
Abstract
Gut dysbiosis is associated with many non-communicable human diseases, but the mechanisms maintaining homeostasis remain incompletely understood. Recent insights suggest that during homeostasis, epithelial hypoxia limits oxygen availability in the colon, thereby maintaining a balanced microbiota that functions as a microbial organ, producing metabolites contributing to host nutrition, immune education and niche protection. Dysbiosis is characterized by a shift in the microbial community structure from obligate to facultative anaerobes, suggesting oxygen as an important ecological driver of microbial organ dysfunction. The ensuing disruption of gut homeostasis can lead to non- communicable disease because microbiota-derived metabolites are either depleted or generated at harmful concentrations. This Opinion article describes the concept that host control over the microbial ecosystem in the colon is critical for the composition and function of our microbial organ, which provides a theoretical framework for linking microorganisms to non-communicable diseases.
Similar content being viewed by others
Main
The germ theory triggered a scientific revolution during the time of Robert Koch (1843–1910) and Louis Pasteur (1822–1895) by guiding efforts to identify bacterial pathogens as leading causes of communicable diseases (Box 1). By developing culture-dependent methods for studying microorganisms, Louis Pasteur laid the foundation for a new discipline, bacteriology. Similarly, recent advances in culture-independent methods for analysing microorganisms have opened up a new field of study, microbiota research, that parallels the remarkable advances made during the 'golden age' of microbiology in the 19th century. Whereas the initial use of culture-dependent methods by Louis Pasteur and his contemporaries established a microbial cause for many communicable human diseases, recent advances in culture-independent methods have made it possible to associate gut-associated microbial communities with many non-communicable human diseases, including obesity1,2, arthritis3, asthma4,5, cardiovascular disease6, cancer7,8,9, diabetes10 and even neurological disorders11,12. However, research into the microbial origin of non-communicable human diseases differs in one important aspect from the studies performed at the end of the 19th century: the identification of pathogens as causes of communicable human diseases was guided by Louis Pasteur's germ theory, whereas contemporary studies on the role of the gut microbiota in health and disease lack an overarching theoretical framework for guidance (Box 2) and therefore remain heavily focused on technological development, census taking and predicting physiological potential.
The germ theory influenced initial studies on the microbial origin of non-communicable diseases as investigators used 16S ribosomal RNA gene profiling to identify pathogens linked to an illness, which is understandable given the focus on the contribution of microorganisms to disease. However, the realization that most non-communicable diseases are not associated with a pathogen resulted in a conceptual crisis, calling the term 'pathogen' into question13,14,15,16. We would argue that the concept of a pathogen is still valid for studying communicable diseases, such as tuberculosis, but it does not provide a theoretical framework that adequately captures the contribution of microbial communities to non-communicable diseases (Box 2). In other words, rather than omitting the term 'pathogen', the core concept of the germ theory, we should be mindful of using it in the context of communicable diseases while independently developing a new theory to guide studies on the microbial origin of non-communicable diseases. In this Opinion article, we discuss the most recent findings of microbiota research in search of a useful theoretical framework that adequately captures the contribution of gut-associated microbial communities to human non-communicable diseases and its implication for guiding future research efforts.
The microbial organ of the large bowel
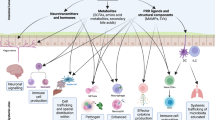
More than 2 millennia have passed since Hippocrates (circa 460–370 bce) commented “all disease begins in the gut”, but in light of recent data revealing how microorganisms that inhabit our body affect human health, this quote feels more timely than ever17. The gut microbiota, the largest microbial community inhabiting our body, fulfils many functions important for human physiology, including nutrition, development of the immune system and niche protection against enteric pathogens18 (Fig. 1). The microbiota of the large bowel is of particular importance and forms the focus of this Opinion article because in the colon, bacterial density reaches a staggering 1011 organisms per gram, making this bacterial community the principal source of microbial metabolites in the human body. In turn, these metabolites are responsible for the beneficial effects of the gut microbiota on host physiology18. It was thus proposed in 2005 that the gut-associated microbial community of the large bowel represents a 'microbial organ' (Ref. 19) because this collection of microorganisms resides as a structural unit and has a common function, which is the production of metabolites at concentrations that promote health. Although this idea is generally accepted17, it has never become an alternative to the germ theory. One aspect that has curbed the use of the microbial organ concept as a guide for microbiota research is the daunting complexity of our gut-associated microbial community. Comparison of the gut microbiota composition between different individuals reveals very little overlap on the species level20, thus making it problematic to define what a 'balanced microbial community' or a functional microbial organ should look like. As a result of limited progress in understanding how a balanced microbial community is maintained during gut homeostasis, the microbial organ concept has long remained elusive, thus curtailing its use as a conceptual framework.

Hypoxia of the colonic epithelial cells (colonocytes) lining the mucosal surface maintains luminal anaerobiosis, which drives the composition of the gut microbiota towards a dominance of obligate anaerobes. Butyrate produced by this microbial organ is oxidized by the colonic epithelium to carbon dioxide (CO2) to preserve epithelial hypoxia (<1% oxygen (O2)), thereby maintaining gut homeostasis. Dividing colonocytes at the bottom of crypts obtain energy through anaerobic glycolysis (that is, the fermentation of glucose to lactate) and are thus not hypoxic. A healthy diet ensures that the microbial organ produces metabolites that function in nutrition, immune development and niche protection. For simplicity, only examples of metabolites are shown. The O2 concentration is indicated by the red colouring of the schematic.
Our poor understanding of gut homeostasis has shifted the focus to aspects that are easier to grasp, such as the association of disease with the presence or absence of individual microorganisms. Specifically, the concept that disease can result from a loss of beneficial commensal bacteria or an overgrowth of resident bacteria that have the potential to cause disease, termed 'pathobionts', is a commonly used guide for contemporary microbiota research21,22,23,24. The term 'keystone species' has been proposed for pathobionts that instigate inflammation even when they are present as quantitatively minor components of the gut microbiota25. The quest to identify pathobionts by profiling microbial communities in health and disease follows in the footsteps of the search for pathogens during the golden age of microbiology (Box 1), illustrating the marked influence that the germ theory continues to exert on microbiota research. However, although a focus on individual microorganisms is appropriate for studying pathogens that cause communicable diseases, this viewpoint is too narrow to fully capture the contribution of microbial community ecology to health and disease (Box 2). Recent insights into how the host controls the gut ecosystem to ensure homeostasis suggest that it is time to revisit the usefulness of the microbial organ concept as a comprehensive guide for microbiota research.
Invertebrate models reveal a striking control of microbial community composition by the host, as species-specific antimicrobial peptides are believed to be the selective force behind the different bacterial communities associated with closely related species of the cnidarian Hydra26, whereas the light organ of Euprymna scolopes is associated with a single bacterial species, Aliivibrio fischeri27.At first glance, host control over the composition of the gut microbiota is less evident in the mammalian gut because the diversity of this microbial community in adults does not suggest selection for any particular bacterial species20. There is evidence that a mammalian host directs the composition of the gut microbiota during infancy through milk oligosaccharides28 and to some degree throughout life by epithelial release of antimicrobials, such as defensins29,30. It is also well established that anaerobiosis is preserved in the mammalian large bowel, but the idea that this global ecosystem control is imposed by the host emerged only recently. We now know that mammalian hosts restrict oxygen availability in the lumen of the large bowel, thereby driving the composition of the microbial community towards a dominance of obligate anaerobes, which is critical for maintaining gut homeostasis31. Epithelial cells are maintained in a state of hypoxia (<1% oxygen) at the colonic surface, which limits the diffusion of oxygen from the colonic mucosa into the intestinal lumen, an attribute that preserves anaerobiosis in the large bowel32. In turn, anaerobiosis ensures a dominance of obligate anaerobic bacteria that belong to the Bacteroidetes phylum and Clostridia class33. This feature is conserved between individuals, although microbial communities tolerate an intimidating degree of species diversity20. Constraining oxygen availability is critical because under anaerobic conditions, our microbial organ self-assembles into a community of microorganisms that stably coexist34 by forming an anaerobic trophic network35. Each position in this trophic network represents a nutrient niche that can be occupied by any microorganism that discharges the required metabolic functions. Each occupant of a nutrient niche is predicted to be able to use a few limiting resources better than any other member within the microbial community, and the abundance of these limiting resources determines the abundance of the respective occupant, a concept known as the nutrient niche hypothesis36. The formation of this anaerobic trophic network generates metabolite profiles that are similar among individuals, regardless of the species composition of the microbial community.
During gut homeostasis, our microbial organ hydrolyses and ferments complex dietary carbohydrates (fibre) that escape degradation by host enzymes in the upper gastrointestinal tract into smaller compounds, with the short-chain fatty acids acetate, propionate and butyrate being the most abundant metabolites produced during gut homeostasis37 (Fig. 1). On the basis of their main metabolic functions, obligate anaerobic bacteria that degrade complex carbohydrates can be grouped into butyrate-producing bacteria, saccharolytic bacteria, sulfate-reducing bacteria and acetate-producing bacteria (acetogens)38. Saccharolytic bacteria produce hydrogen, which is consumed by hydrogenotrophic microorganisms, including sulfate-reducing bacteria, acetogens and methanogens39. Obligate anaerobic microorganisms that belong to these metabolic groupings produce a multitude of metabolites in addition to short-chain fatty acids, as has been elegantly reviewed elsewhere40. Importantly, the majority of microbiota-derived short-chain fatty acids is absorbed by the host41 and contributes to host nutrition42. Evolutionarily, anaerobiosis could be viewed as a host-driven strategy to limit the ability of the microbiota to compete nutritionally with the host for compounds produced by the microbial fermentation of fibre. In the presence of oxygen, facultative anaerobic bacteria can catabolize fermentation products to carbon dioxide. By limiting oxygen availability, the host ensures that obligate anaerobes contribute to its own nutrition by producing metabolites through the fermentation of fibre while at the same time limiting catabolism of fermentation products to carbon dioxide by facultative anaerobic bacteria. Through this mechanism, the host maintains control over the microbial ecosystem without imposing constraints on the species composition of the microbial community.
Microbiota-derived short-chain fatty acids also contribute to immune development43,44,45,46,47,48 and gut homeostasis3,31,49,50. The latter process requires a continuous supply of microbiota-derived short-chain fatty acids to stimulate the intracellular butyrate sensor peroxisome proliferator-activated receptor-γ (PPARγ) in colonic epithelial cells31 and to promote maturation and expansion of regulatory T cells in the colonic mucosa45,46,47,48,50. Regulatory T cells and epithelial PPARγ signalling cooperate to drive the metabolism of the colonic surface towards mitochondrial β-oxidation31, a process that consumes large quantities of oxygen, thereby rendering epithelial cells at the colonic surface hypoxic32. Hence, epithelial hypoxia ensures that obligate anaerobes produce short-chain fatty acids, which in turn induce epithelial hypoxia, thereby driving a virtuous cycle that maintains gut homeostasis and balances the composition of the microbial community31 (Fig. 1).
The picture emerging from these studies is that during gut homeostasis, the host controls its microbial organ in the large bowel through mechanisms that include limiting oxygen availability in this ecosystem. The latter provides a benefit by curbing the ability of facultative anaerobic microorganisms to compete with the host for nutrients and by ensuring the production of microbial fermentation products that promote nutrition, immune development and gut homeostasis.
Microbial organ dysfunction
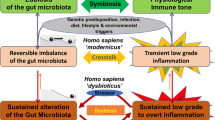
Based on the above model of microbial organ function (Fig. 1), we propose that dysbiosis is a state of microbial organ dysfunction, a condition in which the gut-associated microbial community becomes a liability because the host no longer maintains proper control over the ecosystem. Microbial organ dysfunction can produce disease because metabolites are either depleted or produced at a harmful level21,22,23,24. As the gut microbiota can produce many different metabolites that are capable of acting both locally and on cells in distant tissues40, microbial organ dysfunction can result in metabolite imbalances that affect numerous organs, thereby possibly explaining the broad spectrum of non-communicable diseases associated with dysbiosis17.
Importantly, disruption of the virtuous cycle that maintains anaerobiosis in the colon31 (Fig. 1) impairs the ability of the host to limit the flow of oxygen into the gut lumen, which gives rise to the most consistent and robust ecological pattern observed during gut dysbiosis, a shift in the microbial community structure from obligate to facultative anaerobes51,52. An expansion of facultative anaerobic bacteria of the phylum Proteobacteria has been described in individuals consuming a Western-style diet53,54, undergoing antibiotic therapy55 or suffering from irritable bowel syndrome56,57, inflammatory bowel disease58, metabolic syndrome59, necrotizing enterocolitis60 or colorectal cancer61. An expansion of Proteobacteria is also observed in animal models of chemically induced colitis62,63, genetically induced colitis62,64, colorectal cancer61, infection-induced colitis62,65,66 or antibiotic-mediated loss of colonization resistance67,68,69. The regularity with which a disruption of gut homeostasis produces an expansion of Proteobacteria suggests that this is a microbial signature of gut dysbiosis70. Below, we discuss recent data supporting the concept that increased availability of oxygen or other exogenous electron acceptors constitutes the main ecological driver of expansion of Proteobacteria.
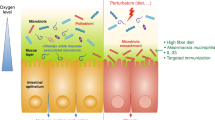
Increased oxygen availability in the colon can ensue from disruption of the gut microbiota, which can be triggered by either antibiotic therapy or intestinal inflammation (Fig. 2). Depletion of gut microorganisms during antibiotic therapy reduces the production of microbiota-derived metabolites, as indicated by a marked drop in the concentration of short-chain fatty acids that are crucial for the maintenance of gut homeostasis71. Intestinal inflammation induced by enteric pathogens, such as Salmonella serovars (phylum Proteobacteria), causes transepithelial migration of neutrophils that deplete Clostridia72,73, thereby lowering short-chain fatty acid levels in the colon74. The depletion of short-chain fatty acids by antibiotics or host neutrophils shifts the energy metabolism of colonic epithelial cells from mitochondrial β-oxidation to anaerobic glycolysis31,75. As the latter pathway does not consume oxygen76, epithelial oxygenation increases, which in turn increases the levels of oxygen emanating from the epithelial surface. An antibiotic-mediated increase in oxygen availability may explain why treatment of mice with streptomycin increases the redox potential in the caecum to levels that approximate an aerobic broth culture71. The consequent disruption in anaerobiosis drives an expansion of facultative anaerobic Proteobacteria regardless of their pathogenic potential31,74. As oxygen originates from the host epithelium, an expansion of Proteobacteria could be viewed as a microbial signature of epithelial dysfunction31.

Antibiotic treatment or the migration of neutrophils into the intestinal lumen during severe intestinal inflammation can cause the disruption of the microbiota, which leads to a depletion of short-chain fatty acids, thereby shifting the metabolism of mature colonic epithelial cells (colonocytes) from mitochondrial β-oxidation to anaerobic glycolysis (that is, the fermentation of glucose to lactate). The resulting increase in epithelial oxygenation disrupts anaerobiosis, which drives an expansion of facultative anaerobic Proteobacteria, a microbial signature of dysbiosis. Neutrophils that encounter bacteria (for example, during transepithelial migration into the lumen) undergo a respiratory burst and become hypoxic (<1% oxygen (O2))124. The O2 concentration is indicated by the red colouring of the schematic.
A second mechanism that can lead to increased oxygen availability in the colon is induction of excessive epithelial repair mechanisms (Fig. 3). Ulcerative colitis is associated with an expansion of Proteobacteria70 that is indicative of a disruption in anaerobiosis51. Consistent with this idea, recent work from animal models of ulcerative colitis, including dextran sulfate sodium (DSS)-induced colitis and Citrobacter rodentium-induced colitis, suggests that the mechanism driving expansion of Proteobacteria is increased oxygen availability in the colon77,78. Epithelial damage induced during DSS treatment or C. rodentium infection triggers colonic crypt hyperplasia, an excessive epithelial repair response that results in crypt elongation, a reduction of goblet cell numbers and the appearance of undifferentiated epithelial cells at the luminal surface79,80. The presence of undifferentiated epithelial cells increases oxygenation of the colonic surface78 because this cell type obtains energy through anaerobic glycolysis76. The consequent disruption in anaerobiosis drives an expansion of Proteobacteria in the colon through aerobic respiration78. Importantly, as discussed below, dysbiosis can lead to disease by mechanisms that are not linked to the expansion of Proteobacteria6,81,82. However, an expansion of this taxon is relevant because it indicates an increased bioavailability of host-derived respiratory electron acceptors, which points to an impaired ability of the host to control the gut ecosystem as an important driver of dysbiosis.

Epithelial damage triggers epithelial repair responses, which are characterized by crypt elongation in the presence of dividing colonocytes at the epithelial surface. These immature dividing colonocytes obtain energy through anaerobic glycolysis (that is, the fermentation of glucose to lactate), which renders the colonic surface normoxic (3–10% oxygen (O2)). The resulting disruption of anaerobiosis drives an expansion of facultative anaerobic Proteobacteria, a microbial signature of dysbiosis. The O2 concentration is indicated by the red colouring of the schematic.
Although mild intestinal inflammation increases epithelial oxygenation31,74 (Figs 2,3), severe intestinal inflammation can render the mucosal surface hypoxic. During severe intestinal inflammation, necrosis of the upper mucosa leads to loss of the epithelial surface and formation of a pseudomembrane that is composed of necrotic tissue, neutrophils and fibrin83. This pseudomembrane becomes hypoxic because neutrophils covering the surface undergo a respiratory burst that depletes oxygen84. Although the mucosal surface is hypoxic, these pathological changes drive an expansion of Proteobacteria, such as Salmonella enterica65 and Yersinia enterocolitica85, as the neutrophil respiratory burst generates electron acceptors for anaerobic bacterial respiration, such as tetrathionate, which promote growth of these enteric pathogens. Thus, depletion of short-chain fatty acids (Fig. 2), colonic crypt hyperplasia (Fig. 3) and pseudomembrane formation each drive an expansion of Proteobacteria by increasing the availability of host-derived exogenous respiratory electron acceptors in the lumen of the large bowel.
It is important to keep in mind that although increased availability of respiratory electron acceptors contributes to the expansion of Proteobacteria, it may not be the only driver. For instance, antibiotic treatment increases the availability of monosaccharides, which have an important role in the observed expansion of Proteobacteria following antibiotic treatment86,87. Furthermore, whereas some Proteobacteria are pathogens or pathobionts, most members of this taxon are commensal microorganisms, and the expansion of such commensals does not drive disease but does represent a biomarker for dysbiotic ecosystem disruption. In other words, although an increase in the abundance of Proteobacteria may be observed during dysbiosis, disease might be caused by additional metabolite imbalances that are not linked to electron acceptor availability or the presence of Proteobacteria. The exact nature of the metabolite imbalance that develops during dysbiosis depends on the factors that trigger the disruption of the ecosystem. For example, the antibiotic-mediated disruption of the microbiota drives an expansion of Proteobacteria, but antibiotic-associated colitis is caused by an expansion of toxin-producing Clostridium difficile, an opportunistic pathogen that is an obligate anaerobe88. Antibiotic treatment lowers colonization resistance against C. difficile by depleting other obligate anaerobic members of the microbiota that are producers of secondary bile acids89,90, which are metabolites that inhibit growth of vegetative C. difficile cells91 (Fig. 1). Restoration of a balanced community of obligate anaerobic bacteria by faecal microbiota transplantation can restore microbial organ function and thus provides an effective treatment for antibiotic-associated colitis92,93. Indeed, inoculation of the secondary bile acid-producing Clostridium scindens was shown to restore levels of secondary bile acids in antibiotic-treated mice, thereby re-establishing colonization resistance against C. difficile81.
Another example is consumption of a Western-style diet, which is associated with an expansion of Proteobacteria53,54 but is linked to disease because dietary inputs alter the metabolite profile produced by the gut microbiota. A Western-style diet is associated with obesity, a condition that increases the risk of atherosclerosis94. A link between diet and cardiovascular disease is the consumption of l-carnitine and choline, both of which are abundant in red meat, a common component of a Western-style diet, and are converted by the gut microbiota to trimethylamine (TMA)6,95. TMA is absorbed by the host and oxidized by human hepatic flavin-containing monooxygenases to TMA N-oxide (TMAO), a metabolite that increases the risk of atherosclerosis6,82. Pathways to convert l-carnitine and choline into TMA are present in bacteria that belong to various phyla6,95,96. Thus, although antibiotic treatment and the consumption of a Western-style diet both disrupt gut homeostasis, as indicated by an expansion of Proteobacteria, each is linked to disease through a different metabolite imbalance.
The germ-organ theory
Recent findings on the mechanisms that maintain or disrupt gut homeostasis can be assembled into the following theoretical framework to describe microbial organ physiology. During gut homeostasis, the host controls the microbial ecosystem97 by limiting luminal oxygen availability in the colon, an ecological input that is critical for maintaining a balanced microbial community31. This balanced microbial community functions as a microbial organ, producing metabolites that maintain a virtuous cycle to preserve epithelial hypoxia (Fig. 1). Microbial organ dysfunction represents an ecosystem disturbance, commonly involving a shift in the microbial community from obligate to facultative anaerobes, which is indicative of a defect in the ability of the host to limit the flow of oxygen and other exogenous electron acceptors into the gut lumen. This loss of host control disrupts the microbial ecosystem, thereby contributing to non-communicable diseases because a microbiota-derived metabolite is either depleted or produced at harmful levels. We propose the term 'germ-organ theory' to describe the concept that host control over the microbial ecosystem in the large bowel is critical for the composition and function of the microbial organ, whereas disruption of host control triggers microbial organ dysfunction, which provides a link between the gut microbiota and non-communicable diseases. The focus on host control over the microbial ecosystem sets the concept of the germ-organ theory apart from the germ theory, which pictures the pathogen in control (Box 2).
Conclusions
Broadening the concepts guiding microbiota research towards an ecological perspective of microbial organ function puts the spotlight on questions that are currently just 'wallflowers' in contemporary microbiota research. For instance, the germ-organ theory suggests that non-communicable diseases associated with an expansion of Proteobacteria can be linked to impaired host control over the gut ecosystem. One example of impaired host control is epithelial dysfunction, which leads to a disruption of anaerobiosis in the large bowel31. We do not know whether depleting short-chain fatty acids (Fig. 2) or inducing excessive epithelial repair (Fig. 3) are the only mechanisms increasing epithelial oxygenation in the colon. More research is needed to unravel the causes of epithelial dysfunction for the various non-communicable diseases associated with expansion of Proteobacteria. The focus on changes in host physiology that impair control over the gut ecosystem represents a paradigm shift from the current focus on identifying pathobionts or microbial biomarkers of disease within the gut microbiota. Identifying causes of epithelial dysfunction is relevant because the germ-organ theory points to host signalling pathways as a potential target for intervention strategies. Such approaches would be aimed at rebalancing the gut microbiota by limiting the availability of exogenous electron acceptors and restoring epithelial hypoxia, which could be used to complement faecal microbiota transfer or to develop alternative treatment strategies.
Additionally, the germ-organ theory advocates for obtaining a more complete understanding of the composition and function of our microbial organ. We know embarrassingly little about the identity of limiting resources that define each nutrient niche within our microbial organ or the metabolic functions that make a specific microorganism a suitable occupant of a particular nutrient niche. Shedding light on these crucial knowledge gaps will be necessary to incorporate an improved understanding of trophic networks into the germ-organ theory, thereby increasing its power to predict additional mechanisms through which the host controls the ecosystem. Ecosystem drivers in addition to oxygen levels might explain dysbiosis in settings where expansion of Proteobacteria is not observed, but the identities of such drivers remain unknown. Tackling this question will require a 'new bacteriology' (Ref. 98) to conduct mechanistic studies on microbial physiology in its natural context of a gut-associated microbial community.
As we venture forward, it is helpful to reflect on the skill sets best suited to tackle challenges that lie ahead. The germ-organ theory instructs a shift from microbial community profiling towards understanding host-mediated control of microbial organ ecology. Although the profiling approaches required to tackle this question can be performed by service facilities, mechanistic follow-up studies aimed at understanding trophic networks, the influence of host physiology on the microbial ecosystem and the role that microbiota-derived metabolites have in health and disease will require expertise in both bacterial physiology and immunology, which can be obtained only by attracting scientists from these fields to enter microbiota research. Researchers in the field of bacterial pathogenesis seem particularly well prepared to enter this arena as their research exposes them to bacterial physiology as well as to the interaction of pathogens with the immune system, thus providing an ideal background for mechanistic microbiota research. Alternatively, collaboration between scientists engaged in bioinformatics, bacterial physiology and ecology, cell biology and immunology could accelerate decryption of the mechanistic basis for microbial contributions to non-communicable diseases. Although funding agencies have not provided major incentives for bacterial physiologists and immunologists to enter microbiota research, history suggests that this might be a move in the right direction as we stand at the threshold of a 'second golden age of microbiology'.
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Turnbaugh, P. J. et al. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 444, 1027–1031 (2006).
Henao-Mejia, J. et al. Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature 482, 179–185 (2012).
Maslowski, K. M. et al. Regulation of inflammatory responses by gut microbiota and chemoattractant receptor GPR43. Nature 461, 1282–1286 (2009).
Russell, S. L. et al. Early life antibiotic-driven changes in microbiota enhance susceptibility to allergic asthma. EMBO Rep. 13, 440–447 (2012).
Trompette, A. et al. Gut microbiota metabolism of dietary fiber influences allergic airway disease and hematopoiesis. Nat. Med. 20, 159–166 (2014).
Wang, Z. et al. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature 472, 57–63 (2011).
Castellarin, M. et al. Fusobacterium nucleatum infection is prevalent in human colorectal carcinoma. Genome Res. 22, 299–306 (2012).
Kostic, A. D. et al. Genomic analysis identifies association of Fusobacterium with colorectal carcinoma. Genome Res. 22, 292–298 (2012).
Yoshimoto, S. et al. Obesity-induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature 499, 97–101 (2013).
Cani, P. D. et al. Changes in gut microbiota control metabolic endotoxemia-induced inflammation in high-fat diet-induced obesity and diabetes in mice. Diabetes 57, 1470–1481 (2008).
Hsiao, E. Y. et al. Microbiota modulate behavioral and physiological abnormalities associated with neurodevelopmental disorders. Cell 155, 1451–1463 (2013).
Minter, M. R. et al. Antibiotic-induced perturbations in gut microbial diversity influences neuro-inflammation and amyloidosis in a murine model of Alzheimer's disease. Sci. Rep. 6, 30028 (2016).
Methot, P. O. & Alizon, S. What is a pathogen? Toward a process view of host-parasite interactions. Virulence 5, 775–785 (2014).
Casadevall, A. & Pirofski, L. A. Microbiology: Ditch the term pathogen. Nature 516, 165–166 (2014).
Altmann, D. & Boyton, R. Nomenclature: Replace 'pathogens' with 'perceptogens'. Nature 518, 35 (2015).
Sultana, S., Sarker, S. A. & Brussow, H. What happened to Koch's postulates in diarrhea? Environ. Microbiol. https://doi.org/10.1111/1462-2920.13787 (2017).
Cani, P. D. Gut microbiota — at the intersection of everything? Nat. Rev. Gastroenterol. Hepatol. 14, 321–322 (2017).
Lopez, C. A., Kingsbury, D. D., Velazquez, E. M. & Baumler, A. J. Collateral damage: microbiota-derived metabolites and immune function in the antibiotic era. Cell Host Microbe 16, 156–163 (2014).
O'Hara, A. M. & Shanahan, F. The gut flora as a forgotten organ. EMBO Rep. 7, 688–693 (2006).
Tap, J. et al. Towards the human intestinal microbiota phylogenetic core. Environ. Microbiol. 11, 2574–2584 (2009).
Shreiner, A., Huffnagle, G. B. & Noverr, M. C. The “Microflora Hypothesis” of allergic disease. Adv. Exp. Med. Biol. 635, 113–134 (2008).
Packey, C. D. & Sartor, R. B. Commensal bacteria, traditional and opportunistic pathogens, dysbiosis and bacterial killing in inflammatory bowel diseases. Curr. Opin. Infect. Dis. 22, 292–301 (2009).
Round, J. L. & Mazmanian, S. K. The gut microbiota shapes intestinal immune responses during health and disease. Nat. Rev. Immunol. 9, 313–323 (2009).
Bjorksten, B. in Microbial–Host Interaction: Tolerance versus Allergy. 64th Nestlé Nutrition Institute Workshop, Pediatric Program (eds Brandtzaeg, P., Isolauri, E. & Prescott, S. L.) 11–22 (Sydney, 2008).
Hajishengallis, G., Darveau, R. P. & Curtis, M. A. The keystone-pathogen hypothesis. Nat. Rev. Microbiol. 10, 717–725 (2012).
Franzenburg, S. et al. Distinct antimicrobial peptide expression determines host species-specific bacterial associations. Proc. Natl Acad. Sci. USA 110, E3730–E3738 (2013).
Ruby, E. G. Lessons from a cooperative, bacterial-animal association: the Vibrio fischeri-Euprymna scolopes light organ symbiosis. Annu. Rev. Microbiol. 50, 591–624 (1996).
Garrido, D., Dallas, D. C. & Mills, D. A. Consumption of human milk glycoconjugates by infant-associated bifidobacteria: mechanisms and implications. Microbiology 159, 649–664 (2013).
Salzman, N. H. et al. Enteric defensins are essential regulators of intestinal microbial ecology. Nat. Immunol. 11, 76–83 (2010).
Hayase, E. et al. R-Spondin1 expands Paneth cells and prevents dysbiosis induced by graft-versus-host disease. J. Exp. Med. https://doi.org/10.1084/jem.20170418 (2017).
Byndloss, M. X. et al. Microbiota-activated PPAR-γ-signaling inhibits dysbiotic Enterobacteriaceae expansion. Science 357, 570–575 (2017).
Furuta, G. T. et al. Hypoxia-inducible factor 1-dependent induction of intestinal trefoil factor protects barrier function during hypoxia. J. Exp. Med. 193, 1027–1034 (2001).
Eckburg, P. B. et al. Diversity of the human intestinal microbial flora. Science 308, 1635–1638 (2005).
Faith, J. J. et al. The long-term stability of the human gut microbiota. Science 341, 1237439 (2013).
Rivera-Chavez, F. & Baumler, A. J. The pyromaniac inside you: Salmonella metabolism in the host gut. Annu. Rev. Microbiol. 69, 31–48 (2015).
Freter, R., Brickner, H., Fekete, J., Vickerman, M. M. & Carey, K. E. Survival and implantation of Escherichia coli in the intestinal tract. Infect. Immun. 39, 686–703 (1983).
Roy, C. C., Kien, C. L., Bouthillier, L. & Levy, E. Short-chain fatty acids: ready for prime time? Nutr. Clin. Pract. 21, 351–366 (2006).
Fischbach, M. A. & Sonnenburg, J. L. Eating for two: how metabolism establishes interspecies interactions in the gut. Cell Host Microbe 10, 336–347 (2011).
Nakamura, N., Lin, H. C., McSweeney, C. S., Mackie, R. I. & Gaskins, H. R. Mechanisms of microbial hydrogen disposal in the human colon and implications for health and disease. Annu. Rev. Food Sci. Technol. 1, 363–395 (2010).
Nicholson, J. K. et al. Host-gut microbiota metabolic interactions. Science 336, 1262–1267 (2012).
den Besten, G. et al. The role of short-chain fatty acids in the interplay between diet, gut microbiota, and host energy metabolism. J. Lipid Res. 54, 2325–2340 (2013).
Velazquez, O. C., Lederer, H. M. & Rombeau, J. L. Butyrate and the colonocyte. Production, absorption, metabolism, and therapeutic implications. Adv. Exp. Med. Biol. 427, 123–134 (1997).
Millard, A. L. et al. Butyrate affects differentiation, maturation and function of human monocyte-derived dendritic cells and macrophages. Clin. Exp. Immunol. 130, 245–255 (2002).
Wang, B., Morinobu, A., Horiuchi, M., Liu, J. & Kumagai, S. Butyrate inhibits functional differentiation of human monocyte-derived dendritic cells. Cell. Immunol. 253, 54–58 (2008).
Atarashi, K. et al. Induction of colonic regulatory T cells by indigenous Clostridium species. Science 331, 337–341 (2011).
Arpaia, N. et al. Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature 504, 451–455 (2013).
Furusawa, Y. et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature 504, 446–450 (2013).
Smith, P. M. et al. The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science 341, 569–573 (2013).
Chang, P. V., Hao, L., Offermanns, S. & Medzhitov, R. The microbial metabolite butyrate regulates intestinal macrophage function via histone deacetylase inhibition. Proc. Natl Acad. Sci. USA 111, 2247–2252 (2014).
Singh, N. et al. Activation of Gpr109a, receptor for niacin and the commensal metabolite butyrate, suppresses colonic inflammation and carcinogenesis. Immunity 40, 128–139 (2014).
Rigottier-Gois, L. Dysbiosis in inflammatory bowel diseases: the oxygen hypothesis. ISME J. 7, 1256–1261 (2013).
Rivera-Chavez, F., Lopez, C. A. & Baumler, A. J. Oxygen as a driver of gut dysbiosis. Free Radic. Biol. Med. 105, 93–101 (2017).
Devkota, S. et al. Dietary-fat-induced taurocholic acid promotes pathobiont expansion and colitis in Il10−/− mice. Nature 487, 104–108 (2012).
Martinez-Medina, M. et al. Western diet induces dysbiosis with increased E coli in CEABAC10 mice, alters host barrier function favouring AIEC colonisation. Gut 63, 116–124 (2014).
Vollaard, E. J., Clasener, H. A. & Janssen, A. J. Co-trimoxazole impairs colonization resistance in healthy volunteers. J. Antimicrob. Chemother. 30, 685–691 (1992).
Carroll, I. M., Ringel-Kulka, T., Siddle, J. P. & Ringel, Y. Alterations in composition and diversity of the intestinal microbiota in patients with diarrhea-predominant irritable bowel syndrome. Neurogastroenterol. Motil. 24, 521–530 (2012).
Krogius-Kurikka, L. et al. Microbial community analysis reveals high level phylogenetic alterations in the overall gastrointestinal microbiota of diarrhoea-predominant irritable bowel syndrome sufferers. BMC Gastroenterol. 9, 95 (2009).
Morgan, X. C. et al. Dysfunction of the intestinal microbiome in inflammatory bowel disease and treatment. Genome Biol. 13, R79 (2012).
Ferreira, R. B. et al. The intestinal microbiota plays a role in Salmonella-induced colitis independent of pathogen colonization. PLoS ONE 6, e20338 (2011).
Normann, E., Fahlen, A., Engstrand, L. & Lilja, H. E. Intestinal microbial profiles in extremely preterm infants with and without necrotizing enterocolitis. Acta Paediatr. 102, 129–136 (2013).
Arthur, J. C. et al. Intestinal inflammation targets cancer-inducing activity of the microbiota. Science 338, 120–123 (2012).
Lupp, C. et al. Host-mediated inflammation disrupts the intestinal microbiota and promotes the overgrowth of Enterobacteriaceae. Cell Host Microbe 2, 204 (2007).
Winter, S. E. et al. Host-derived nitrate boosts growth of E. coli in the inflamed gut. Science 339, 708–711 (2013).
Garrett, W. S. et al. Enterobacteriaceae act in concert with the gut microbiota to induce spontaneous and maternally transmitted colitis. Cell Host Microbe 8, 292–300 (2010).
Winter, S. E. et al. Gut inflammation provides a respiratory electron acceptor for Salmonella. Nature 467, 426–429 (2010).
Haag, L. M. et al. Intestinal microbiota shifts towards elevated commensal Escherichia coli loads abrogate colonization resistance against Campylobacter jejuni in mice. PLoS ONE 7, e35988 (2012).
Bohnhoff, M., Drake, B. L. & Miller, C. P. Effect of streptomycin on susceptibility of intestinal tract to experimental Salmonella infection. Proc. Soc. Exp. Biol. Med. 86, 132–137 (1954).
Saito, K. Studies on the habitation of pathogenic Escherichia coli in the intestinal tract of mice. I. Comparative experiments on the habitation of each type of resistant pathogenic Escherichia coli under an administration of streptomycin [Japanese]. Paediatr. Jpn. 65, 385–393 (1961).
Saito, K. Studies on the habitation of pathogenic Escherichia coli in the intestinal tract of mice. II. Experimental inoculation of type 055 Escherichia coli after long-term administration of streptomycin [Japanese]. Paediatr. Jpn. 65, 394–399 (1961).
Shin, N. R., Whon, T. W. & Bae, J. W. Proteobacteria: microbial signature of dysbiosis in gut microbiota. Trends Biotechnol. 33, 496–503 (2015).
Meynell, G. G. Antibacterial mechanisms of the mouse gut. II. The role of Eh and volatile fatty acids in the normal gut. Br. J. Exp. Pathol. 44, 209–219 (1963).
Gill, N. et al. Neutrophil elastase alters the murine gut microbiota resulting in enhanced Salmonella colonization. PLoS ONE 7, e49646 (2012).
Deatherage Kaiser, B. L. et al. A multi-omic view of host-pathogen-commensal interplay in -mediated intestinal infection. PLoS ONE 8, e67155 (2013).
Rivera-Chavez, F. et al. Depletion of butyrate-producing Clostridia from the gut microbiota drives an aerobic luminal expansion of Salmonella. Cell Host Microbe 19, 443–454 (2016).
Kelly, C. J. et al. Crosstalk between microbiota-derived short-chain fatty acids and intestinal epithelial HIF augments tissue barrier function. Cell Host Microbe 17, 662–671 (2015).
Fan, Y. Y. et al. A bioassay to measure energy metabolism in mouse colonic crypts, organoids, and sorted stem cells. Am. J. Physiol. Gastrointest. Liver Physiol. 309, G1–G9 (2015).
Hughes, E. R. et al. Microbial respiration and formate oxidation as metabolic signatures of inflammation-associated dysbiosis. Cell Host Microbe 21, 208–219 (2017).
Lopez, C. A. et al. Virulence factors enhance Citrobacter rodentium expansion through aerobic respiration. Science 353, 1249–1253 (2016).
Ahmed, I. et al. Critical roles of Notch and Wnt/β-catenin pathways in the regulation of hyperplasia and/or colitis in response to bacterial infection. Infect. Immun. 80, 3107–3121 (2012).
Chandrakesan, P. et al. Novel changes in NF-κB activity during progression and regression phases of hyperplasia: role of MEK, ERK, and p38. J. Biol. Chem. 285, 33485–33498 (2010).
Buffie, C. G. et al. Precision microbiome reconstitution restores bile acid mediated resistance to Clostridium difficile. Nature 517, 205–208 (2015).
Bennett, B. J. et al. Trimethylamine-N-oxide, a metabolite associated with atherosclerosis, exhibits complex genetic and dietary regulation. Cell Metab. 17, 49–60 (2013).
Tsolis, R. M., Adams, L. G., Ficht, T. A. & Baumler, A. J. Contribution of Salmonella typhimurium virulence factors to diarrheal disease in calves. Infect. Immun. 67, 4879–4885 (1999).
Campbell, E. L. et al. Transmigrating neutrophils shape the mucosal microenvironment through localized oxygen depletion to influence resolution of inflammation. Immunity 40, 66–77 (2014).
Kamdar, K. et al. Genetic and metabolic signals during acute enteric bacterial infection alter the microbiota and drive progression to chronic inflammatory disease. Cell Host Microbe 19, 21–31 (2016).
Ng, K. M. et al. Microbiota-liberated host sugars facilitate post-antibiotic expansion of enteric pathogens. Nature 502, 96–99 (2013).
Faber, F. et al. Host-mediated sugar oxidation promotes post-antibiotic pathogen expansion. Nature 534, 697–699 (2016).
Kelly, C. P., Pothoulakis, C. & LaMont, J. T. Clostridium difficile colitis. N. Engl. J. Med. 330, 257–262 (1994).
Sorg, J. A. & Sonenshein, A. L. Bile salts and glycine as cogerminants for Clostridium difficile spores. J. Bacteriol. 190, 2505–2512 (2008).
Theriot, C. M. et al. Antibiotic-induced shifts in the mouse gut microbiome and metabolome increase susceptibility to Clostridium difficile infection. Nat. Commun. 5, 3114 (2014).
Wilson, K. H. Efficiency of various bile salt preparations for stimulation of Clostridium difficile spore germination. J. Clin. Microbiol. 18, 1017–1019 (1983).
van Nood, E. et al. Duodenal infusion of donor feces for recurrent Clostridium difficile. N. Engl. J. Med. 368, 407–415 (2013).
Weingarden, A. R. et al. Microbiota transplantation restores normal fecal bile acid composition in recurrent Clostridium difficile infection. Am. J. Physiol. Gastrointest. Liver Physiol. 306, G310–G319 (2014).
Fung, T. T. et al. Association between dietary patterns and plasma biomarkers of obesity and cardiovascular disease risk. Am. J. Clin. Nutr. 73, 61–67 (2001).
Zhu, Y. et al. Carnitine metabolism to trimethylamine by an unusual Rieske-type oxygenase from human microbiota. Proc. Natl Acad. Sci. USA 111, 4268–4273 (2014).
Rath, S., Heidrich, B., Pieper, D. H. & Vital, M. Uncovering the trimethylamine-producing bacteria of the human gut microbiota. Microbiome 5, 54 (2017).
Foster, K. R., Schluter, J., Coyte, K. Z. & Rakoff-Nahoum, S. The evolution of the host microbiome as an ecosystem on a leash. Nature 548, 43–51 (2017).
Pedron, T., Nigro, G. & Sansonetti, P. J. From homeostasis to pathology: decrypting microbe-host symbiotic signals in the intestinal crypt. Philos. Trans. R. Soc. Lond. B. Biol. Sci. https://doi.org/10.1098/rstb.2015.0500 (2016).
Bordenave, G. Louis Pasteur (1822–1895). Microbes Infect. 5, 553–560 (2003).
Pasteur, L. Etudes Sur La Maladie Des Vers A Soie. Vol. 1,2 (Gauthier-Villars, 1870).
Hansen, G. H. A. On the etiology of leprosy. Br. Foreign Med. Chir. Rev. 55, 459–489 (1875).
Losch, F. Massenhafte entwickelung von amöben im dickdarm. Arch. Pathol. Anat. Physiol. 65, 196–211 (1875).
Koch, R. Untersuchungen über Bakterien, V. Die aetiologie der milzbrandkrankheit, begründet auf der entwicklungsgeschichte des Bacillus anthracis. Beiträge Biol. Pflanzen 2, 277–308 (1877).
Eberth, K. J. Die organismen in den organen bei typhus abdominalis [German]. Virchows Arch. 81, 58–74 (1880).
Pasteur, L. Note sur la maladie nouvelle provoquee par la salive d'un enfant mort de la rage [French]. C. R. Acad. Sci. 92, 159–165 (1881).
Sternberg, G. M. A fatal form of septicaemia, produced by the injection of human saliva. An experimental research. Bull. Nat. Board Health USA 2, 781–783 (1881).
Koch, R. Die aetiologie der tuberkulose. Berliner Klinische Wochenschrift 15, 221–230 (1882).
Koch, R. Der zweite bericht der deutschen cholerakommission. Dt. Med. Wochenschr. 9, 743–744 (1883).
Koch, R. Ueber die cholerabakterien. Dt. Med. Wochenschr. 10, 725–728 (1884).
Gaffky, G. Zur ätiologie des abdominaltyphus. Mitteilungen Kaiserlichen Gesundheitsamt 2, 372–420 (1884).
Loeffler, F. Untersuchungen über die bedeutung der mikroorganismen für die entstehung der diphtherie beim menschen, bei der taube und beim kalbe. Mitth. a. d. Kaiserl. Gesundheitsamtes 2, 421–499 (1884).
Nicolaier, A. Ueber infectiösen tetanus. Dt. Med. Wochenschr. 10, 842–844 (1884).
Yersin, A. La peste bubonique à Hong-Kong. Ann. l'Institut Pasteur 8, 662–667 (1894).
Kitasato, S. The bacillus of bubonic plague. Lancet 144, 428–430 (1894).
Shiga, K. Ueber den dysenterie-bacillus (Bacillus dysenteriae). Zentralbl Bakteriol Orig 24, 913–918 (1898).
Roux, E., Y. A. Contribution à l'étude de la diphtérie Ann. Inst. Pasteur 2, 421–499 (1888).
Conradi, H. Über lösliche, durch aseptische autolyse erhaltene giftstoffe von ruhr- und typhus-bazillen. Dtsch. Med. Wochenschr. 29, 26–28 (1903).
Neisser, M. & Shiga, K. Ueber freie receptoren von typhus- und dysenteriebazillen und über das dysenterietoxin. Dtsch. Med. Wochenschr. 29, 61–62 (1903).
von Behring, E. & Kitasato, S. Ueber das zustandekommen der diphtherie — immunität und der tetanus — immunität bei tieren. Dt. Med. Wochenschr. 16, 1113–1114 (1890).
von Behring, E. Untersuchungen über das zustandekommen de diphtherie — immunität bei tieren. Dt. Med. Wochenschr. 16, 1145–1148 (1890).
Pasteur, L., Roux, E. & Chamberland, C. Summary report of the experiments conducted at Pouilly-le-Fort, near Melun, on the anthrax vaccination, 1881. C. R. Acad. Sci. 92, 1378–1383 (1881).
Pasteur, L. Methode pour prevenir l rage apres morsure. C. R. Acad. Sci. 101, 765–772 (1885).
Pfeiffer, R. & Kolle, W. Experimentele untersuchungen zur frage der schutzimpfungen des menschen gegen den typhus abdominalis. Dtsch. Med. Wochenschr. 22, 735–737 (1896).
Jennewein, J. et al. Low-oxygen tensions found in Salmonella-infected gut tissue boost Salmonella replication in macrophages by impairing antimicrobial activity and augmenting Salmonella virulence. Cell. Microbiol. 17, 1833–1847 (2015).
Acknowledgements
Work in the authors' laboratory was supported by public health service grants AI112445, AI112949 and AI096528 (A.J.B.).
Author information
Authors and Affiliations
Contributions
M.X.B. and A.J.B. substantially contributed to the discussion of content and review and editing of the manuscript before submission.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
About this article

Cite this article
Byndloss, M., Bäumler, A. The germ-organ theory of non-communicable diseases. Nat Rev Microbiol 16, 103–110 (2018). https://doi.org/10.1038/nrmicro.2017.158
Published:
Issue Date:
DOI: https://doi.org/10.1038/nrmicro.2017.158
This article is cited by
-
Mechanisms of medicinal, pharmaceutical, and immunomodulatory action of probiotics bacteria and their secondary metabolites against disease management: an overview
Folia Microbiologica (2024)
-
Metabolic Interaction Between Host and the Gut Microbiota During High-Fat Diet-Induced Colorectal Cancer
Journal of Microbiology (2024)
-
Association of gut microbial dysbiosis with disease severity, response to therapy and disease outcomes in Indian patients with COVID-19
Gut Pathogens (2023)
-
Triangulating nutrigenomics, metabolomics and microbiomics toward personalized nutrition and healthy living
Human Genomics (2023)
-
Gut instincts in neuroimmunity from the eighteenth to twenty-first centuries
Seminars in Immunopathology (2022)
