
Key Points
-
Healthy mitochondria are essential for normal kidney function; mitochondrial cytopathies can result in renal disease and mitochondrial damage has a role in the pathophysiology of acute kidney injury (AKI)
-
Although mitochondrial diseases are characterized by maternal inheritance, many mitochondrial disorders are caused by mutations in nuclear genes and are inherited according to classic Mendelian rules
-
Most mitochondrial diseases with kidney involvement cause tubular defects; however, mutations in the coenzyme Q10 biosynthesis pathway and the mtDNA 3243 A>G mutation primarily cause glomerular disease
-
Diagnosis of genetic mitochondrial disorders increasingly relies on new sequencing techniques, but thorough biochemical and clinical characterization of patients is essential to guide these analyses
-
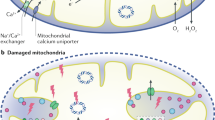
In AKI, mitochondrial dysfunction precedes and participates in the physiopathology of tissue damage; mitochondrial biogenesis might represent a crucial step in the recovery phase
-
Potential therapies that target mitochondrial dynamics, mitophagy and/or mitochondrial biogenesis might limit renal damage during AKI and promote recovery of kidney function
Abstract
Mitochondria are increasingly recognized as key players in genetic and acquired renal diseases. Most mitochondrial cytopathies that cause renal symptoms are characterized by tubular defects, but glomerular, tubulointerstitial and cystic diseases have also been described. For example, defects in coenzyme Q10 (CoQ10) biosynthesis and the mitochondrial DNA 3243 A>G mutation are important causes of focal segmental glomerulosclerosis in children and in adults, respectively. Although they sometimes present with isolated renal findings, mitochondrial diseases are frequently associated with symptoms related to central nervous system and neuromuscular involvement. They can result from mutations in nuclear genes that are inherited according to classic Mendelian rules or from mutations in mitochondrial DNA, which are transmitted according to more complex rules of mitochondrial genetics. Diagnosis of mitochondrial disorders involves clinical characterization of patients in combination with biochemical and genetic analyses. In particular, prompt diagnosis of CoQ10 biosynthesis defects is imperative because of their potentially reversible nature. In acute kidney injury (AKI), mitochondrial dysfunction contributes to the physiopathology of tissue injury, whereas mitochondrial biogenesis has an important role in the recovery of renal function. Potential therapies that target mitochondrial dysfunction or promote mitochondrial regeneration are being developed to limit renal damage during AKI and promote repair of injured tissue.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
$209.00 per year
only $17.42 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout




Similar content being viewed by others


References
Andersson, S. G. et al. The genome sequence of Rickettsia prowazekii and the origin of mitochondria. Nature 396, 133–140 (1998).
Frey, T. G. & Mannella, C. A. The internal structure of mitochondria. Trends Biochem. Sci. 25, 319–324 (2000).
Anderson, S. et al. Sequence and organization of the human mitochondrial genome. Nature 290, 457–465 (1981).
Schon, E. A., DiMauro, S. & Hirano, M. Human mitochondrial DNA: roles of inherited and somatic mutations. Nat. Rev. Genet. 13, 878–890 (2012).
Opalinska, M. & Meisinger, C. Metabolic control via the mitochondrial protein import machinery. Curr. Opin. Cell Biol. 33, 42–48 (2015).
Dimmer, K. S. & Scorrano, L. (De)constructing mitochondria: what for? Physiology (Bethesda) 21, 233–241 (2006).
Scorrano, L. et al. A distinct pathway remodels mitochondrial cristae and mobilizes cytochrome c during apoptosis. Dev. Cell 2, 55–67 (2002).
Acin-Perez, R., Fernandez-Silva, P., Peleato, M. L., Perez-Martos, A. & Enriquez, J. A. Respiratory active mitochondrial supercomplexes. Mol. Cell 32, 529–539 (2008).
Ghezzi, D. & Zeviani, M. Assembly factors of human mitochondrial respiratory chain complexes: physiology and pathophysiology. Adv. Exp. Med. Biol. 748, 65–106 (2012).
DiMauro, S., Schon, E. A., Carelli, V. & Hirano, M. The clinical maze of mitochondrial neurology. Nat. Rev. Neurol. 9, 429–444 (2013).
Doimo, M. et al. Genetics of coenzyme q10 deficiency. Mol. Syndromol. 5, 156–162 (2014).
Nguyen, T. P. et al. Molecular characterization of the human COQ5 C-methyltransferase in coenzyme Q10 biosynthesis. Biochim. Biophys. Acta 1841, 1628–1638 (2014).
Allen, J. W. Cytochrome c biogenesis in mitochondria — systems III and V. FEBS J. 278, 4198–4216 (2011).
DiMauro, S. & Schon, E. A. Mitochondrial disorders in the nervous system. Annu. Rev. Neurosci. 31, 91–123 (2008).
Mayr, J. A. et al. Spectrum of combined respiratory chain defects. J. Inherit. Metab. Dis. 38, 4198–4216 (2015).
Gorman, G. S. et al. Prevalence of nuclear and mitochondrial DNA mutations related to adult mitochondrial disease. Ann. Neurol. 77, 753–759 (2015).
Petruzzella, V. et al. Deep sequencing unearths nuclear mitochondrial sequences under Leber's hereditary optic neuropathy-associated false heteroplasmic mitochondrial DNA variants. Hum. Mol. Genet. 21, 3753–3764 (2012).
Giordano, C. et al. Pathogenesis of the deafness-associated A1555G mitochondrial DNA mutation. Biochem. Biophys. Res. Commun. 293, 521–529 (2002).
Doimo, M. et al. Effect of vanillic acid on COQ6 mutants identified in patients with coenzyme Q10 deficiency. Biochim. Biophys. Acta 1842, 1–6 (2014).
Emma, F., Bertini, E., Salviati, L. & Montini, G. Renal involvement in mitochondrial cytopathies. Pediatr. Nephrol. 27, 539–550 (2012).
Quinzii, C. M. et al. Reactive oxygen species, oxidative stress, and cell death correlate with level of CoQ10 deficiency. FASEB J. 24, 3733–3743 (2010).
Morison, I. M. et al. A mutation of human cytochrome c enhances the intrinsic apoptotic pathway but causes only thrombocytopenia. Nat. Genet. 40, 387–389 (2008).
De Rocco, D. et al. Mutations of cytochrome c identified in patients with thrombocytopenia THC4 affect both apoptosis and cellular bioenergetics. Biochim. Biophys. Acta 1842, 269–274 (2014).
Desbats, M. A. et al. Primary coenzyme Q10 deficiency presenting as fatal neonatal multiorgan failure. Eur. J. Hum. Genet. 23, 1254–1258 (2015).
Emma, F., Montini, G., Salviati, L. & Dionisi-Vici, C. Renal mitochondrial cytopathies. Int. J. Nephrol. 2011, 609213 (2011).
Bodemer, C. et al. Hair and skin disorders as signs of mitochondrial disease. Pediatrics 103, 428–433 (1999).
Niaudet, P. & Rotig, A. The kidney in mitochondrial cytopathies. Kidney Int. 51, 1000–1007 (1997).
Deetjen, P. Measurement of metabolism during renal work. Int. J. Biochem. 12, 243–244 (1980).
Thurau, K. Renal Na-reabsorption and O2-uptake in dogs during hypoxia and hydrochlorothiazide infusion. Proc. Soc. Exp. Biol. Med. 106, 714–717 (1961).
Ogier, H. et al. de Toni-Fanconi-Debré syndrome with Leigh syndrome revealing severe muscle cytochrome c oxidase deficiency. J. Pediatr. 112, 734–739 (1988).
Niaudet, P. et al. Deletion of the mitochondrial DNA in a case of de Toni-Debré-Fanconi syndrome and Pearson syndrome. Pediatr. Nephrol. 8, 164–168 (1994).
Rotig, A. Renal disease and mitochondrial genetics. J. Nephrol. 16, 286–292 (2003).
Morris, A. A. et al. Neonatal Fanconi syndrome due to deficiency of complex III of the respiratory chain. Pediatr. Nephrol. 9, 407–411 (1995).
Au, K. M. et al. Mitochondrial DNA deletion in a girl with Fanconi's syndrome. Pediatr. Nephrol. 22, 136–140 (2007).
Kuwertz-Broking, E. et al. Renal Fanconi syndrome: first sign of partial respiratory chain complex IV deficiency. Pediatr. Nephrol. 14, 495–498 (2000).
Mochizuki, H. et al. Mitochondrial encephalomyopathies preceded by de-Toni-Debré-Fanconi syndrome or focal segmental glomerulosclerosis. Clin. Nephrol. 46, 347–352 (1996).
De Meirleir, L. et al. Clinical and diagnostic characteristics of complex III deficiency due to mutations in the BCS1L gene. Am. J. Med. Genet. A 121A, 126–131 (2003).
Liu, H. M. et al. A novel 3670-base pair mitochondrial DNA deletion resulting in multi-systemic manifestations in a child. Pediatr. Neonatol. 53, 264–268 (2012).
Tzoufi, M. et al. A rare case report of simultaneous presentation of myopathy, Addison's disease, primary hypoparathyroidism, and Fanconi syndrome in a child diagnosed with Kearns–Sayre syndrome. Eur. J. Pediatr. 172, 557–561 (2013).
Pitchon, E. M. et al. Patient with Fanconi syndrome (FS) and retinitis pigmentosa (RP) caused by a deletion and duplication of mitochondrial DNA (mtDNA). Klin. Monbl Augenheilkd 224, 340–343 (2007).
Mori, K., Narahara, K., Ninomiya, S., Goto, Y. & Nonaka, I. Renal and skin involvement in a patient with complete Kearns–Sayre syndrome. Am. J. Med. Genet. 38, 583–587 (1991).
Topaloglu, R. et al. Two new cases with Pearson syndrome and review of Hacettepe experience. Turk. J. Pediatr. 50, 572–576 (2008).
O'Toole, J. F. Renal manifestations of genetic mitochondrial disease. Int. J. Nephrol. Renovasc. Dis. 7, 57–67 (2014).
Duncan, A. J. et al. A nonsense mutation in COQ9 causes autosomal-recessive neonatal-onset primary coenzyme Q10 deficiency: a potentially treatable form of mitochondrial disease. Am. J. Hum. Genet. 84, 558–566 (2009).
Lee, Y. S. et al. Mitochondrial tubulopathy: the many faces of mitochondrial disorders. Pediatr. Nephrol. 16, 710–712 (2001).
Gilbert, R. D. & Emms, M. Pearson's syndrome presenting with Fanconi syndrome. Ultrastruct. Pathol. 20, 473–475 (1996).
Ezgu, F. et al. Severe renal tubulopathy in a newborn due to BCS1L gene mutation: effects of different treatment modalities on the clinical course. Gene 528, 364–366 (2013).
Gai, X. et al. Mutations in FBXL4, encoding a mitochondrial protein, cause early-onset mitochondrial encephalomyopathy. Am. J. Hum. Genet. 93, 482–495 (2013).
Tucker, E. J. et al. Next-generation sequencing in molecular diagnosis: NUBPL mutations highlight the challenges of variant detection and interpretation. Hum. Mutat. 33, 411–418 (2012).
Matsutani, H. et al. Partial deficiency of cytochrome c oxidase with isolated proximal renal tubular acidosis and hypercalciuria. Child Nephrol. Urol. 12, 221–224 (1992).
Martin-Hernandez, E. et al. Renal pathology in children with mitochondrial diseases. Pediatr. Nephrol. 20, 1299–1305 (2005).
Emma, F. et al. 'Bartter-like' phenotype in Kearns–Sayre syndrome. Pediatr. Nephrol. 21, 355–360 (2006).
Goto, Y. et al. Renal tubular involvement mimicking Bartter syndrome in a patient with Kearns–Sayre syndrome. J. Pediatr. 116, 904–910 (1990).
Visapaa, I. et al. GRACILE syndrome, a lethal metabolic disorder with iron overload, is caused by a point mutation in BCS1L. Am. J. Hum. Genet. 71, 863–876 (2002).
Wilson, F. H. et al. A cluster of metabolic defects caused by mutation in a mitochondrial tRNA. Science 306, 1190–1194 (2004).
Reilly, R. F. & Ellison, D. H. Mammalian distal tubule: physiology, pathophysiology, and molecular anatomy. Physiol. Rev. 80, 277–313 (2000).
McCormick, J. A. & Ellison, D. H. Distal convoluted tubule. Compr. Physiol. 5, 45–98 (2015).
Simon, D. B. et al. Gitelman's variant of Bartter's syndrome, inherited hypokalaemic alkalosis, is caused by mutations in the thiazide-sensitive Na–Cl cotransporter. Nat. Genet. 12, 24–30 (1996).
Soltoff, S. P. ATP and the regulation of renal cell function. Annu. Rev. Physiol. 48, 9–31 (1986).
Klootwijk, E. D. et al. Mistargeting of peroxisomal EHHADH and inherited renal Fanconi's syndrome. N. Engl. J. Med. 370, 129–138 (2014).
Jefferson, J. A., Alpers, C. E. & Shankland, S. J. Podocyte biology for the bedside. Am. J. Kidney Dis. 58, 835–845 (2011).
Muller-Deile, J. & Schiffer, M. The podocyte power-plant disaster and its contribution to glomerulopathy. Front. Endocrinol. (Lausanne) 5, 209 (2014).
Saleem, M. A. 100 ways to kill a podocyte. Nephrol. Dial. Transplant. http://dx.doi.org/10.1093/ndt/gfu363 (2015).
Higgins, G. C. & Coughlan, M. T. Mitochondrial dysfunction and mitophagy: the beginning and end to diabetic nephropathy? Br. J. Pharmacol. 171, 1917–1942 (2014).
Che, R., Yuan, Y., Huang, S. & Zhang, A. Mitochondrial dysfunction in the pathophysiology of renal diseases. Am. J. Physiol. Renal Physiol. 306, F367–F378 (2014).
Kawakami, T. et al. Deficient autophagy results in mitochondrial dysfunction and FSGS. J. Am. Soc. Nephrol. 26, 1040–1052 (2015).
Montini, G., Malaventura, C. & Salviati, L. Early coenzyme Q10 supplementation in primary coenzyme Q10 deficiency. N. Engl. J. Med. 358, 2849–2850 (2008).
Heeringa, S. F. et al. COQ6 mutations in human patients produce nephrotic syndrome with sensorineural deafness. J. Clin. Invest. 121, 2013–2024 (2011).
Ashraf, S. et al. ADCK4 mutations promote steroid-resistant nephrotic syndrome through CoQ10 biosynthesis disruption. J. Clin. Invest. 123, 5179–5189 (2013).
Gasser, D. L. et al. Focal segmental glomerulosclerosis is associated with a PDSS2 haplotype and, independently, with a decreased content of coenzyme Q10 . Am. J. Physiol. Renal Physiol. 305, F1228–1238 (2013).
Quinzii, C. et al. A mutation in para-hydroxybenzoate-polyprenyl transferase (COQ2) causes primary coenzyme Q10 deficiency. Am. J. Hum. Genet. 78, 345–349 (2006).
Lopez, L. C. et al. Leigh syndrome with nephropathy and CoQ10 deficiency due to decaprenyl diphosphate synthase subunit 2 (PDSS2) mutations. Am. J. Hum. Genet. 79, 1125–1129 (2006).
Vasta, V., Merritt, J. L. 2nd, Saneto, R. P. & Hahn, S. H. Next-generation sequencing for mitochondrial diseases: a wide diagnostic spectrum. Pediatr. Int. 54, 585–601 (2012).
Salviati, L. et al. Infantile encephalomyopathy and nephropathy with CoQ10 deficiency: a CoQ10-responsive condition. Neurology 65, 606–608 (2005).
Diomedi-Camassei, F. et al. COQ2 nephropathy: a newly described inherited mitochondriopathy with primary renal involvement. J. Am. Soc. Nephrol. 18, 2773–2780 (2007).
McCarthy, H. J. et al. Simultaneous sequencing of 24 genes associated with steroid-resistant nephrotic syndrome. Clin. J. Am. Soc. Nephrol. 8, 637–648 (2013).
Dinwiddie, D. L. et al. Diagnosis of mitochondrial disorders by concomitant next-generation sequencing of the exome and mitochondrial genome. Genomics 102, 148–156 (2013).
Scalais, E. et al. Early myoclonic epilepsy, hypertrophic cardiomyopathy and subsequently a nephrotic syndrome in a patient with CoQ10 deficiency caused by mutations in para-hydroxybenzoate-polyprenyl transferase (COQ2). Eur. J. Paediatr. Neurol. 17, 625–630 (2013).
The Multiple-System Atrophy Research Collaboration. Mutations in COQ2 in familial and sporadic multiple-system atrophy. N. Engl. J. Med. 369, 233–244 (2013).
Rotig, A. et al. Quinone-responsive multiple respiratory-chain dysfunction due to widespread coenzyme Q10 deficiency. Lancet 356, 391–395 (2000).
Rahman, S., Clarke, C. F. & Hirano, M. 176th ENMC International Workshop: diagnosis and treatment of coenzyme Q10 deficiency. Neuromuscul. Disord. 22, 76–86 (2012).
Mollet, J. et al. Prenyldiphosphate synthase, subunit 1 (PDSS1) and OH-benzoate polyprenyltransferase (COQ2) mutations in ubiquinone deficiency and oxidative phosphorylation disorders. J. Clin. Invest. 117, 765–772 (2007).
Peng, M. et al. Primary coenzyme Q deficiency in Pdss2 mutant mice causes isolated renal disease. PLoS Genet. 4, e1000061 (2008).
Saiki, R. et al. Coenzyme Q10 supplementation rescues renal disease in Pdss2kd/kd mice with mutations in prenyl diphosphate synthase subunit 2. Am. J. Physiol. Renal Physiol. 295, F1535–F1544 (2008).
Falk, M. J. et al. Probucol ameliorates renal and metabolic sequelae of primary CoQ deficiency in Pdss2 mutant mice. EMBO Mol. Med. 3, 410–427 (2011).
Lopez, L. C. et al. Treatment of CoQ10 deficient fibroblasts with ubiquinone, CoQ analogs, and vitamin C: time- and compound-dependent effects. PLoS ONE 5, e11897 (2010).
Quinzii, C. M. et al. Tissue-specific oxidative stress and loss of mitochondria in CoQ-deficient Pdss2 mutant mice. FASEB J. 27, 612–621 (2013).
Freyer, C. et al. Rescue of primary ubiquinone deficiency due to a novel COQ7 defect using 2,4-dihydroxybensoic acid. J. Med. Genet. 52, 779–783 (2015).
Desbats, M. A., Lunardi, G., Doimo, M., Trevisson, E. & Salviati, L. Genetic bases and clinical manifestations of coenzyme Q10 (CoQ10) deficiency. J. Inherit. Metab. Dis. 38, 145–156 (2015).
Korkmaz, E. et al. ADCK4-associated glomerulopathy causes adolescence-onset FSGS. J. Am. Soc. Nephrol. http://dx.doi.org/10.1681/ASN.2014121240 (2015).
Mollet, J. et al. CABC1 gene mutations cause ubiquinone deficiency with cerebellar ataxia and seizures. Am. J. Hum. Genet. 82, 623–630 (2008).
Lagier-Tourenne, C. et al. ADCK3, an ancestral kinase, is mutated in a form of recessive ataxia associated with coenzyme Q10 deficiency. Am. J. Hum. Genet. 82, 661–672 (2008).
Luna-Sanchez, M. et al. The clinical heterogeneity of coenzyme Q10 deficiency results from genotypic differences in the Coq9 gene. EMBO Mol. Med. 7, 670–687 (2015).
Pavlakis, S. G., Phillips, P. C., DiMauro, S., De Vivo, D. C. & Rowland, L. P. Mitochondrial myopathy, encephalopathy, lactic acidosis, and strokelike episodes: a distinctive clinical syndrome. Ann. Neurol. 16, 481–488 (1984).
Mehrazin, M. et al. Longitudinal changes of mtDNA A3243G mutation load and level of functioning in MELAS. Am. J. Med. Genet. A 149A, 584–587 (2009).
Seidowsky, A. et al. Renal involvement in MELAS syndrome — a series of 5 cases and review of the literature. Clin. Nephrol. 80, 456–463 (2013).
Hall, A. M. et al. The urinary proteome and metabonome differ from normal in adults with mitochondrial disease. Kidney Int. 87, 610–622 (2015).
Wilichowski, E., Pouwels, P. J., Frahm, J. & Hanefeld, F. Quantitative proton magnetic resonance spectroscopy of cerebral metabolic disturbances in patients with MELAS. Neuropediatrics 30, 256–263 (1999).
Salviati, L. et al. Novel SURF1 mutation in a child with subacute encephalopathy and without the radiological features of Leigh syndrome. Am. J. Med. Genet. A 128A, 195–198 (2004).
Suomalainen, A. et al. FGF-21 as a biomarker for muscle-manifesting mitochondrial respiratory chain deficiencies: a diagnostic study. Lancet Neurol. 10, 806–818 (2011).
Gropman, A. L. Neuroimaging in mitochondrial disorders. Neurotherapeutics 10, 273–285 (2013).
Bricout, M. et al. Brain imaging in mitochondrial respiratory chain deficiency: combination of brain MRI features as a useful tool for genotype/phenotype correlations. J. Med. Genet. 51, 429–435 (2014).
Leigh, P. N., Al-Sarraj, S. & DiMauro, S. Subacute necrotising encephalomyelopathy (Leigh's disease; Leigh syndrome). J. Neurol. Neurosurg. Psychiatry 86, 363–365 (2015).
Sundaram, C. et al. Contribution of muscle biopsy and genetics to the diagnosis of chronic progressive external opthalmoplegia of mitochondrial origin. J. Clin. Neurosci. 18, 535–538 (2011).
Trevisson, E., DiMauro, S., Navas, P. & Salviati, L. Coenzyme Q deficiency in muscle. Curr. Opin. Neurol. 24, 449–456 (2011).
Spinazzi, M., Casarin, A., Pertegato, V., Salviati, L. & Angelini, C. Assessment of mitochondrial respiratory chain enzymatic activities on tissues and cultured cells. Nat. Protoc. 7, 1235–1246 (2012).
Montero, R. et al. Analysis of coenzyme Q10 in muscle and fibroblasts for the diagnosis of CoQ10 deficiency syndromes. Clin. Biochem. 41, 697–700 (2008).
Payne, B. A., Gardner, K., Coxhead, J. & Chinnery, P. F. Deep resequencing of mitochondrial DNA. Methods Mol. Biol. 1264, 59–66 (2015).
Shanske, S. et al. Varying loads of the mitochondrial DNA A3243G mutation in different tissues: implications for diagnosis. Am. J. Med. Genet. A 130A, 134–137 (2004).
Yubero, D. et al. Molecular diagnosis of coenzyme Q10 deficiency. Expert Rev. Mol. Diagn. 15, 1049–1059 (2015).
Sadowski, C. E. et al. A single-gene cause in 29.5% of cases of steroid-resistant nephrotic syndrome. J. Am. Soc. Nephrol. 26, 1279–1289 (2015).
Tran, M. et al. PGC-1α promotes recovery after acute kidney injury during systemic inflammation in mice. J. Clin. Invest. 121, 4003–4014 (2011).
Brooks, C., Wei, Q., Cho, S. G. & Dong, Z. Regulation of mitochondrial dynamics in acute kidney injury in cell culture and rodent models. J. Clin. Invest. 119, 1275–1285 (2009).
Manny, J. et al. Structural changes in the perfused canine kidney exposed to the direct action of endotoxin. Isr. J. Med. Sci. 16, 153–161 (1980).
Trump, B. F. et al. The application of electron microscopy and cellular biochemistry to the autopsy. Hum. Pathol. 6, 499–516 (1975).
Takasu, O. et al. Mechanisms of cardiac and renal dysfunction in patients dying of sepsis. Am. J. Respir. Crit. Care Med. 187, 509–517 (2013).
Parekh, D. J. et al. Tolerance of the human kidney to isolated controlled ischemia. J. Am. Soc. Nephrol. 24, 506–517 (2013).
Zsengeller, Z. K. et al. Cisplatin nephrotoxicity involves mitochondrial injury with impaired tubular mitochondrial enzyme activity. J. Histochem. Cytochem. 60, 521–529 (2012).
Funk, J. A. & Schnellmann, R. G. Persistent disruption of mitochondrial homeostasis after acute kidney injury. Am. J. Physiol. Renal Physiol. 302, F853–F864 (2012).
Zhang, Q. et al. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 464, 104–107 (2010).
Feldkamp, T., Kribben, A., Roeser, N. F., Senter, R. A. & Weinberg, J. M. Accumulation of nonesterified fatty acids causes the sustained energetic deficit in kidney proximal tubules after hypoxia-reoxygenation. Am. J. Physiol. Renal Physiol. 290, F465–F477 (2006).
Weinberg, J. M., Venkatachalam, M. A., Roeser, N. F. & Nissim, I. Mitochondrial dysfunction during hypoxia/reoxygenation and its correction by anaerobic metabolism of citric acid cycle intermediates. Proc. Natl Acad. Sci. USA 97, 2826–2831 (2000).
Bienholz, A. et al. Substrate modulation of fatty acid effects on energization and respiration of kidney proximal tubules during hypoxia/reoxygenation. PLoS ONE 9, e94584 (2014).
Li, S. et al. Transgenic expression of proximal tubule peroxisome proliferator-activated receptor-α in mice confers protection during acute kidney injury. Kidney Int. 76, 1049–1062 (2009).
Tannenbaum, J., Purkerson, M. L. & Klahr, S. Effect of unilateral ureteral obstruction on metabolism of renal lipids in the rat. Am. J. Physiol. 245, F254–F262 (1983).
Zager, R. A., Johnson, A. C. & Hanson, S. Y. Renal tubular triglyercide accumulation following endotoxic, toxic, and ischemic injury. Kidney Int. 67, 111–121 (2005).
Cocheme, H. M. et al. Mitochondrial targeting of quinones: therapeutic implications. Mitochondrion 7, S94–S102 (2007).
Kelso, G. F. et al. Selective targeting of a redox-active ubiquinone to mitochondria within cells: antioxidant and antiapoptotic properties. J. Biol. Chem. 276, 4588–4596 (2001).
Zhao, K. et al. Cell-permeable peptide antioxidants targeted to inner mitochondrial membrane inhibit mitochondrial swelling, oxidative cell death, and reperfusion injury. J. Biol. Chem. 279, 34682–34690 (2004).
Szeto, H. H. et al. Mitochondria-targeted peptide accelerates ATP recovery and reduces ischemic kidney injury. J. Am. Soc. Nephrol. 22, 1041–1052 (2011).
Mukhopadhyay, P. et al. Mitochondrial-targeted antioxidants represent a promising approach for prevention of cisplatin-induced nephropathy. Free Radic. Biol. Med. 52, 497–506 (2012).
Tang, W. X., Wu, W. H., Qiu, H. Y., Bo, H. & Huang, S. M. Amelioration of rhabdomyolysis-induced renal mitochondrial injury and apoptosis through suppression of Drp-1 translocation. J. Nephrol. 26, 1073–1082 (2013).
Morigi, M. et al. Sirtuin 3-dependent mitochondrial dynamic improvements protect against acute kidney injury. J. Clin. Invest. 125, 715–726 (2015).
Ishihara, M. et al. Sestrin-2 and BNIP3 regulate autophagy and mitophagy in renal tubular cells in acute kidney injury. Am. J. Physiol. Renal Physiol. 305, F495–F509 (2013).
Jiang, M. et al. Autophagy in proximal tubules protects against acute kidney injury. Kidney Int. 82, 1271–1283 (2012).
Puigserver, P. et al. A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell 92, 829–839 (1998).
Sweeney, T. E., Suliman, H. B., Hollingsworth, J. W. & Piantadosi, C. A. Differential regulation of the PGC family of genes in a mouse model of Staphylococcus aureus sepsis. PLoS ONE 5, e11606 (2010).
Sweeney, T. E., Suliman, H. B., Hollingsworth, J. W., Welty-Wolf, K. E. & Piantadosi, C. A. A toll-like receptor 2 pathway regulates the Ppargc1a/b metabolic co-activators in mice with Staphylococcal aureus sepsis. PLoS ONE 6, e25249 (2011).
Rasbach, K. A. & Schnellmann, R. G. PGC-1α over-expression promotes recovery from mitochondrial dysfunction and cell injury. Biochem. Biophys. Res. Commun. 355, 734–739 (2007).
Jesinkey, S. R. et al. Formoterol restores mitochondrial and renal function after ischemia-reperfusion injury. J. Am. Soc. Nephrol. 25, 1157–1162 (2014).
Wrigley, A., Wilkinson, S. & Appleby, J. B. Mitochondrial replacement: ethics and identity. Bioethics 29, 631–638 (2015).
Schon, E. A. et al. A direct repeat is a hotspot for large-scale deletion of human mitochondrial DNA. Science 244, 346–349 (1989).
Glerum, D. M. & Tzagoloff, A. Isolation of a human cDNA for heme A:farnesyltransferase by functional complementation of a yeast cox10 mutant. Proc. Natl Acad. Sci. USA 91, 8452–8456 (1994).
Lim, S. C. et al. Mutations in LYRM4, encoding iron–sulfur cluster biogenesis factor ISD11, cause deficiency of multiple respiratory chain complexes. Hum. Mol. Genet. 22, 4460–4473 (2013).
Cogliati, S. et al. Mitochondrial cristae shape determines respiratory chain supercomplexes assembly and respiratory efficiency. Cell 155, 160–171 (2013).
de Brito, O. M. & Scorrano, L. Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature 456, 605–610 (2008).
Hirano, M., Lagier-Tourenne, C., Valentino, M. L., Marti, R. & Nishigaki, Y. Thymidine phosphorylase mutations cause instability of mitochondrial DNA. Gene 354, 152–156 (2005).
Valnot, I. et al. A mutation in the human heme A:farnesyltransferase gene (COX10) causes cytochrome c oxidase deficiency. Hum. Mol. Genet. 9, 1245–1249 (2000).
Tay, S. K. et al. Unusual clinical presentations in four cases of Leigh disease, cytochrome C oxidase deficiency, and SURF1 gene mutations. J. Child Neurol. 20, 670–674 (2005).
de Lonlay, P. et al. A mutant mitochondrial respiratory chain assembly protein causes complex III deficiency in patients with tubulopathy, encephalopathy and liver failure. Nat. Genet. 29, 57–60 (2001).
Tucker, E. J. et al. Mutations in the UQCC1-interacting protein, UQCC2, cause human complex III deficiency associated with perturbed cytochrome b protein expression. PLoS Genet. 9, e1004034 (2013).
Magner, M. et al. TMEM70 deficiency: long-term outcome of 48 patients. J. Inherit. Metab. Dis. 38, 417–426 (2015).
Saada, A. et al. Antenatal mitochondrial disease caused by mitochondrial ribosomal protein (MRPS22) mutation. J. Med. Genet. 44, 784–786 (2007).
Nakajima, J. et al. A novel homozygous YARS2 mutation causes severe myopathy, lactic acidosis, and sideroblastic anemia 2. J. Hum. Genet. 59, 229–232 (2014).
Belostotsky, R. et al. Mutations in the mitochondrial seryl-tRNA synthetase cause hyperuricemia, pulmonary hypertension, renal failure in infancy and alkalosis, HUPRA syndrome. Am. J. Hum. Genet. 88, 193–200 (2011).
Bourdon, A. et al. Mutation of RRM2B, encoding p53-controlled ribonucleotide reductase (p53R2), causes severe mitochondrial DNA depletion. Nat. Genet. 39, 776–780 (2007).
Prasad, C. et al. Exome sequencing reveals a homozygous mutation in TWINKLE as the cause of multisystemic failure including renal tubulopathy in three siblings. Mol. Genet. Metab. 108, 190–194 (2013).
El-Hattab, A. W. & Scaglia, F. Mitochondrial DNA depletion syndromes: review and updates of genetic basis, manifestations, and therapeutic options. Neurotherapeutics 10, 186–198 (2013).
Carrozzo, R. et al. SUCLA2 mutations are associated with mild methylmalonic aciduria, Leigh-like encephalomyopathy, dystonia and deafness. Brain 130, 862–874 (2007).
Dimmock, D. P. et al. Clinical and molecular features of mitochondrial DNA depletion due to mutations in deoxyguanosine kinase. Hum. Mutat. 29, 330–331 (2008).
Acknowledgements
Mitochondrial studies in S.M.P.'s laboratory are supported by R01-DK0950972. Studies in L.S.'s laboratory are supported by Telethon grants 14187 and 13222.
Author information
Authors and Affiliations
Contributions
All authors researched the data, made a substantial contribution to discussion of the content, wrote the article and reviewed and/or edited the manuscript before submission.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
About this article

Cite this article
Emma, F., Montini, G., Parikh, S. et al. Mitochondrial dysfunction in inherited renal disease and acute kidney injury. Nat Rev Nephrol 12, 267–280 (2016). https://doi.org/10.1038/nrneph.2015.214
Published:
Issue Date:
DOI: https://doi.org/10.1038/nrneph.2015.214
This article is cited by
-
Hyporeninemic hypoaldosteronism in RMND1-related mitochondrial disease
Pediatric Nephrology (2024)
-
β-Aminoisobutyric acid (L-BAIBA) is a novel regulator of mitochondrial biogenesis and respiratory function in human podocytes
Scientific Reports (2023)
-
Inhibition of TMEM16A improves cisplatin-induced acute kidney injury via preventing DRP1-mediated mitochondrial fission
Acta Pharmacologica Sinica (2023)
-
Renal oximetry for early acute kidney injury detection in neonates with hypoxic ischemic encephalopathy receiving therapeutic hypothermia
Pediatric Nephrology (2023)
-
Molecular mechanisms underlying the renal protective effects of coenzyme Q10 in acute kidney injury
Cellular & Molecular Biology Letters (2022)
