
Abstract
To identify novel signaling pathways necessary for rhabdomyosarcoma (RMS) survival, we performed a loss-of-function screen using an inducible small hairpin RNA (shRNA) library in an alveolar and an embryonal RMS cell line. This screen identified CRKL expression as necessary for growth of alveolar RMS and embryonal RMS both in vitro and in vivo. We also found that CRKL was uniformly highly expressed in both RMS cell lines and tumor tissue. As CRKL is a member of the CRK adapter protein family that contains an SH2 and two SH3 domains and is involved in signal transduction from multiple tyrosine kinase receptors, we evaluated CRKL interaction with multiple tyrosine kinase receptor signaling pathways in RMS cells. While we saw no interaction of CRKL with IGFIR, MET or PI3KAKT/mTOR pathways, we determined that CRKL signaling was associated with SRC family kinase (SFK) signaling, specifically with YES kinase. Inhibition of SFK signaling with dasatinib or another SFK inhibitor, sarcatinib, suppressed RMS cell growth in vitro and in vivo. These data identify CRKL as a novel critical component of RMS growth. This study also demonstrates the use of functional screening to identify a potentially novel therapeutic target and treatment approach for these highly aggressive pediatric cancers.
Similar content being viewed by others


Introduction
Rhabdomyosarcoma (RMS) is the most common soft-tissue sarcoma of childhood. The tumor is separated into two major histological subtypes. Alveolar RMS is characterized by the tumor-specific PAX3/7-FOXO1A translocation,1, 2 and embryonal RMS demonstrates loss of heterozygosity of 11p15,3 suggesting the presence of a tumor suppressor gene in this region. Both alveolar and embryonal RMS share many common features. These include positive immunohistochemical staining for markers of skeletal muscle differentiation, similar clinical presentation and similar treatment modalities and therapeutic interventions. Despite the success in improving overall prognosis after four decades of clinical trials, long-term survival for patients with metastatic RMS remains poor (<30%) and prognosis has not improved significantly in the past 15 years.4 Thus, there is a critical need to identify new molecular targets for patients with RMS. We reasoned that a large-scale loss-of-function screen would be an effective first step in this process and proceeded to adapt the inducible RNA interference screen developed by Ngo et al.5 We report the use of this approach for identification of SRC-CRKL-YES pathway as a novel RMS target necessary for tumor growth in vitro and in vivo.
Results
Identification of CRKL as a determinant of RMS cell growth
We infected both an embryonal (RD) and an alveolar (RH30) RMS cell line with a bar-coded retrovirus small hairpin RNA (shRNA) library targeting ∼5000 genes with three targeted shRNAs per gene. Each cell line was then split into two batches and either treated with doxycycline (shRNA-induced) or not treated (shRNA-non-induced) for a period of 21 days. DNA was recovered from the cells and differentially labeled with Cy3 dye (shRNA-induced) or Cy5 dye (shRNA-non-induced) and hybridized to a DNA microarray consisting of anti-barcode oligonucleotides. This allowed us to quantify the resulting ratio (representing presence of bar code) of all 15 000 bar codes. Of these, 395 bar codes (representing distinct shRNA constructs) were found to be more than twofold reduced in both RD and RH30 cells following shRNA induction, suggesting that knockdown of the corresponding gene was associated with decreased cell survival or proliferation over 21 days. We also utilized data from four lymphoma cell lines previously reported using this same shRNA library, including OCI-Ly3, OCI-Ly10, OCI-Ly7 and OCI-Ly19.5 shRNA constructs that were underexpressed in both RMS and lymphoma cell lines were excluded, further narrowing our list to 40 shRNA constructs (Supplementary Table S1) that represent more RMS-specific targets. Of these 40 candidates, we chose to characterize the role of CRKL because of its known function in signaling pathways and because multiple CRKL shRNA constructs in our initial screen scored as significant inhibitors of RMS cell growth.
In both RD and RH30 cells, induction of several independent shRNA constructs targeting CRKL resulted in a 4–31-fold decrease in barcode intensity (data not shown). Initial validation studies demonstrated that all shRNA clones targeting CRKL affected cell growth in vitro. We chose the shRNA targeting bp1305 of CRKL that yielded the greatest in vitro cell growth inhibition (Supplementary Table S1) for additional experiments unless otherwise specified.
CRKL is highly expressed in RMS cell lines, xenografts and human tumor samples
We next sought to determine the expression level of CRKL in RMS cell lines, as well as in tumor specimens. We initially examined RNA microarray data (Oncogenomic data base, http://home.ccr.cancer.gov/oncology/oncogenomics/) and found high levels of CRKL expression in the majority of RMS cell lines, xenografts and human tumor samples in both alveolar and embryonal subtypes (Figure 1a). In contrast, all non-RMS soft-tissue sarcoma tumor samples demonstrated low levels of CRKL expression. In addition, with the exception of normal cerebellar and uterine tissue, other normal organs had very low levels of CRKL expression (Figure 1a). We also noted both total CRKL and pCRKL protein levels were highly expressed in three RMS cell lines (RD, Rh30,and Rh4) and in three osteosarcoma cell lines (HOS, MNNG HOS, U2) examined (Figure 1b). To confirm that CRKL expression is common in both ERMS and ARMS tumors, we performed a tissue microarray analysis for pCRKL expression in >150 tumor samples. In both ERMS and ARMS, >90% of tumors expressed pCRKL, compared with normal skeletal muscle where pCRKL expression was negative in the same tissue microarray analysis (Figures 1c–h). The mechanism of CRKL overexpression remains unclear, as we were unable to observe any CNAs at 22q11 where CRKL has been mapped, nor did we observe any mutations in CRKL in five RMS tumors subjected to whole exome sequencing (Khan personal communication). The enhanced sensitivity of RMS to CRKL knockdown was demonstrated by lack of growth inhibition of three osteosarcoma cell lines exposed to shCRKL compared with RMS cells (Figure 1i). We also demonstrated that oncogene transformed normal human cells, HEK293T, were not inhibited by the two shCRKL sequences compared with the shGFP control (Figure 1j).

CRKL cDNA expression profile. (a) cDNA expression profile of two different CRKL probes showing expression levels in human RMS (ERMS, embryonal RMS, ARMS, alveolar RMS) cell lines (C) xenograft tumors of human cell lines (X) and human tumor tissue (T). For comparison, non-RMS tumors and normal human tissue is shown. (http://www.ncbi.nlm.nih.gov/sites/entrez). (b) Phospho-CRKL and total CRKL protein are highly expressed in RMS cell lines and osteosarcoma cell lines. Anti-phospho-Tyr207-CRKL, anti-CRKL and anti-beta actin were used to probe the blots. (c, d) Low power view of tissue microarray immunohistochemistry stained for pCRKL in 81 out of 90 ARMS (c) and 57 out of 61 ERMS (d). (e–h) High power view (80 × ) examples of positive staining ERMS (e), ARMS (f) samples, a negative ARMS sample (g), and negative staining in 14 out of 14 normal skeletal muscle (h). (i) Osteosarcoma cell lines were not inhibited by the two different shCRKL sequences (948 and 1305) contrary to Rh30. Cells were visualizing with NEAT stain. (j) In the HEK293T cell line, neither shCRKL sequence inhibits the cell growth compared with the shGFP control as by MTS assay.
Inhibition of CRKL inhibits RMS cell growth in vitro
To verify that the shRNA to CRKL decreased CRKL protein levels, RD and RH30 cells expressing the tetracycline repressor were infected with an shRNA targeting CRKL. Cells were grown in the presence or absence of doxycycline and cell lysates were collected after 72 h of doxycycline exposure. Western blot analysis demonstrated that both RD and RH30 cells have markedly decreased levels of CRKL protein expression when the shRNA construct is expressed in the presence of doxycycline (Figure 2a).

CRKL knockdown inhibits RMS cell growth. shCRKL knockdown CRKL expression only in the shCRKL Rh30 (a) and shCRKL RD (a) in the presence of doxycycline (25 ng/μl) compared with shRNA Control cells ± doxycycline and Rh30 and RD minus doxycycline. Note doxycycline did not affect CRKL expression in the control. (b) RD cells were treated with doxycycline and MTS assays were read at different days. Each data point represents an average of six wells. Exogenous CRKL expression in the RD cell (RD CRKL) resulted in increased proliferation compared with the RD control cell (RD Control). Proliferation was inhibited in the RD cell line with induction of shCRKL (RD shCRKL). (c) Rh30 cells with shCRKL induction (Rh30 shCRKL) inhibited proliferation compared with the control (Rh30 Control) and exogenous CRKL in Rh30 (Rh30 CRKL) enhanced proliferation compared with the control. All experiments were performed at least three times. (d, e) IncuCyte measured time to reach confluence in RD (d) and Rh30 (e) cell lines were significantly inhibited with shCRKL induction (▴) compared with their uninduced control ( ). Each data point represented an average of 24 wells for control and for doxycycline group in both cell lines.
). Each data point represented an average of 24 wells for control and for doxycycline group in both cell lines.
To confirm the role of CRKL in the growth of RMS cells, we evaluated the effect of CRKL expression on cell growth in standard tissue culture assays. Cells were seeded in 96-well plates and analyzed 24, 48 and 120 h after either overexpression of exogenous CRKL (blue line), expression of a control shRNA (black line) or expression of CRKL shRNA (red line). Growth of both RD and RH30 cells were markedly inhibited after five days in the presence of CRKL shRNA expression (P=0.001 for both cell lines). In addition, overexpression of exogenous wild-type CRKL (Supplementary Figure S1) led to enhancement of cell growth in both cell lines (P<0.001 for both cell lines) (Figures 2b and c). We confirmed the results with an independent cell proliferation assay and found that silencing CRKL decreased proliferation in both cell lines (Figures 2d and e). These data suggest that knockdown of CRKL using shRNA leads to a reduction in RMS cell growth. CRKL knockdown was associated with G1 arrest in both cell lines (Supplementary Figure S2) with no apparent increase in apoptosis (data not shown).
Antiproliferative effect of CRKL shRNA is target specific
In order to ensure that the induction of CRKL shRNA was specific for the CRKL target and not related to off-target effects, we used an additional shRNA (Sigma, St Louis, MO, USA) targeting the 3′-untranslated region (UTR) of the CRKL mRNA. Although this 3′-UTR shRNA targets endogenous CRKL mRNA, it is not able to target an exogenous recombinant CRKL that lacks the 3′-UTR (Figure 3a). Therefore, the antiproliferative effects of the 3′-UTR shRNA construct should be reversed by the addition of recombinant exogenous CRKL. Proliferation assays demonstrate that RH30 cells (Rh30 control) expressing the 3′-UTR CRKL shRNA and a control rescue vector (CMV lacZ) have a significant decrease in cell number compared with RH30 cells (Rh30 rescue) expressing the 3′-UTR CRKL shRNA and rescued with the CMV CRKL complementary DNA (cDNA) that lacks the 3′-UTR target (CMV CRKL) (Figure 3b). This effect was visualized easily following eosin/methylene blue staining (Figure 3c). Cells were transfected with a control shRNA (panels 1 and 3) or a 3′-UTR CRKL shRNA (panels 2 and 4). In addition, cells received either a CMV LacZ plasmid (panels 1 and 2) or a CMV CRKL cDNA that lacks the 3′-UTR target (panels 3 and 4). The antiproliferative effect of CRKL knockdown was apparent (panel 2) and the addition of exogenous CRKL almost completely reversed this growth inhibition (panel 4). Furthermore, we discovered that the embryonal cell line, RD, had a deletion in the 3′-UTR that directly overlapped the region targeted by the 3′-UTR shRNA. Thus the lack of off-target effects would predict that RD cell growth should not be affected by this shRNA and that is precisely what we observed (data not shown). These data strongly suggest that the effects on cell growth seen with the CRKL shRNA were specific to CRKL and not due to off-target effects.

Specificity of CRKL knock-down (a) To determine if the CRKL affect on cell proliferation was specific, recombinant CRKL was made without its 3′-UTR and a shCRKL targeting the 3′-UTR was delivered to the Rh30 cell line. (b) Control cells expressing CMV LacZ are growth inhibited by CRKL 3′-UTR shRNA (Rh30 control) while cells expressing CMV CRKL (lacks the 3′-UTR) are rescued from the effect of CRKL 3′-UTR shRNA (Rh30 rescue). RLU, relative light unit. (c) Staining illustrating CMV LacZ (C1) and CMV CRKL (C3), grown in the absence of the CRKL targeting shRNA, and LacZ (C2) or CMV CRKL (C4) grown in the presence of the 3′-UTR CRKL shRNA. Cells grown in the presence of 3′-UTR shCRKL and the rescue CMV CRKL are not inhibited (C4) compared with the cells grown in the presence of the 3′-UTR shCRKL and the control vector (C2).
Silencing CRKL inhibits RMS xenograft growth in vivo
To determine whether CRKL expression would affect growth of RMS tumors in vivo, we performed xenograft experiments in Rh30 cells expressing either the inducible CRKL shRNA or CRKL cDNA. Cells were injected orthotopically into the gastrocnemius musculature of the hind leg of Nude Scid-Beige mice who were fed either a normal diet (CRKL shRNA-non-induced) or a doxycycline diet (CRKL shRNA-induced). Resulting tumors were measured and animals were killed when tumors reached 2 cm in largest dimension. Mice bearing xenograft tumors with an inducible CRKL shRNA fed doxycycline grew tumors at a much slower rate compared with controls (CRKL shRNA without doxycycline, CMV CRKL, or control Rh30 cells (Figure 4a). Doxycycline treatment alone had no effect on parental Rh30 and RD tumors in vivo (data not shown). Also note that the uninduced CRKL shRNA xenografts grew at rates that were indistinguishable from either the control or CRKL cDNA expressing xenografts. We then performed follow-up xenograft studies in both RD and Rh30 cells. Cohorts of 10 mice were included in each of the three groups, including CRKL cDNA expressing cells, control cells (shRNA targeting GFP) and CRKL shRNA cells, and all mice were fed doxycycline. In both cell lines, the mean number of days for tumors to reach 2 cm in the longest dimension was significantly longer in the cohort expressing the CRKL shRNA for RD cells (P=0.0006) and for Rh30 cells (P=0.0004) (Figures 4b and c). Each dot represents the time to reach 2 cm for an individual mouse. Furthermore RD cells that overexpressed CRKL reached a size of 2 cm at a significantly faster rate than the control cells (P=0.001). Similar trends were observed in Rh30 expressing CRKL cDNA; however, they did not reach statistical significance compared with the control cells (P=0.4). In these xenograft experiments all the tumors were allowed to reach 2 cm before the tumors were harvested for western blots analysis. Four RMS xenograft tumors from the shControl and four from the doxycycline-treated shCRKL-induced group were probed for pCRKL. The results demonstrated that in the shCRKL RD xenograft tumors 50% of the samples had decreased pCRKL expression compared with the shControl and in the shCRKL Rh30 xenograft tumors all of the shCRKL samples had decreased pCRKL compared with the shControl (Supplementary Figure S3). Thus, the delayed growth of the shCRKL-induced xenografts was clearly associated with decreased CRKL expression in the tumors, and it is likely that the eventual growth in these tumors was due either to only partial CRKL knockdown, or growth of a cell population where shCRKL was not expressed.

Inhibition of CRKL inhibits RMS xenograft tumor growth. (a) Doxycycline induction of shCRKL inhibited xenograft tumor growth compared with the uninduced xenograft tumor. Animals were killed when the tumor reached 2 cm in largest dimension. shCRKL tumor ( ) without induction grew faster than shCRKL with doxycycline induction (
) without induction grew faster than shCRKL with doxycycline induction ( ). shCRKL tumor without induction grew similar to the control (
). shCRKL tumor without induction grew similar to the control ( ) and the CMV CRKL (
) and the CMV CRKL ( ). (b) RD RMS primary xenograft tumor growth was significantly inhibited with doxycycline-induced shRNA expression of CRKL compared with controls. Each dot represents the number of days xenograft tumor took to reach 2 cm for individual mouse. Note that RD cells expressing exogenous CRKL cDNA grew faster than control vector expressing cells. (c) Rh30 tumor growth was significantly inhibited with doxycycline-induced shRNA to CRKL compared with controls. Note that Rh30 expressing exogenous CRKL tumors grew slightly faster compared with controls but did not reach statistical significance.
). (b) RD RMS primary xenograft tumor growth was significantly inhibited with doxycycline-induced shRNA expression of CRKL compared with controls. Each dot represents the number of days xenograft tumor took to reach 2 cm for individual mouse. Note that RD cells expressing exogenous CRKL cDNA grew faster than control vector expressing cells. (c) Rh30 tumor growth was significantly inhibited with doxycycline-induced shRNA to CRKL compared with controls. Note that Rh30 expressing exogenous CRKL tumors grew slightly faster compared with controls but did not reach statistical significance.
Association of CRKL and the SFK member YES
As CRKL is a member of the CRK adapter protein family containing one SH2 and two SH3 domains involved in transducing signals from several receptor tyrosine kinases,6 we sought to identify the signaling pathway or pathways likely to be involved in the mechanism of CRKL-associated growth in RMS cells. Therefore, we evaluated the potential interaction of CRKL with a number of kinase signaling pathways. While we saw no evidence of interaction between CRKL signaling and the IGF-1R (Supplementary Figure S4), MET (Supplementary Figure S5), or PI3K/AKTt/mTOR pathways (Supplementary Figure S6), we observed a direct association of loss of CRKL function and suppression of SRC family kinases (SFK). Namely, suppression of CRKL by CRKL shRNA was associated with a decrease in phospho-SFK (Figure 5a). We attempted to identify the specific SFK that associated with CRKL by first screening for SFK members that were expressed in RMS. We screened for SRC, FYN, LYN, HCK and YES expression. All SFK screened were expressed with the exception of LYN, which was expressed only in Rh30 cells (Supplementary Figure S7), and HCK, which was not expressed in either cell line (data not shown). SFK immunoprecipitation (IP) and antiphosphotyrosine western blot demonstrated that phosphorylation of YES was decreased with loss of CRKL expression (Figure 5b upper panel) in both cell lines. However, no changes in tyrosine phosphorylation were detected in FYN or SRC IP (note that FYN is weakly phosphorylated, while SRC is highly phosphorylated in both cell lines (Figure 5b upper panel). Total FYN, SRC and YES were all immunoprecipitated with their respective antibodies for both cell lines (Figure 5b lower panel). These data suggest that decreasing CRKL protein expression leads a decrease in the phosphorylation and activation of YES.

Association between CRKL and YES. (a) Decreased CRKL with doxycycline induction of shCRKL leads to a decrease in activated SFK (pY416) in both RD and Rh30 cell lines. Beta-actin served as loading control. (b) IP of three SFK, included FYN, SRC and YES, with mouse antibodies targeting the respective SFK from RD and Rh30 cell lines with or without CRKL knockdown. Rabbit anti-SRC or YES was used to probe the respective SFK IP blots and mouse anti-FYN was used for the FYN IP (lower panel). SFK IP blots were probed with mouse anti-phospho-tyrosine (upper panel). In both cell lines, FYN was weakly tyrosine phosphorylated compared with SRC and YES and only YES tyrosine phosphorylation was diminished with CRKL knockdown. Experiments in a and b were replicated at least twice.
SRC kinase inhibitors decrease RMS cell growth
Based on the YES kinase data, we sought to determine whether treatment of RMS cells with SFK inhibitors could impede RMS tumor growth. Dasatinib inhibited growth of both RD and Rh30 cell lines (Figures 6a and b). Similar results were seen when an additional embryonal RMS cell line, Rh36, and an additional alveolar RMS cell line, Rh41 were tested (data not shown). Dasatinib decreased phosphorylation of tyrosine 207 on CRKL and decreased phosphorylation of tyrosine 416 on SFK (Figure 6c). While the functional significance of Y207 phosphorylation is unclear, it is noteworthy that SFK inhibitors affect this phosphorylation. Virtually identical results were obtained when the cells were treated with another SFK inhibitor saracatinib (Supplementary Figure S8). Of note, the ABL kinase inhibitor imatinib did not affect CRKL phosphorylation suggesting that ABL kinase does not phoshorylate CRKL in RMS cells (Supplementary Figure S9). This is consistent with western blot analysis that failed to detect any activated ABL with phospho-tyrosine 245 and 412 antibodies (Cell Signaling, Boston, MA, USA) in RD and Rh30 cell lines (data not shown).

Inhibition of RMS cell growth in vitro with dasatinib exposure. The RD (a) and Rh30 (b) cell lines were treated with dasatinib at different doses for 72 h. Each data point represents an average of six wells. (c) Dasatinib treatment decreased phosphorylation of SFK (tyr 416), as well as phosphorylation of CRKL (tyr207) at 250 nm for 72 h. (d) and (e) show that RD and Rh30 xenograft tumor growth is inhibited by dasatinib treatment. Western blot analysis of the tumors obtained from four mice in each of the control and the treated groups showed dasatinib treatment inhibits CRKL phosphorylation in both RD xenografts (f) and Rh30 xenografts (g) tumors. Normal mouse muscle shows minimal baseline pCRKL expression (f). (h) Cells transfected with mutant src (ck-src) are resistant to dasatinib inhibibition of CRKL phosphorylation and resistances to dasatinib growth inhibition (i and j).
Dasatinib inhibits RMS xenograft growth
To determine whether SFK inhibition could inhibit RMS growth in vivo, we tested the efficacy of dasatinib in RMS xenografts. RMS cells were injected orthotopically into the gastrocnemius musculature of the hind leg of Scid-Beige mice and the tumors were allowed to grow until palpable before starting dasatinib treatment. Cohorts of 20 mice were randomized once tumors were palpable to either receive the buffer control or the dasatinib, 100 mg/Kg orally by gavage, 6 days per week, for each cell line (RD and Rh30). Dasatinib treatment led to significant tumor growth inhibition of RMS xenografts by day 35 for RD and by day 52 for Rh30, respectively, from the start of treatment compared with control treated groups (Figures 6d and e). The high variability in Rh30 xenografts data was due to a wide distribution of tumor volumes among the Rh30 tumor bearing mice at the start of treatment, whereas the RD tumor volumes were more uniform. To confirm that RMS tumor growth inhibition was due to dasatinib inhibition of CRKL, western blot analysis of the tumors at the end of this experiment showed that phosphorylation of CRKL was completely inhibited in four different xenograft tumors sampled for both cell lines compared with four sampled untreated tumors (Figures 6f and g). Normal mouse gastrocnemius muscle is shown as a baseline value of phospho-CRKL (Figure 6f). The finding that dasatinib treatment led to decreased pCRKL suggested that SRC may be contributing to phosphorylation of CRKL. To determine whether SRC itself was involved, we transfected a chicken src,7 (Addgene plasmid 26 980) gene containing a T338I mutation that renders it resistant to dasatinib into both RD and Rh30 cells. Following transfection of the mutant gene, both cell lines were exposed for 48 h to 250 nM dasatinib. While control cells treated with dasatinib showed marked decrease in pCRKL, as well as decreased cell number, the cells containing the dasatinib resistant SRC construct demonstrated both increased pCRKL compared with controls, as well as deceased affect on cell proliferation (Figures 6h–j). These data strongly suggest that SRC kinase itself is at least partially responsible for phosphorylation of CRKL in RMS cells.
Inhibition of YES directly decreases RMS cell growth
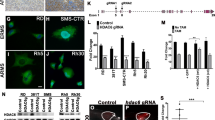
To further implicate YES directly in RMS cell growth, we sought to determine if knocking down YES expression directly would also inhibit cell growth. We identified two independent shRNA YES sequences, 9 and 11, delivered in lentiviral vectors that knockdown YES expression (Figure 7a). Note that there is minimal effect of shYES on either pY207CRKL or pY416 SFK. Both shRNA vectors induced significant in vitro growth inhibition of RD and Rh30 cells (Figures 7b and c). Expression of YES protein in RMS cell lines is easily detected as shown in Figure 7d and the YES mRNA is highly expressed in RMS compared with normal tissue and non-RMS tissue (Figure 7e). These data demonstrate that YES is highly expressed in both ERMS and ARMS, and that YES expression has a role in survival of RMS cells. Furthermore, as knockdown of YES expression does not alter phosphorylation of CRKL, but inhibits growth, it is likely that YES activation is downstream of CRKL.

Lentiviral shYES inhibit RMS cell growth. (a) shYES sequences 9 and 11 both inhibit YES expression in RD and Rh30 cells. shRNA sequence 9 and 11 gave >90% knockdown of YES by western analysis compared with the shControl. Neither pY416 SFK nor pCRKL were altered by YES knockdown. RD (b) and Rh30 (c) cell lines were growth inhibited by shYES compared with cells treated with lentivirus shControl. Experiments were repeated twice. (d) Western blot for YES and CRKL expression in a panel of RMS cell lines. (e) Heat map of YES RNA expression in human RMS tumors, xenograft tumors and cell lines compared with normal and non-RMS tissue.
Discussion
Using a loss of function high throughput shRNA screen, we uncovered a previously unidentified role for CRKL in the growth of RMS cells both in vitro and in vivo. CRKL is a member of the Crk adapter protein family, whose members contain one SH2 domain and one or two SH3 domains. These adapter proteins has a role in intracellular signaling pathways and are known to transduce signals downstream of several receptor tyrosine kinases.6 CRKL has been shown to be the major protein phosphorylated by the BCR–ABL oncogene in neutrophils from patients with CML.8, 9, 10 CRKL is located on 22q11 and has been implicated in the manifestations of DiGeorge/velocardiofacial syndrome caused by deletion of 22q11.11, 12
There have been previous reports of IGF-1R signaling mediated thru IRS-4/CRKII/CRKL activity in fibroblasts.13 However, we have been unable to demonstrate a direct alteration of CRKL upon blockade of the IGF-1R in RMS cells (Supplementary Figure S4). This may simply be a result of other signaling pathways that interact with CRKL becoming more active upon IGF-1R blockade, or it may be that in RMS cells CRKL mediated signaling is independent of the IGF-1R. Another pathway that has been implicated in both RMS and CRKL is HGF/MET signaling. Upon activation, the HGF receptor c-MET has been shown to bind and phosphorylate GAB-1, which in turn associates with CRKL.14, 15 It has recently been demonstrated that shRNAs targeting c-MET can inhibit the growth of both embryonal and alveolar RMS.16 However, we were unable to demonstrate a direct alteration of CRKL tyrosine phosphorylation, of SFK tyrosine phosphorylation, and of RMS cell growth with MET/ALK inhibitor crizotinib (Supplementary Figure S5). RMS cell lines grown in serum-free or charcoal striped serum also did not affect phosphorylation of CRKL (Supplementary Figure S10). Perhaps not surprisingly then, we found no evidence that altering CRKL signaling in RMS had any effect on the PI3K/AKT/mTor pathway (Supplementary Figure S6). Rather, we demonstrate that CRKL and YES are intimately linked to RMS growth. Namely, shRNA-induced downregulation of CRKL led to a decrease in phospho-YES, and SFK inhibition with dasatinib or saracatinib led to a decrease in phospho-CRKL. Inhibition of either CRKL using shRNA, or SFK using small molecule inhibitors led to growth suppression of RMS in vitro and in vivo. It is worth noting that shYES did not score positively in our initial shRNA screen, even though it was included. The sequences used in the screen were different from the sequences we used to inhibit YES in subsequent studies. Thus we believe the failure to pick up YES in our initial shRNA screen is likely due to the myriad technical issues that one deals with in carrying out such screens. To date, we have found no evidence of a direct interaction between CRKL and YES, and work is ongoing to identify possible mechanisms to explain this association. We do show that direct downregulation of YES using shRNA inhibits RMS cell growth but does not affect CRKL phosphorylation (Figure 7a) suggesting that YES itself does not alter CRKL phosphorylation. Rather, SRC itself appears to contribute to phosphorylation of CRKL (Figure 6h). These data suggest that CRKL may serve as a signaling ‘node’ for SFK proliferative pathways that are critical for RMS survival but not in osteosarcoma cell lines or in non-sarcoma cell line such as HEK293T (Figures 1i and j). Support for such a nodal function is provided by a recent reports where CRKL was identified as the top scored gene in NSCLC when combining gene amplification regions with loss-of-function screening.17 Of particular note is that these investigators subsequently showed that CRKL-induced transformation of NSCLC cells was at least partially due to activation of SFK (Cheung et al. 2011).18
In summary, our loss of function ‘Achilles’ heel’ approach has identified CRKL/YES as critical interrelated pathways necessary for RMS cell growth and survival and suggests a potential therapeutic role of SFK inhibition in the treatment of RMS. The availability of SFK inhibitors such as dasatinib for patient use suggests it may be of interest to further investigate a potential role of SFK inhibition as a target for RMS therapy.
Materials and Methods
Cell lines
The RD, Rh30, HOS and U2, sarcoma cell lines were obtained from ATCC (Manassas,VA, USA). Rh41 and Rh4 are RMS cell lines that express the PAX-3-FOXO1 translocation and were a gift from Dr PJ Houghton. The cell lines were cultured in RPMI1640, 100 units/l penicillin and 100 μg/ml streptomycin, 2 mM glutamine (Invitrogen, Grand Island, NY, USA), and 10% heat inactivated fetal bovine serum (Sigma). 293T cells and lymphoma cell lines OCI-Ly3, OCI-Ly10, OCI-Ly7 and OCI-Ly19 were previously described.5
Lipofectamine 2000 and retrovirus transduction
All Lipofectamine 2000 (Invitrogen) transfections were performed with DNA to Lipofectamine at 1:1 w/v ratio and selection started 48 h post transfection. All retrovirus transductions used 8 μg/ml of polybrene (Millipore, Billerica, MA, USA).
Doxycycline-inducible cell line preparation
RD and Rh30 doxycycline-inducible cell lines were engineered as described.5 The clones that met criteria for tight regulation and good induction were used in the shRNA library screen. To maximize performance of the assay, we chose clones that were infected at the highest levels.
Transduction of shRNA barcode library
Bar-coded, shRNA expression constructs targeting ∼5000 genes, 15 000 shRNAs, were packaged as described.5, 19
Tissue Microarrays
Tissue microarrays containing samples of either ERMS tumors or ARMS with normal skeletal muscle control samples were obtained from the Children’s Oncology group with codes for each spot. Each of the tissue microarrays were immunostained with anti-Phospho-CRKL (Y207) antibody (Cell Signaling) at a dilution of 1:50. Chromagenic detection was performed following standard protocols (Dako, Carpinteria, CA, USA).
Retroviral Packaging and transduction
A mixture of shRNA retroviral constructs containing the retroviral helper plasmids, pHIT/EA6 × 3*, pHIT60 (kindly provided by Stephen Goff),20 and lymphoma cell lines were transduced by spin infection as described.5 Detail is provided in Supplementary data.
Rescue experiment
CRKL cDNA was constructed as described in Supplementary data.
Lentivirus with shCRKL targeting the CRKL 3′-UTR was purchased from Sigma (TRCN0000006378) and RMS cell lines were infected at 5:1 lentivirus to cell ratio with polybrene (8 μg/ml). Infected cells were cultured for 11 days then cell number was quantitated by ATP assay (Promega, Madison, WI, USA). Cells were also plated in 96-well plates at 3000 cells per well and visualizing with NEAT staining (AstralDiagnostics, West Deptford, NJ, USA).
In vitro confirmation experiments
shCRKL cell lines were created for validation by infecting RD and Rh30 with retrovirus encoding the shRNA targeting CRKL at bp948 of NM_005207 (detail in Supplementary data).
Cell proliferation assay
Three independent methods for measuring cell proliferation were done. Cell proliferation was determined by 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium assay according to the manufacturer’s recommendations (Promega). The plate was read on a Molecular Device VERSAmax plate reader. Cell proliferation kinetics were monitored and recorded with the Incucyte system (Essen BioScience, Ann Arbor, MI, USA). ATP proliferation assays were done according to the manufacturer’s recommendations (Promega). For detail see Supplementary data.
SRC inhibitor experiments
For the dasatinib (LC Lab, Woburn, MA, USA) proliferation assays, cells were plated as described above. The control group was DMSO and the dasatinib group was treated with 250 nM dasatinib diluted in DMSO. For doxycycline induction, the control was media alone. Proliferation assays were done at 48, 72, and 120 h post treatment using the 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium assay. Identical studies were performed using a second SFK inhibitor, saracatinib (AZD 0530, Astra-Zeneca, London, UK) at a concentration of 5 μM. To evaluate the effect of Src specifically on pCRKL, we transfected a mutant (T338I ) chicken (Gallus gallus) src, (Addgene plasmid 26 980, Cambridge, MA, USA) and a control empty vector pBABE-Hygro retroviral vector (Cell Biolab, San Diego, CA, USA). Mutant chicken src or empty vector were transfected with HilyMax (Dojindo, Rockville, MD, USA), 5:1 DNA:lipid ratio, into RMS cell lines. Cells were selected with hygromycin, 250 μg/ml, 48 h post transfection, for 5 days then expanded. Cell proliferation was measured using a MTS assay 72 h post treatment with 250 nM dasatinib. Western blot analysis was done at 48 h post dasatinib treatment (250 nM).
Western blot and IP
One to two million cells were plated in 10-cm plates and treated with either doxycycline, 25 ng/ml, or dasatinib for 48–72 h. Cell were harvested with 1 × cell lysis buffer (Cell Signaling) plus protease and phosphortase inhibitor (Roche, Indianapolis, IN, USA). Antibodies against CRKL, P-Y207-CRKL, P-Y416-Src, YES, anti-FYN were from Cell Signaling. Anti-YES used for IP was from Wako (Richmond, VA, USA). Mouse anti-SRC and anti-FYN antibodies for IP were from Santa cruz biotechnology (Santa cruz, CA, USA). For IP, total protein of 0.5–0.8 mg was used with overnight incubation at 4 °C followed by western blot analysis for phospho-tyrosine (Millipore) and SFK. For IP blots, the secondary antibodies were horseradish peroxidase-conjugated light chain specific (Jackson Immuno Research, West Grove, PA, USA). Normal mouse immunoglobulin G from Sigma was used as IP control.
In vivo xenograft growth
Protocols for animal care were reviewed and approved by the National Cancer Institute Animal Care and Use Committee. The xenograft experimental protocol is provided in the Supplementary Data.
ShYES lentivirus
Cells were plated at 1000 cells per well and 5:1 lentivirus particles to cell ratio was used for transduction of shYES, with 5 μg/μl of polybrene. Twenty-four hours post infection the media was replaced with fresh media and 72 h post infection the media was replaced with media containing 5 μg/ml of puromycin. MTS assay was read when shRNA control was near 50–70% confluence or 11 days post infection. Lentivirus shYES clones 9 and 11 were obtained from Sigma (TRCN0000001609 and TRCN0000001611, respectively).
References
Shapiro DN, Sublett JE, Li B, Downing JR, Naeve CW . Fusion of PAX3 to a member of the forkhead family of transcription factors in human alveolar rhabdomyosarcoma. Cancer Res 1993; 53: 5108–5112.
Davis RJ, D’Cruz CM, Lovell MA, Biegel JA, Barr FG . Fusion of PAX7 to FKHR by the variant t(1;13)(p36;q14) translocation in alveolar rhabdomyosarcoma. Cancer Res 1994; 54: 2869–2872.
Scrable HJ, Witte DP, Lampkin BC, Cavenee WK . Chromosomal localization of the human rhabdomyosarcoma locus by mitotic recombination mapping. Nature 1987; 329: 645–647.
Oberlin O, Rey A, Lyden E, Bisogno G, Stevens MC, Meyer WH et al. Prognostic factors in metastatic rhabdomyosarcomas: results of a pooled analysis from United States and European cooperative groups. J Clin Oncol 2008; 26: 2384–2389.
Ngo VN, Davis RE, Lamy L, Yu X, Zhao H, Lenz G et al. A loss-of-function RNA interference screen for molecular targets in cancer. Nature 2006; 441: 106–110.
Feller SM . Crk family adaptors-signalling complex formation and biological roles. Oncogene 2001; 20: 6348–6371.
Zhang XH, Wang Q, Gerald W, Hudis CA, Norton L, Smid M et al. Latent bone metastasis in breast cancer tied to Src-dependent survival signals. Cancer Cell 2009; 16: 67–78.
Oda T, Heaney C, Hagopian JR, Okuda K, Griffin JD, Druker BJ . Crkl is the major tyrosine-phosphorylated protein in neutrophils from patients with chronic myelogenous leukemia. J Biol Chem 1994; 269: 22925–22928.
Nichols GL, Raines MA, Vera JC, Lacomis L, Tempst P, Golde DW . Identification of CRKL as the constitutively phosphorylated 39-kD tyrosine phosphoprotein in chronic myelogenous leukemia cells. Blood 1994; 84: 2912–2918.
ten Hoeve J, Arlinghaus RB, Guo JQ, Heisterkamp N, Groffen J . Tyrosine phosphorylation of CRKL in Philadelphia+ leukemia. Blood 1994; 84: 1731–1736.
ten Hoeve J, Morris C, Heisterkamp N, Groffen J . Isolation and chromosomal localization of CRKL, a human crk-like gene. Oncogene 1993; 8: 2469–2474.
Guris DL, Fantes J, Tara D, Druker BJ, Imamoto A . Mice lacking the homologue of the human 22q11.2 gene CRKL phenocopy neurocristopathies of DiGeorge syndrome. Nat Genet 2001; 27: 293–298.
Koval AP, Karas M, Zick Y, LeRoith D . Interplay of the proto-oncogene proteins CrkL and CrkII in insulin-like growth factor-I receptor-mediated signal transduction. J Biol Chem 1998; 273: 14780–14787.
Sakkab D, Lewitzky M, Posern G, Schaeper U, Sachs M, Birchmeier W et al. Signaling of hepatocyte growth factor/scatter factor (HGF) to the small GTPase Rap1 via the large docking protein Gab1 and the adapter protein CRKL. J Biol Chem 2000; 275: 10772–10778.
Arai A, Aoki M, Weihua Y, Jin A, Miura O . CrkL plays a role in SDF-1-induced activation of the Raf-1/MEK/Erk pathway through Ras and Rac to mediate chemotactic signaling in hematopoietic cells. Cell Signal 2006; 18: 2162–2171.
Taulli R, Scuoppo C, Bersani F, Accornero P, Forni PE, Miretti S et al. Validation of met as a therapeutic target in alveolar and embryonal rhabdomyosarcoma. Cancer Res 2006; 66: 4742–4749.
Luo B, Cheung HW, Subramanian A, Sharifnia T, Okamoto M, Yang X et al. Highly parallel identification of essential genes in cancer cells. Proc Natl Acad Sci USA. 2008; 105: 20380–20385.
Cheung HW, Du J, Boehm JS, He F, Weir BA, Wang X et al. Amplification of CRKL induces transformation and epidermal growth factor receptor inhibitor resistance in human non-small cell lung cancers. Cancer discovery 2011; 1: 608–625.
Berns K, Hijmans EM, Mullenders J, Brummelkamp TR, Velds A, Heimerikx M et al. A large-scale RNAi screen in human cells identifies new components of the p53 pathway. Nature 2004; 428: 431–437.
Markowitz D, Goff S, Bank A . A safe packaging line for gene transfer: separating viral genes on two different plasmids. J Virol 1988; 62: 1120–1124.
Acknowledgements
This work was supported through funding to the Intramural Research Program of the National Cancer Institute. We would also like to thank the many patients and their families who continue to contribute to and inform our research. We thank Dr Su Young Kim experiments, Elena Kuznetsova, Dr Arnulfo Mendoza, and Dr Melinda G Hollingshead for advice and help with xenograft experiment. We also thank the Children’s Oncology Group Soft-Tissue Sarcoma Committee for providing the tissue microarrays for analysis of CRKL expression. We thank Joan Massague for the sharing the Addgene plasmid 26 980.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on the Oncogene website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Yeung, C., Ngo, V., Grohar, P. et al. Loss-of-function screen in rhabdomyosarcoma identifies CRKL-YES as a critical signal for tumor growth. Oncogene 32, 5429–5438 (2013). https://doi.org/10.1038/onc.2012.590
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/onc.2012.590
Keywords
This article is cited by
-
A renaissance for YES in cancer
Oncogene (2023)
-
RBM15 promotes hepatocellular carcinoma progression by regulating N6-methyladenosine modification of YES1 mRNA in an IGF2BP1-dependent manner
Cell Death Discovery (2021)
-
Up-regulation of CRKL by microRNA-335 methylation is associated with poor prognosis in gastric cancer
Cancer Cell International (2017)
-
miR-140-5p suppresses the proliferation, migration and invasion of gastric cancer by regulating YES1
Molecular Cancer (2017)
-
Pharmacological targeting of the ephrin receptor kinase signalling by GLPG1790 in vitro and in vivo reverts oncophenotype, induces myogenic differentiation and radiosensitizes embryonal rhabdomyosarcoma cells
Journal of Hematology & Oncology (2017)
