antibodies have the advantage of targeting CD4+ and CD8+ T cells to restore the function of the adaptive immune system. By re-engaging CD4+ T cells, both recombinant IL-7 and PD1-specific antibodies will have effects not only on adaptive immune cells but also indirectly on monocytes and macrophages. PD1- and PDL1-specific antibodies will prevent and/or reverse T cell exhaustion. Thus, the net effects of these antibodies will be to prevent decreased IFNγ production, T cell apoptosis and decreased CD8+ T cell cytotoxicity. IL-7 will block T cell apoptosis, induce T cell proliferation, increase IFNγ production by T cells, increase T cell receptor (TCR) diversity and increase T cell trafficking by increasing the expression of cell integrins such as lymphocyte function-associated antigen 1 (LFA1). The application of immunotherapeutic agents will depend on the use of various biomarkers or tests of immune function to document that patients have entered the immunosuppressive phase of sepsis.
In clinical trials, recombinant IL-7 has been used to treat patients with idiopathic lymphopenia and with lymphopenia-driven diseases, including patients who are infected with HIV and have persistently low lymphocyte numbers despite effective antiretroviral therapy152. Clinical studies in over 250 patients have shown that IL-7 is safe and well tolerated148. The use of IL-7 in clinical trials of sepsis is supported by data from animal models of sepsis131,150,153 (Box 1). In mice with polymicrobial sepsis due to peritonitis, treatment with IL-7 improved T cell viability, trafficking and IFNγ production, and restored the delayed-type hypersensitivity response to recall antigens150. Similar beneficial effects of IL-7 were observed in a murine fungal sepsis model that reproduces the delayed secondary infections that frequently affect patients in ICUs130. Importantly, in these mouse experiments, there was also a more than twofold improvement in survival. The ability of IL-7 to reverse the defects in lymphocyte function that occur in patients with sepsis has been tested in an in vitro model154. Ex vivo IL-7 treatment of T cells from patients with sepsis corrected sepsis-induced defects, including CD4+ and CD8+ T cell proliferation abnormalities, decreased IFNγ production, impaired phosphorylation of signal transducer and activator of transcription 5 (STAT5) and decreased BCL-2 levels154. These data indicate that the IL-7 signalling pathway remains fully operative during sepsis and that IL-7 reverses crucial sepsis-induced defects in T cell function.
PD1- and PDL1-specific antibodies. Another approach that holds great potential in reversing immunosuppression in sepsis involves blockade of the co-inhibitory molecules PD1 and PDL1 (Fig. 4). As discussed previously, PD1 and PDL1 are widely expressed on immune effector cells, endothelial cells and bronchial epithelial cells in patients with sepsis12. Blockade of PD1–PDL1 signalling improves survival in clinically relevant animal models of bacterial sepsis135,136,137 and markedly decreases mortality in primary and secondary fungal sepsis caused by C . albicans155. Thus, PD1 expressed by lymphocytes or PDL1 expressed by monocytes could function as potential biomarkers for the selection of patients with sepsis as candidates for therapy. A recent important study showed that in vitro blockade of PD1 improved IFNγ production and decreased apoptosis of T cells from patients with active M. tuberculosis infections156. A second major finding in this study was that when patients with tuberculosis were effectively treated, the number of PD1-expressing T cells decreased and inversely correlated with the IFNγ T cell response against M. tuberculosis. We believe that this work has major implications for the broader field of sepsis because of the similarities between active tuberculosis and protracted sepsis.
IFNγ. A key immunological defect in sepsis is decreased production of IFNγ, which is a cytokine that is essential for the activation of innate immunity (Fig. 4). Small clinical trials using recombinant IFNγ have been carried out in sepsis. Treatment with recombinant IFNγ reversed monocyte dysfunction in patients with sepsis whose monocytes had decreased HLA-DR expression and produced reduced amounts of TNF in response to LPS. Out of nine patients with severe sepsis who were treated with IFNγ to improve monocyte function, eight survived157. Most recently, IFNγ treatment in a patient with protracted Staphylococcus aureus sepsis caused an increase in monocyte HLA-DR expression and in the numbers of IL-17-expressing CD4+ T cells, and led to the eradication of the bacteria158. Furthermore, IFNγ is effective in treating fungal sepsis in patients with chronic granulomatous disease159. Finally, in a recent trial, patients with HIV who had cryptococcal meningitis and who were treated with IFNγ showed enhanced clearing of fungi from the cerebrospinal fluid compared to control individuals160. Thus, IFNγ might be effective if targeted to patients with sepsis who have entered the immunosuppressive phase, as identified by decreased monocyte HLA-DR expression and/or the development of fungal sepsis. A clinical trial of IFNγ is currently underway in the Netherlands in patients with sepsis who have been determined to have entered the immunosuppressive phase of the disorder (ClinicalTrials.gov number: NCT01649921).
Although IFNγ offers real promise as a potential immunotherapy in sepsis because of its ability to restore monocyte function, it will not correct the fundamental defect in T cells that is a major pathological abnormality in sepsis. In the authors' opinion, recombinant IL-7 and PD1-specific antibodies are more likely to be of benefit to patients with sepsis because of their favourable effects on the functions of CD4+ and CD8+ T cells: they both restore IFNγ production in sepsis and have many other beneficial effects on various other immune effector cells.
G-CSF and GM-CSF. Given the many defects in neutrophil function that occur in sepsis, investigators carried out two randomized clinical trials using recombinant granulocyte colony-stimulating factor (G-CSF), which is a drug that increases neutrophil numbers and function, in patients with sepsis resulting from hospital-acquired and community-acquired pneumonia80,81. G-CSF had already been shown to be highly beneficial in decreasing the incidence of sepsis in patients who have abnormally low absolute neutrophil numbers owing to chemotherapy or radiation therapy. Although neutrophil numbers are typically increased in sepsis, investigators postulated that administration of G-CSF to further increase neutrophil numbers might improve pathogen killing. Although white blood cell numbers increased in these patients, there was no effect on overall survival161,162. These two clinical studies indicate that it is unlikely that drugs that enhance neutrophil numbers or function will be of benefit in patients with non-neutropenia-associated sepsis.
Another immunotherapy that is being actively investigated in sepsis is the administration of GM-CSF, which is a cytokine that accelerates the production of neutrophils, monocytes and macrophages11 (Fig. 4). Treatment of ventilator-dependent patients with sepsis who had entered the immunosuppressive phase of the disorder, as identified by persistent decreases in monocyte HLA-DR expression, with recombinant GM-CSF, resulted in the restoration of HLA-DR expression, fewer days on the ventilator and fewer days in the ICU163. In addition, treatment of immunosuppressed paediatric patients with sepsis with recombinant GM-CSF restored TNF production and reduced the acquisition of nosocomial infections11.
Biomarker-guided therapy. A prerequisite for the application of immunotherapy in sepsis is the proper selection of patients. Various biomarkers should help in deciphering whether the patient is in the hyperinflammatory versus the hypoinflammatory phase of the disorder (Fig. 1). Indeed, immunotherapy could worsen the outcome by causing an over-exuberant inflammatory response if applied during the wrong phase of the disorder. Readers are referred to numerous recent reviews that discuss the use of various biomarkers in sepsis91,164,165. Box 2 provides a list of clinical and laboratory findings that could be used to identify immunosuppressed patients who might benefit from immunotherapies. Potential biomarkers include some currently available parameters, such as decreased monocyte HLA-DR expression and increased circulating IL-10 concentrations (both of which assess innate immune function and can be used to stratify patients for GM-CSF or IFNγ treatments), and decreased absolute CD4+ T cell numbers and increased percentage of TReg cells (both of which assess adaptive immunity and can be used to stratify patients for IL-7 therapy). Ideally, ex vivo functional testing of circulating immune cells remains the gold standard for the evaluation of immunity because it directly measures the capacity of cells to respond to pathogens.
Two hallmarks of sepsis-induced immunosuppression have been identified: the decreased capacity of monocytes to produce pro-inflammatory cytokines (mainly TNF) in response to endotoxin challenge; and decreased lymphocyte proliferation. Currently, these two characteristics are thought to provide a reasonable assessment of immune effector cell status but remain barely usable in routine clinical monitoring owing to methodological challenges, such as long incubation time, lengthy cell purification procedures and low standardization. A major challenge is to develop automated or standardized biomarkers that can be used in routine daily practice. More precise cellular monitoring might be possible through the use of innovative therapies, such as monoclonal antibody-based therapies targeting PD1- and CTLA4-expressing cells. Quantifying the mRNA for a panel of candidate genes in whole blood might enable clinicians to identify immunosuppressed patients and to provide the appropriate treatment. In the authors' opinion, the ideal method for identifying immunosuppressed patients would be a combination of cell phenotypic assays (for example, measuring HLA-DR or PD1 expression), functional assays (for example, measuring TNF production in whole blood) and genomic assays166.
Conclusions
The current understanding of the immune response to sepsis is controversial — some investigators suggest that sepsis-induced morbidity and mortality is the result of persistent immune activation with accompanying inflammation, whereas others assert that it is the result of immunosuppression. We support the idea that sepsis-induced immunosuppression with T cell exhaustion is a major abnormality. As discussed in this Review, sepsis induces numerous defects in host innate and adaptive immunity such that, if the invading organisms are not promptly eliminated, the host becomes more susceptible to intractable infection or to new secondary infections. Methods for identifying when patients have entered an immunosuppressive phase of sepsis and for detecting particular defects in immunity will enable the application of potent new immunotherapies that have shown great promise in animal models of sepsis and in early clinical studies of various infectious disorders. In particular, recombinant IL-7 and PD1-specific antibodies reverse fundamental immune defects in sepsis, lead to improved survival in multiple clinically relevant animal models of sepsis, are clinically well tolerated and have an ability to improve immunity in patients with cancer and chronic viral infections. Immunotherapy could therefore represent the next major advance in the treatment of infectious disease and sepsis.
References
Vincent, J. L., Opal, S. M., Marshall, J. C. & Tracey, K. J. Sepsis definitions: time for change. Lancet 381, 774–775 (2013).
Martin, G. S., Mannino, D. M. & Moss, M. The effect of age on the development and outcome of adult sepsis. Crit. Care Med. 34, 15–21 (2006).
Cohen, J., Opal, S. & Calandra, T. Sepsis studies need new direction. Lancet Infect. Dis. 12, 503–505 (2012).
Wenzel, R. P. & Edmond, M. B. Septic shock--evaluating another failed treatment. N. Engl. J. Med. 366, 2122–2124 (2012).
Ward, P. A. New approaches to the study of sepsis. EMBO Mol. Med. 4, 1234–1243 (2012).
Angus, D. C. The search for effective therapy for sepsis: back to the drawing board? JAMA 306, 2614–2615 (2011).
Hotchkiss, R. S., Monneret, G. & Payen, D. Immunosuppression in sepsis: a novel understanding of the disorder and a new therapeutic approach. Lancet Infect. Dis. 13, 260–268 (2013).
Hotchkiss, R. S. & Karl, I. E. The pathophysiology and treatment of sepsis. N. Engl. J. Med. 348, 138–150 (2003). This review article presents one of the first conceptual theories for a new understanding of the immunological basis of sepsis and points to potential new therapeutic approaches based on the immunological phase of sepsis.
Hotchkiss, R. S. & Opal, S. Immunotherapy for sepsis — a new approach against an ancient foe. N. Engl. J. Med. 363, 87–89 (2010).
Payen, D., Monneret, G. & Hotchkiss, R. Immunotherapy — a potential new way forward in the treatment of sepsis. Crit. Care 17, 118 (2013).
Hall, M. W. et al. Immunoparalysis and nosocomial infection in children with multiple organ dysfunction syndrome. Intensive Care Med. 37, 525–532 (2011).
Boomer, J. S. et al. Immunosuppression in patients who die of sepsis and multiple organ failure. JAMA 306, 2594–2605 (2011). This study is the first to document considerable defects in immunity in tissues from patients dying of sepsis. It determines that T cell exhaustion is an important mechanism of immunosuppression in this disorder.
Ward, P. A. Immunosuppression in sepsis. JAMA 306, 2618–2619 (2011).
Torgersen, C. et al. Macroscopic postmortem findings in 235 surgical intensive care patients with sepsis. Anesth. Analg. 108, 1841–1847 (2009).
Otto, G. P. et al. The late phase of sepsis is characterized by an increased microbiological burden and death rate. Crit. Care 15, R183 (2011).
Hodi, F. S. et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 363, 711–723 (2010).
Topalian, S. L. et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N. Engl. J. Med. 366, 2443–2454 (2012). This study confirms that immunotherapy represents a major breakthrough in the treatment of cancer, a disorder that shares many immune defects with sepsis.
Munford, R. S. & Pugin, J. Normal responses to injury prevent systemic inflammation and can be immunosuppressive. Am. J. Respir. Crit. Care Med. 163, 316–321 (2001). One of the first studies to demonstrate that both pro-inflammatory and anti-inflammatory processes occur rapidly after sepsis.
Xiao, W. et al. A genomic storm in critically injured humans. J. Exp. Med. 208, 2581–2590 (2011).
Stearns-Kurosawa, D. J., Osuchowski, M. F., Valentine, C., Kurosawa, S. & Remick, D. G. The pathogenesis of sepsis. Annu. Rev. Pathol. 6, 19–48 (2011).
Geiger, H., de Haan, G. & Florian, M. C. The ageing haematopoietic stem cell compartment. Nature Rev. Immunol. 13, 376–389 (2013).
van Dissel, J. T., van Langevelde, P., Westendorp, R. G., Kwappenberg, K. & Frölich, M. Anti-inflammatory cytokine profile and mortality in febrile patients. Lancet 351, 950–953 (1998).
Ertel, W. et al. Downregulation of proinflammatory cytokine release in whole blood from septic patients. Blood 85, 1341–1347 (1995).
Munoz, C. et al. Dysregulation of in vitro cytokine production by monocytes during sepsis. J. Clin. Invest. 88, 1747–1754 (1991).
Rigato, O. & Salomao, R. Impaired production of interferon-γ and tumor necrosis factor-α but not of interleukin 10 in whole blood of patients with sepsis. Shock 19, 113–116 (2003).
Wong, H. R. Genome-wide expression profiling in pediatric septic shock. Pediatr. Res. 73, 564–569 (2013).
Hotchkiss, R. S. et al. Apoptotic cell death in patients with sepsis, shock, and multiple organ dysfunction. Crit. Care Med. 27, 1230–1251 (1999). This study was the first to demonstrate that apoptosis causes massive death and depletion of immune effector cells in patients with sepsis.
Hotchkiss, R. S. et al. Sepsis-induced apoptosis causes progressive profound depletion of B and CD4+ T lymphocytes in humans. J. Immunol. 166, 6952–6963 (2001).
Hotchkiss, R. S. et al. Depletion of dendritic cells, but not macrophages, in patients with sepsis. J. Immunol. 168, 2493–2500 (2002).
Felmet, K. A., Hall, M. W., Clark, R. S., Jaffe, R. & Carcillo, J. A. Prolonged lymphopenia, lymphoid depletion, and hypoprolactinemia in children with nosocomial sepsis and multiple organ failure. J. Immunol. 174, 3765–3772 (2005).
Toti, P. et al. Spleen depletion in neonatal sepsis and chorioamnionitis. Am. J. Clin. Pathol. 122, 765–771 (2004).
Hotchkiss, R. S. et al. Rapid onset of intestinal epithelial and lymphocyte apoptotic cell death in patients with trauma and shock. Crit. Care Med. 28, 3207–3217 (2000).
Chang, K. C. et al. Multiple triggers of cell death in sepsis: death receptor and mitochondrial-mediated apoptosis. FASEB J. 21, 708–719 (2007).
Hotchkiss, R. S. et al. Overexpression of Bcl-2 in transgenic mice decreases apoptosis and improves survival in sepsis. J. Immunol. 162, 4148–4156 (1999).
Chung, C. S. et al. Inhibition of Fas/Fas ligand signaling improves septic survival: differential effects on macrophage apoptotic and functional capacity. J. Leukoc. Biol. 74, 344–351 (2003).
Oberholzer, C. et al. Targeted adenovirus-induced expression of IL-10 decreases thymic apoptosis and improves survival in murine sepsis. Proc. Natl Acad. Sci. USA 98, 11503–11508 (2001).
Iwata, A. et al. Over-expression of Bcl-2 provides protection in septic mice by a trans effect. J. Immunol. 171, 3136–3141 (2003).
Voll, R. E. et al. Immunosuppressive effects of apoptotic cells. Nature 390, 350–351 (1997).
Green, D. R. & Beere, H. M. Apoptosis. Gone but not forgotten. Nature 405, 28–29 (2000).
Tamayo, E. et al. Evolution of neutrophil apoptosis in septic shock survivors and nonsurvivors. J. Crit. Care 27, 415.e1–415.e11 (2012).
Drifte, G., Dunn-Siegrist, I., Tissieres, P. & Pugin, J. Innate immune functions of immature neutrophils in patients with sepsis and severe systemic inflammatory response syndrome. Crit. Care Med. 41, 820–832 (2013).
Alves-Filho, J. C., Spiller, F. & Cunha, F. Q. Neutrophil paralysis in sepsis. Shock 34 (Suppl. 1), 15–21 (2010).
Kovach, M. A. & Standiford, T. J. The function of neutrophils in sepsis. Curr. Opin. Infect. Dis. 25, 321–327 (2012).
Cummings, C. J. et al. Expression and function of the chemokine receptors CXCR1 and CXCR2 in sepsis. J. Immunol. 162, 2341–2346 (1999).
Stephan, F. et al. Impairment of polymorphonuclear neutrophil functions precedes nosocomial infections in critically ill patients. Crit. Care Med. 30, 315–322 (2002).
Delano, M. J. et al. Sepsis induces early alterations in innate immunity that impact mortality to secondary infection. J. Immunol. 186, 195–202 (2011).
Altrichter, J. et al. Extracorporeal cell therapy of septic shock patients with donor granulocytes: a pilot study. Crit. Care 15, R82 (2011).
Kasten, K. R., Muenzer, J. T. & Caldwell, C. C. Neutrophils are significant producers of IL-10 during sepsis. Biochem. Biophys. Res. Commun. 393, 28–31 (2010).
Pillay, J. et al. A subset of neutrophils in human systemic inflammation inhibits T cell responses through Mac-1. J. Clin. Invest. 122, 327–336 (2012).
Delano, M. J. et al. MyD88-dependent expansion of an immature GR-1+CD11b+ population induces T cell suppression and Th2 polarization in sepsis. J. Exp. Med. 204, 1463–1474 (2007). This study identified MDSCs as important mediators of immunosuppression in sepsis.
Makarenkova, V. P., Bansal, V., Matta, B. M., Perez, L. A. & Ochoa, J. B. CD11b+/Gr-1+ myeloid suppressor cells cause T cell dysfunction after traumatic stress. J. Immunol. 176, 2085–2094 (2006).
Cuenca, A. G. et al. A paradoxical role for myeloid-derived suppressor cells in sepsis and trauma. Mol. Med. 17, 281–292 (2011).
Brudecki, L., Ferguson, D. A., McCall, C. E. & El Gazzar, M. Myeloid-derived suppressor cells evolve during sepsis and can enhance or attenuate the systemic inflammatory response. Infect. Immun. 80, 2026–2034 (2012).
Dumitru, C. A., Moses, K., Trellakis, S., Lang, S. & Brandau, S. Neutrophils and granulocytic myeloid-derived suppressor cells: immunophenotyping, cell biology and clinical relevance in human oncology. Cancer Immunol. Immunother. 61, 1155–1167 (2012).
Guisset, O. et al. Decrease in circulating dendritic cells predicts fatal outcome in septic shock. Intensive Care Med. 33, 148–152 (2007).
Riccardi, F. et al. Flow cytometric analysis of peripheral blood dendritic cells in patients with severe sepsis. Cytometry B Clin. Cytom. 80, 14–21 (2011).
Poehlmann, H., Schefold, J. C., Zuckermann-Becker, H., Volk, H. D. & Meisel, C. Phenotype changes and impaired function of dendritic cell subsets in patients with sepsis: a prospective observational analysis. Crit. Care 13, R119 (2009).
Dreschler, K. et al. Altered phenotype of blood dendritic cells in patients with acute pneumonia. Respiration 83, 209–217 (2012).
Grimaldi, D. et al. Profound and persistent decrease of circulating dendritic cells is associated with ICU-acquired infection in patients with septic shock. Intensive Care Med. 37, 1438–1446 (2011).
Pastille, E. et al. Modulation of dendritic cell differentiation in the bone marrow mediates sustained immunosuppression after polymicrobial sepsis. J. Immunol. 186, 977–986 (2011).
Faivre, V. et al. Human monocytes differentiate into dendritic cells subsets that induce anergic and regulatory T cells in sepsis. PLoS ONE 7, e47209 (2012).
Gautier, E. L. et al. Enhanced dendritic cell survival attenuates lipopolysaccharide-induced immunosuppression and increases resistance to lethal endotoxic shock. J. Immunol. 180, 6941–6946 (2008).
Bohannon, J., Cui, W., Sherwood, E. & Toliver-Kinsky, T. Dendritic cell modification of neutrophil responses to infection after burn injury. J. Immunol. 185, 2847–2853 (2010).
Toliver-Kinsky, T. E., Cui, W., Murphey, E. D., Lin, C. & Sherwood, E. R. Enhancement of dendritic cell production by fms-like tyrosine kinase-3 ligand increases the resistance of mice to a burn wound infection. J. Immunol. 174, 404–410 (2005).
Toliver-Kinsky, T. E., Lin, C. Y., Herndon, D. N. & Sherwood, E. R. Stimulation of hematopoiesis by the Fms-like tyrosine kinase 3 ligand restores bacterial induction of Th1 cytokines in thermally injured mice. Infect. Immun. 71, 3058–3067 (2003).
Benjamim, C. F., Hogaboam, C. M., Lukacs, N. W. & Kunkel, S. L. Septic mice are susceptible to pulmonary aspergillosis. Am. J. Pathol. 163, 2605–2617 (2003).
Benjamim, C. F., Lundy, S. K., Lukacs, N. W., Hogaboam, C. M. & Kunkel, S. L. Reversal of long-term sepsis-induced immunosuppression by dendritic cells. Blood 105, 3588–3595 (2005).
Roquilly, A. et al. TLR-4 agonist in post-haemorrhage pneumonia: role of dendritic and natural killer cells. Eur. Respir. J. http://dx.doi.org/10.1183/09031936.00152612 (2013).
Scumpia, P. O. et al. CD11c+ dendritic cells are required for survival in murine polymicrobial sepsis. J. Immunol. 175, 3282–3286 (2005).
Cavaillon, J. M. & Adib-Conquy, M. Bench-to-bedside review: endotoxin tolerance as a model of leukocyte reprogramming in sepsis. Crit. Care 10, 233 (2006).
Biswas, S. K. & Lopez-Collazo, E. Endotoxin tolerance: new mechanisms, molecules and clinical significance. Trends Immunol. 30, 475–487 (2009).
Zhang, X. & Morrison, D. C. Lipopolysaccharide structure-function relationship in activation versus reprogramming of mouse peritoneal macrophages. J. Leukoc. Biol. 54, 444–450 (1993).
Monneret, G. et al. Persisting low monocyte human leukocyte antigen-DR expression predicts mortality in septic shock. Intensive Care Med. 32, 1175–1183 (2006).
Rossato, M. et al. IL-10-induced microRNA-187 negatively regulates TNF-α, IL-6, and IL-12p40 production in TLR4-stimulated monocytes. Proc. Natl Acad. Sci. USA 109, E3101–E3110 (2012).
Ishii, M. et al. Epigenetic regulation of the alternatively activated macrophage phenotype. Blood 114, 3244–3254 (2009).
Carson, W. F., Cavassani, K. A., Dou, Y. & Kunkel, S. L. Epigenetic regulation of immune cell functions during post-septic immunosuppression. Epigenetics 6, 273–283 (2011).
Turrel-Davin, F. et al. mRNA-based approach to monitor recombinant gamma-interferon restoration of LPS-induced endotoxin tolerance. Crit. Care 15, R252 (2011).
Chen, J. & Ivashkiv, L. B. IFN-γ abrogates endotoxin tolerance by facilitating Toll-like receptor-induced chromatin remodeling. Proc. Natl Acad. Sci. USA 107, 19438–19443 (2010).
Monneret, G. et al. The anti-inflammatory response dominates after septic shock: association of low monocyte HLA-DR expression and high interleukin-10 concentration. Immunol. Lett. 95, 193–198 (2004).
Gogos, C. A., Drosou, E., Bassaris, H. P. & Skoutelis, A. Pro- versus anti-inflammatory cytokine profile in patients with severe sepsis: a marker for prognosis and future therapeutic options. J. Infect. Dis. 181, 176–180 (2000).
Hynninen, M. et al. Predictive value of monocyte histocompatibility leukocyte antigen-DR expression and plasma interleukin-4 and -10 levels in critically ill patients with sepsis. Shock 20, 1–4 (2003).
Oberholzer, A., Oberholzer, C. & Moldawer, L. L. Interleukin-10: a complex role in the pathogenesis of sepsis syndromes and its potential as an anti-inflammatory drug. Crit. Care Med. 30, S58–S63 (2002).
Sfeir, T., Saha, D. C., Astiz, M. & Rackow, E. C. Role of interleukin-10 in monocyte hyporesponsiveness associated with septic shock. Crit. Care Med. 29, 129–133 (2001).
Muehlstedt, S. G., Lyte, M. & Rodriguez, J. L. Increased IL-10 production and HLA-DR suppression in the lungs of injured patients precede the development of nosocomial pneumonia. Shock 17, 443–450 (2002).
Randow, F. et al. Mechanism of endotoxin desensitization: involvement of interleukin 10 and transforming growth factor β. J. Exp. Med. 181, 1887–1892 (1995).
Muenzer, J. T. et al. Characterization and modulation of the immunosuppressive phase of sepsis. Infect. Immun. 78, 1582–1592 (2010).
Lukaszewicz, A. C. et al. Monocytic HLA-DR expression in intensive care patients: interest for prognosis and secondary infection prediction. Crit. Care Med. 37, 2746–2752 (2009).
Astiz, M., Saha, D., Lustbader, D., Lin, R. & Rackow, E. Monocyte response to bacterial toxins, expression of cell surface receptors, and release of anti-inflammatory cytokines during sepsis. J. Lab Clin. Med. 128, 594–600 (1996).
Rode, H. N. et al. Lymphocyte function in anergic patients. Clin. Exp. Immunol. 47, 155–161 (1982).
Monneret, G., Venet, F., Pachot, A. & Lepape, A. Monitoring immune dysfunctions in the septic patient: a new skin for the old ceremony. Mol. Med. 14, 64–78 (2008).
Venet, A., Lukaszewica, A., Payen, D., Hotchkiss, R. & Monneret, G. Monitoring the immune response in sepsis: a rational approach to administration of immunoadjuvant therapies. Curr. Opin. Immunol. 25, 477–483 (2013).
Landelle, C. et al. Low monocyte human leukocyte antigen-DR is independently associated with nosocomial infections after septic shock. Intensive Care Med. 36, 1859–1866 (2010).
Chiche, L. et al. The role of natural killer cells in sepsis. J. Biomed. Biotechnol. 2011, 986491 (2011).
Souza-Fonseca-Guimaraes, F., Adib-Conquy, M. & Cavaillon, J. M. Natural killer (NK) cells in antibacterial innate immunity: angels or devils? Mol. Med. 18, 270–285 (2012).
Venet, F. et al. Early assessment of leukocyte alterations at diagnosis of septic shock. Shock 34, 358–363 (2010).
Forel, J. M. et al. Phenotype and functions of natural killer cells in critically-ill septic patients. PLoS ONE 7, e50446 (2012).
Holub, M. et al. Lymphocyte subset numbers depend on the bacterial origin of sepsis. Clin. Microbiol. Infect. 9, 202–211 (2003).
Giamarellos-Bourboulis, E. J. et al. Early changes of CD4-positive lymphocytes and NK cells in patients with severe Gram-negative sepsis. Crit. Care 10, R166 (2006).
Souza-Fonseca-Guimaraes, F. et al. Toll-like receptors expression and interferon-γ production by NK cells in human sepsis. Crit. Care 16, R206 (2012).
Puente, J. et al. In vitro studies of natural killer cell activity in septic shock patients. Response to a challenge with α-interferon and interleukin-2. Int. J. Clin. Pharmacol. Ther. Toxicol. 31, 271–275 (1993).
Georgeson, G. D. et al. Natural killer cell cytotoxicity is deficient in newborns with sepsis and recurrent infections. Eur. J. Pediatr. 160, 478–482 (2001).
Blazar, B. A. et al. Suppression of natural killer-cell function in humans following thermal and traumatic injury. J. Clin. Immunol. 6, 26–36 (1986).
Bender, B. S. et al. Depressed natural killer cell function in thermally injured adults: successful in vivo and in vitro immunomodulation and the role of endotoxin. Clin. Exp. Immunol. 71, 120–125 (1988).
Chiche, L. et al. Interferon-γ production by natural killer cells and cytomegalovirus in critically ill patients. Crit. Care Med. 40, 3162–3169 (2012).
Souza-Fonseca-Guimaraes, F., Parlato, M., Fitting, C., Cavaillon, J. M. & Adib-Conquy, M. NK cell tolerance to TLR agonists mediated by regulatory T cells after polymicrobial sepsis. J. Immunol. 188, 5850–5858 (2012).
Luyt, C. E. et al. Herpes simplex virus lung infection in patients undergoing prolonged mechanical ventilation. Am. J. Respir. Crit. Care Med. 175, 935–942 (2007).
Limaye, A. P. et al. Cytomegalovirus reactivation in critically ill immunocompetent patients. JAMA 300, 413–422 (2008).
Cook, C. H. & Trgovcich, J. Cytomegalovirus reactivation in critically ill immunocompetent hosts: a decade of progress and remaining challenges. Antiviral Res. 90, 151–159 (2011).
Chiche, L., Forel, J. M. & Papazian, L. The role of viruses in nosocomial pneumonia. Curr. Opin. Infect. Dis. 24, 152–156 (2011).
Wesselkamper, S. C. et al. NKG2D is critical for NK cell activation in host defense against Pseudomonas aeruginosa respiratory infection. J. Immunol. 181, 5481–5489 (2008).
Vantourout, P. & Hayday, A. Six-of-the-best:unique contributions of γδ T cells to immunology. Nature Rev. Immunol. 13, 88–100 (2013).
Andreu-Ballester, J. C. et al. Association of γδ T cells with disease severity and mortality in septic patients. Clin. Vaccine Immunol. 20, 738–746 (2012).
Hotchkiss, R. S. et al. Accelerated lymphocyte death in sepsis occurs by both the death receptor and mitochondrial pathways. J. Immunol. 174, 5110–5118 (2005).
Gouel-Cheron, A., Venet, F., Allaouchiche, B. & Monneret, G. CD4+ T-lymphocyte alterations in trauma patients. Crit. Care 16, 432 (2012).
Le Tulzo, Y. et al. Early circulating lymphocyte apoptosis in human septic shock is associated with poor outcome. Shock 18, 487–494 (2002).
Inoue, S. et al. Reduction of immunocompetent T cells followed by prolonged lymphopenia in severe sepsis in the elderly. Crit. Care Med. 41, 810–819 (2013).
Heffernan, D. S. et al. Failure to normalize lymphopenia following trauma is associated with increased mortality, independent of the leukocytosis pattern. Crit. Care 16, R12 (2012).
Cheadle, W. G. et al. Lymphocyte subset responses to trauma and sepsis. J. Trauma 35, 844–849 (1993).
Venet, F. et al. Increased percentage of CD4+CD25+ regulatory T cells during septic shock is due to the decrease of CD4+CD25− lymphocytes. Crit. Care Med. 32, 2329–2331 (2004).
De, A. K. et al. Induction of global anergy rather than inhibitory Th2 lymphokines mediates posttrauma T cell immunodepression. Clin. Immunol. 96, 52–66 (2000).
Heidecke, C. D. et al. Selective defects of T lymphocyte function in patients with lethal intraabdominal infection. Am. J. Surg. 178, 288–292 (1999).
Wick, M., Kollig, E., Muhr, G. & Koller, M. The potential pattern of circulating lymphocytes TH1/TH2 is not altered after multiple injuries. Arch. Surg. 135, 1309–1314 (2000).
Pachot, A. et al. Longitudinal study of cytokine and immune transcription factor mRNA expression in septic shock. Clin. Immunol. 114, 61–69 (2005).
Venet, F. et al. Regulatory T cell populations in sepsis and trauma. J. Leukoc. Biol. 83, 523–535 (2008).
Carson, W. F.t. et al. Impaired CD4+ T-cell proliferation and effector function correlates with repressive histone methylation events in a mouse model of severe sepsis. Eur. J. Immunol. 40, 998–1010 (2010).
Romani, L. Immunity to fungal infections. Nature Rev. Immunol. 11, 275–288 (2011).
Gow, N. A., van de Veerdonk, F. L., Brown, A. J. & Netea, M. G. Candida albicans morphogenesis and host defence: discriminating invasion from colonization. Nature Rev. Microbiol. 10, 112–122 (2011).
van de Veerdonk, F. L. et al. Deficient Candida-specific T-helper 17 response during sepsis. J. Infect. Dis. 206, 1798–1802 (2012).
Monneret, G., Venet, F., Kullberg, B. J. & Netea, M. G. ICU-acquired immunosuppression and the risk for secondary fungal infections. Med. Mycol. 49 (Suppl. 1), S17–23 (2011).
Unsinger, J. et al. Interleukin-7 ameliorates immune dysfunction and improves survival in a 2-hit model of fungal sepsis. J. Infect. Dis. 206, 606–616 (2012).
Kasten, K. R. et al. Interleukin-7 (IL-7) treatment accelerates neutrophil recruitment through γδ T-cell IL-17 production in a murine model of sepsis. Infect. Immun. 78, 4714–4722 (2010).
Zajac, A. J. et al. Viral immune evasion due to persistence of activated T cells without effector function. J. Exp. Med. 188, 2205–2213 (1998).
Wherry, E. J. T cell exhaustion. Nature Immunol. 12, 492–499 (2011). An outstanding review of the importance of T cell exhaustion in chronic infections and cancer.
Guignant, C. et al. Programmed death-1 levels correlate with increased mortality, nosocomial infection and immune dysfunctions in septic shock patients. Crit. Care 15, R99 (2011).
Huang, X. et al. PD-1 expression by macrophages plays a pathologic role in altering microbial clearance and the innate inflammatory response to sepsis. Proc. Natl Acad. Sci. USA 106, 6303–6308 (2009). This was the first study to show that blocking the PD1 pathway can improve survival in sepsis.
Brahmamdam, P. et al. Delayed administration of anti-PD-1 antibody reverses immune dysfunction and improves survival during sepsis. J. Leukoc. Biol. 88, 233–240 (2010).
Zhang, Y. et al. PD-L1 blockade improves survival in experimental sepsis by inhibiting lymphocyte apoptosis and reversing monocyte dysfunction. Crit. Care 14, R220 (2010).
Monneret, G. et al. Marked elevation of human circulating CD4+CD25+ regulatory T cells in sepsis-induced immunoparalysis. Crit. Care Med. 31, 2068–2071 (2003).
Zanin-Zhorov, A. et al. Heat shock protein 60 enhances CD4+ CD25+ regulatory T cell function via innate TLR2 signaling. J. Clin. Invest. 116, 2022–2032 (2006).
Huang, L. F. et al. Association between regulatory T cell activity and sepsis and outcome of severely burned patients: a prospective, observational study. Crit. Care 14, R3 (2010).
Venet, F. et al. Increased circulating regulatory T cells (CD4+CD25+CD127−) contribute to lymphocyte anergy in septic shock patients. Intensive Care Med. 35, 678–686 (2009).
Carrigan, S. O. et al. Depletion of natural CD4+CD25+ T regulatory cells with anti-CD25 antibody does not change the course of Pseudomonas aeruginosa-induced acute lung infection in mice. Immunobiology 214, 211–222 (2009).
Scumpia, P. O. et al. Treatment with GITR agonistic antibody corrects adaptive immune dysfunction in sepsis. Blood 110, 3673–3681 (2007).
Cavassani, K. A. et al. The post sepsis-induced expansion and enhanced function of regulatory T cells create an environment to potentiate tumor growth. Blood 115, 4403–4411 (2010).
Venet, F. et al. Human CD4+CD25+ regulatory T lymphocytes inhibit lipopolysaccharide-induced monocyte survival through a Fas/Fas ligand-dependent mechanism. J. Immunol. 177, 6540–6547 (2006).
Li, L. & Wu, C. Y. CD4+ CD25+ Treg cells inhibit human memory γδ T cells to produce IFN-γ in response to M tuberculosis antigen ESAT-6. Blood 111, 5629–5636 (2008).
Tiemessen, M. M. et al. CD4+CD25+Foxp3+ regulatory T cells induce alternative activation of human monocytes/macrophages. Proc. Natl Acad. Sci. USA 104, 19446–19451 (2007).
Mackall, C. L., Fry, T. J. & Gress, R. E. Harnessing the biology of IL-7 for therapeutic application. Nature Rev. Immunol. 11, 330–342 (2011). An outstanding review article on a remarkable cytokine that has great potential as an immunotherapeutic agent for reversing immunosuppression in various disorders, including sepsis and cancer.
Perales, M. A. et al. Recombinant human interleukin-7 (CYT107) promotes T-cell recovery after allogeneic stem cell transplantation. Blood 120, 4882–4891 (2012).
Unsinger, J. et al. IL-7 promotes T cell viability, trafficking, and functionality and improves survival in sepsis. J. Immunol. 184, 3768–3779 (2010).
Venet, F. et al. Decreased T-cell repertoire diversity in sepsis: a preliminary study. Crit. Care Med. 41, 111–119 (2013).
Levy, Y. et al. Effects of recombinant human interleukin 7 on T-cell recovery and thymic output in HIV-infected patients receiving antiretroviral therapy: results of a Phase I/IIa randomized, placebo-controlled, multicenter study. Clin. Infect. Dis. 55, 291–300 (2012).
Pellegrini, M. et al. IL-7 engages multiple mechanisms to overcome chronic viral infection and limit organ pathology. Cell 144, 601–613 (2011).
Venet, F. et al. IL-7 restores lymphocyte functions in septic patients. J. Immunol. 189, 5073–5081 (2012). An important study demonstrating that IL-7 restores function in immune effector cells from patients with sepsis.
Chang, K. C. et al. Blockade of the negative co-stimulatory molecules PD-1 and CTLA-4 improves survival in primary and secondary fungal sepsis. Crit. Care 17, R85 (2013).
Singh, A., Mohan, A., Dey, A. B. & Mitra, D. K. Inhibiting the programmed death 1 pathway rescues Mycobacterium tuberculosis-specific interferon γ-producing T cells from apoptosis in patients with pulmonary tuberculosis. J. Infect. Dis. 208, 603–615 (2013).
Docke, W. D. et al. Monocyte deactivation in septic patients: restoration by IFN-γ treatment. Nature Med. 3, 678–681 (1997). One of the first studies to show that therapy with agents that enhance host immunity administered to patients in the immunosuppressive phase of sepsis can improve survival.
Nalos, M. et al. Immune effects of interferon γ in persistent staphylococcal sepsis. Am. J. Respir. Crit. Care Med. 185, 110–112 (2012).
Marciano, B. E. et al. Long-term interferon-γ therapy for patients with chronic granulomatous disease. Clin. Infect. Dis. 39, 692–699 (2004).
Jarvis, J. N. et al. Adjunctive interferon-gamma immunotherapy for the treatment of HIV-associated cryptococcal meningitis: a randomized controlled trial. AIDS 26, 1105–1113 (2012).
Root, R. K. et al. Multicenter, double-blind, placebo-controlled study of the use of filgrastim in patients hospitalized with pneumonia and severe sepsis. Crit. Care Med. 31, 367–373 (2003).
Nelson, S. et al. A randomized controlled trial of filgrastim as an adjunct to antibiotics for treatment of hospitalized patients with community-acquired pneumonia. CAP Study Group. J. Infect. Dis. 178, 1075–1080 (1998).
Meisel, C. et al. Granulocyte-macrophage colony-stimulating factor to reverse sepsis-associated immunosuppression: a double-blind, randomized, placebo-controlled multicenter trial. Am. J. Respir. Crit. Care Med. 180, 640–648 (2009).
Reinhart, K., Bauer, M., Riedemann, N. C. & Hartog, C. S. New approaches to sepsis: molecular diagnostics and biomarkers. Clin. Microbiol. Rev. 25, 609–634 (2012).
Leentjens, J., Kox, M., van der Hoeven, J. G., Netea, M. G. & Pickkers, P. Immunotherapy for the adjunctive treatment of sepsis: from immunosuppression to immunostimulation. Time for a paradigm change? Am. J. Respir. Crit. Care Med. 187, 1287–1293 (2013).
Wong, H. R. Genetics and genomics in pediatric septic shock. Crit. Care Med. 40, 1618–1626 (2012).
Mestas, J. & Hughes, C. C. Of mice and not men: differences between mouse and human immunology. J. Immunol. 172, 2731–2738 (2004).
Seok, J. et al. Genomic responses in mouse models poorly mimic human inflammatory diseases. Proc. Natl Acad. Sci. USA 110, 3507–3512 (2013).
Brahmamdam, P. et al. Targeted delivery of siRNA to cell death proteins in sepsis. Shock 32, 131–139 (2009).
Weber, S. U. et al. Induction of Bim and Bid gene expression during accelerated apoptosis in severe sepsis. Crit. Care 12, R128 (2008).
Acknowledgements
The authors would like to thank P. Swanson (Professor of Pathology, University of Washington School of Medicine in Seattle, Washington, USA) for providing the immunohistochemical images of patient spleens used in figure 2. R.S.H. is supported by US National Institutes of Health grants GM44118 and GM55194. G.M. is supported by funding from the Hospices Civils de Lyon.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Related links
DATABASES
Glossary
- Immunosenescence
-
The decreased function of the immune system with age. In particular, the number of naive T cells decreases as thymic function decreases.
- Myeloid-derived suppressor cell
-
(MDSC). A subset of immune cells that includes mature and immature myeloid cells. They are generated and/or activated during an inflammatory immune response. Through direct interactions and secreted components, they negatively affect T cells, which leads to impaired T cell function.
- Follicular dendritic cells
-
(Follicular DCs). Cells with a dendritic morphology that are present in the lymph nodes. These cells display on their surface intact antigens that are held in immune complexes, and B cells in the lymph nodes can interact with these antigens. Follicular DCs are of non-haematopoietic origin and are not related to DCs.
- Interdigitating DCs
-
Potent antigen-presenting cells that are rich in MHC class II molecules. Interdigitating DCs take up antigens in the periphery and migrate to the paracortical region of the lymph nodes and the spleen, where they interact with T cells.
- Gut-associated lymphoid tissues
-
(GALTs). Lymphoid structures and aggregates that are associated with the intestinal mucosa, specifically the tonsils, Peyer's patches, lymphoid follicles, appendix or coecal patch, and mesenteric lymph nodes. They are enriched in conventional and unconventional lymphocytes and specialized dendritic cell and macrophage subsets.
- Anergy
-
A state of immune unresponsiveness. Anergic B cells or T cells do not respond to their cognate antigens.
- Endotoxin tolerance
-
A transient state of hyporesponsiveness of the host or of cultured macrophages and/or monocytes to lipopolysaccharide (an endotoxin) following previous exposure to lipopolysaccharide.
- Extracorporeal cell therapy
-
A therapy whereby the patient's blood is circulated through a haemodialysis-like device through which particular types of cells, for example, donor granulocytes, are added to the blood and subsequently infused back into the patient. Extracorporeal cell therapy of autologous granulocytes has shown success in small clinical trials in sepsis.
- FMS-related tyrosine kinase 3 ligand
-
(FLT3L). An endogenous cytokine that stimulates the proliferation of stem and progenitor cells through binding to the FLT3 receptor (which is a type III receptor tyrosine kinase member of the platelet-derived growth factor family). FLT3L administration substantially increases the number of dendritic cells in lymphoid and non-lymphoid tissues.
- MicroRNAs
-
Single-stranded RNA molecules of approximately 21–23 nucleotides in length that are thought to regulate the expression of other genes.
- T cell exhaustion
-
The condition of functionally impaired antigen-specific T cells, typified by increased surface expression of programmed cell death 1, which occurs in the context of persistent high antigen load. The defects in effector T cell function include a progressive decrease in their ability to produce cytokines, loss of proliferative capacity and decreased cytotoxicity, and can result in apoptotic cell death.
- Alarmins
-
Endogenous molecules that are released after tissue injury and that promote activation of the innate immune system. Excessive release of alarmins might promote uncontrolled inflammation and exacerbate tissue injury. Conversely, alarmins may have beneficial effects by activating the immune system to combat potential pathogens.
- Small interfering RNA
-
(siRNA). Synthetic RNA molecules of 19–23 nucleotides that are used to 'knock down' (that is, to silence the expression of) a specific gene. This is known as RNA interference and is mediated by the sequence-specific degradation of mRNA.
- Central memory T cells
-
Antigen-experienced T cells that express cell surface receptors that are required for homing to secondary lymphoid organs. These cells are generally thought to be long-lived and can function as the precursors for effector T cells for recall responses.
- Delayed-type hypersensitivity
-
A cellular immune response to antigens that develops over 24–72 hours with the infiltration of T cells and monocytes, and that is dependent on the production of T helper 1 cell-associated cytokines.
- Chronic granulomatous disease
-
An inherited disorder caused by defective oxidase activity in the respiratory burst of phagocytes. It results from mutations in any of four genes that are necessary to generate the superoxide radicals required for normal neutrophil function. Affected patients suffer from increased susceptibility to recurrent infections.
Rights and permissions
About this article
Cite this article
Hotchkiss, R., Monneret, G. & Payen, D. Sepsis-induced immunosuppression: from cellular dysfunctions to immunotherapy. Nat Rev Immunol 13, 862–874 (2013). https://doi.org/10.1038/nri3552
Published:
Issue Date:
DOI: https://doi.org/10.1038/nri3552
This article is cited by
-
Gut microbiota composition and changes in patients with sepsis: potential markers for predicting survival
BMC Microbiology (2024)
-
Clinical description and outcome of overall varicella-zoster virus-related organ dysfunctions admitted in intensive care units: the VAZOREA cohort study
Annals of Intensive Care (2024)
-
ICU-acquired infections in immunocompromised patients
Intensive Care Medicine (2024)
-
The clinical trajectory of peripheral blood immune cell subsets, T-cell activation, and cytokines in septic patients
Inflammation Research (2024)
-
PD-L1 Blockade Improves Survival in Sepsis by Reversing Monocyte Dysfunction and Immune Disorder
Inflammation (2024)
