
Abstract
The p53 tumor suppressor protein is a major sensor of cellular stresses, and upon stabilization, activates or represses many genes that control cell fate decisions. While the mechanism of p53-mediated transactivation is well established, several mechanisms have been proposed for p53-mediated repression. Here, we demonstrate that the cyclin-dependent kinase inhibitor p21 is both necessary and sufficient for the downregulation of known p53-repression targets, including survivin, CDC25C, and CDC25B in response to p53 induction. These same targets are similarly repressed in response to p16 overexpression, implicating the involvement of the shared downstream retinoblastoma (RB)-E2F pathway. We further show that in response to either p53 or p21 induction, E2F4 complexes are specifically recruited onto the promoters of these p53-repression targets. Moreover, abrogation of E2F4 recruitment via the inactivation of RB pocket proteins, but not by RB loss of function alone, prevents the repression of these genes. Finally, our results indicate that E2F4 promoter occupancy is globally associated with p53-repression targets, but not with p53 activation targets, implicating E2F4 complexes as effectors of p21-dependent p53-mediated repression.
Similar content being viewed by others


Introduction
The p53 tumor suppressor protein is activated in response to a variety of cellular stresses, such as DNA damage, oncogene activation, and nucleotide depletion.1, 2 As a bona fide transcription factor, p53 transactivates its target genes in response to these stresses, resulting in cell-cycle arrest, senescence, or apoptosis, to prevent the proliferation of damaged cells.3 The best known examples of such targets include p21 and MDM2.4, 5 More than one hundred p53 direct transcriptional targets have so far been identified.6, 7 Transcriptional activation by p53 is well understood and involves the direct recruitment of p53 tetramers to its response element present on target promoters.8
p53 can also transcriptionally repress the expression of a number of genes.9 Both direct and indirect mechanisms of repression have been proposed. Direct mechanisms involve the recruitment of p53 to the promoter regions of its target genes.1, 10 This has been reported to occur either through the direct binding of p53 to its response element, or through interaction with other transcription factors at their respective binding sites.10 Genes reported to be regulated by p53 through these direct mechanisms include survivin (BIRC5), CDC25C, CDC25B, CHK2, cyclin B, CKS1B, RECQL4, and CDC20.11, 12, 13, 14, 15, 16, 17, 18, 19
Indirect p53-mediated repression has also been implicated through activation of its direct transcriptional target, p21.20 p21, a member of the Ink4a/Cip1 family of cyclin-dependent kinase (CDK) inhibitors, induces cell-cycle arrest by binding and inhibiting CDK4 and CDK6/cyclin D complexes, resulting in de-phosphorylation and activation of the retinoblastoma (RB) pocket proteins that function together with E2F transcription factors to repress the transcription of cell cycle-related genes.21 p53-repression targets regulated by p21 include hTERT, EZH2, and CHK1.22, 23, 24 However, for many p53 target genes, there are conflicting reports as to whether this repression occurs through direct or indirect mechanisms.13, 14, 18, 25, 26, 27 A better understanding of the mechanisms of p53-mediated repression is critical, since many of the genes repressed by p53 contribute to its tumor-suppressor activity by affecting cell-cycle arrest and apoptosis.18, 28, 29, 30 Here, we show that p53-mediated repression occurs indirectly through p21, and is completely independent of p53 binding to target promoters. We provide novel evidence that the mechanism of repression through p21 involves the recruitment of E2F4 repression complexes onto these target promoters. Furthermore, we use data mining to extrapolate these results, suggesting that a similar repression mechanism occurs globally.
Results
p21 expression is necessary for the downregulation of p53-repression targets
To investigate the role of p21 in p53-mediated gene repression, we induced p53 activity in the HCT116 colon carcinoma cell line expressing wild-type (WT) p53 and p21, or its isogenic derivatives containing a somatic knockout of either p53 (HCT116 p53−/−)31 or p21 (HCT116 p21−/−),32 by treating them with doxorubicin or nutlin-3. Doxorubicin activates p53 by inducing DNA double-strand breaks through topoisomerase II inhibition,33 and nutlin-3 stabilizes p53 by inhibiting the interaction between p53 and its negative regulator, MDM2, in the absence of genotoxic stress.1 Doxorubicin and nutlin-3 treatment of HCT116 WT cells led to a G2 and G1 cell-cycle arrest, respectively (Figure 1a). Treatment with doxorubicin also led to a G2 cell-cycle arrest in HCT116 p53−/− and HCT116 p21−/− cells, whereas treatment with nutlin-3 did not induce an arrest in these same cells (Figure 1a). Next, we examined the ability of p53 to activate or repress well-established p53 target genes under these conditions. As shown in Figure 1b, p21 was upregulated by both doxorubicin and nutlin-3 treatments of HCT116 WT cells, but was not induced in either HCT116 p53−/− or HCT116 p21−/− cells. Furthermore, the known p53 direct transcriptional activation targets, MDM2 and PIG3, were equally upregulated in HCT116 WT and HCT116 p21−/− cells with either doxorubicin or nutlin-3 treatment, but were not induced in HCT116 p53−/− cells (Figure 1b).

p21 is required for the downregulation of p53-repression targets. HCT116 WT, HCT116 p53−/−, and HCT116 p21−/− cells were treated with 0.5 μM doxorubicin (Doxo), 20 μM nutlin-3, or vehicle control for 24 h. (a) Cell-cycle analysis. The propidium iodine intensities and the percentages of cells in each cell-cycle phase are indicated. (b, c) Quantitative RT–PCR showing the expression of (b) p53 upregulated target genes and (c) p53-repression target genes. Error bars indicate s.d. of representative experiments performed in triplicate.
Among a number of reported p53-repressed genes, we included survivin, CDC25C, and CDC25B in our analysis based on their strong transcriptional repression in our microarray analysis of p53-induced expression changes and published studies implicating them as direct p53 transcriptional targets with key roles in cell-cycle progression and apoptosis.16, 17, 18, 34 As expected, the expression levels of each of these p53-repression targets decreased in HCT116 WT cells in response to both doxorubicin and nutlin-3 treatments, but not in HCT116 p53−/− cells (Figure 1c). However, this repression did not occur in HCT116 p21−/− cells (Figure 1c). These results indicated that the presence and/or activation of p21 were likely required for p53 to repress these target genes.
Inhibition of p21 abrogates the ability of p53 to repress its target genes
To confirm that p21 was necessary for p53-dependent target gene repression, we knocked down p21 expression by shRNA in EJp53 bladder carcinoma cells (Figures 2b and c), which are p53 null but express exogenous p53 under the control of the tetracycline (tet)-off promoter.35 When p53 was induced in these cells expressing the control shRNA (EJp53-shGFP), they underwent a significant G1 cell-cycle arrest (Figure 2a). In contrast, this arrest was abrogated in EJp53 cells expressing the shRNA against p21 (EJp53-shp21) (Figure 2a). Of note, despite comparable levels of p53 induction and activation of its direct transcriptional target, MDM2 (Figures 2b and c), transcriptional downregulation of the p53-repression targets, survivin, CDC25C, and CDC25B was inhibited in EJp53-shp21 cells (Figure 2d). Thus, we demonstrated in two different cell lines that p21 was absolutely necessary for downregulating these genes in response to p53 activation.

Inhibition of p21 abrogates the ability of p53 to repress its target genes. p53 was induced in EJp53 cells stably expressing either shGFP or shp21 by reducing the tet concentration from 1.0 to 0.02 μg/ml for 24 h. (a) Cell-cycle analysis. The propidium iodine intensities and the percentages of cells in each cell-cycle phase are indicated. (b, c) Quantitative RT–PCR (b) and immunoblots (c) showing the expression of p53 and its upregulated target genes p21 and MDM2. (d) Quantitative RT–PCR of p53 downregulated genes. Error bars indicate s.d. of representative experiments performed in triplicate.
Induction of p21 alone is sufficient for the downregulation of p53-repression targets
To determine whether p21 in the absence of p53 is sufficient for the downregulation of p53-repression targets, we compared the effects of upregulating either p53 or p21 using tet-regulated EJp53 and EJp21 cells, respectively, in which p21 expression was also controlled by the tet-off promoter.35 In each case, removal of tet from the culture medium led to a G1 cell-cycle arrest (Figure 3a). Since EJp21 cells are p53 null,35 the induction of p21 was independent of p53, and as expected, these cells showed no induction of MDM2 (Figures 3b and c). Of note, levels of p21 induction were lower in EJp21 than in EJp53 cells. However, all tested p53-repression targets, including survivin, CDC25C, and CDC25B, were downregulated in both of these cell lines (Figure 3d). Taken together, these results established that p21 induction was both necessary and sufficient for the downregulation of these p53-repression targets.

p21 in the absence of p53 is sufficient for the downregulation of p53-repression targets. p53 and p21 were induced in EJp53 and EJp21 cells, respectively, by the complete removal of tet for the indicated time points. (a) Cell-cycle analysis. The propidium iodine intensities and the percentages of cells in each cell-cycle phase are indicated. (b, c) Quantitative RT–PCR (b) and immunoblots (c) showing the expression of p53 and its upregulated target genes p21 and MDM2. (d) Quantitative RT–PCR of p53-repression target genes. Error bars indicate s.d. of representative experiments performed in triplicate.
Induction of p16 also downregulates p53-repression targets
To determine whether the decreased expression of p53-repression targets is specific to induction of p21, we measured p53 target gene repression in response to activation of p16. p16 (CDKN2A, p16Ink4A), like p21, binds to and inhibits CDK activity, leading to RB de-phosphorylation and repression of E2F target genes.21 We utilized p53-null EJp16 cells, in which p16 expression is regulated by the tet-off promoter.36 Induction of p16 in these cells (Figure 4b) led to a similar G1 cell-cycle arrest (Figure 4a) to that observed with induction of p53 and p21 in EJp53 and EJp21 cells, respectively (Figure 3a). Moreover, expression levels of the p53-repression targets survivin, CDC25C, and CDC25B all decreased in response to p16 induction (Figure 4c) in the absence of any detectable increase in p21 RNA levels (data not shown). These results strongly argued that downregulation of these p53-repression targets by p21 must involve downstream pathways shared with p16.

p16 is sufficient for the downregulation of p53-repression targets. p16 was induced in EJp16 cells by the complete removal of tet for the indicated time points. (a) Cell-cycle analysis. The propidium iodine intensities and the percentages of cells in each cell-cycle phase are indicated. (b, c) Quantitative RT–PCR showing the expression of (b) p16 and (c) p53-repression target genes. Error bars indicate s.d. of representative experiments performed in triplicate.
The RB-E2F pathway mediates p53-dependent transcriptional repression
Even though p21 and p16 CDK inhibitors work independently in blocking cell-cycle progression, they both converge on the RB-E2F pathway.21 To determine whether the E2F pathway is transcriptionally active during p53 induction, we analyzed mRNA levels of several genes established to be regulated through E2Fs, including E2F1, CDC6, CDC2, and cyclin A.37 We observed decreased mRNA levels of each of these genes in response to induction of p53, p21, or p16 (Figure 5a). E2F target gene transcript levels were also reduced in HCT116 WT cells in response to doxorubicin or nutlin-3 treatment, but not in HCT116 p53−/− or HCT116 p21−/− cells (Figure 5b). These findings established that E2F-mediated signaling is repressed in a p21-dependent manner in response to p53 induction, and independently by either p21 or p16 induction. Moreover, the decreased expression of these E2F target genes positively correlated with that of the p53-repression targets, suggesting that their regulation involved a similar mechanism.

Regulation of known E2F target genes by p53, p21, and p16. (a) Quantitative RT–PCR of E2F target genes in EJp53, EJp21, and EJp16 cells. p53, p21, and p16 were induced in their respective cells by the complete removal of tet for 24 h. (b) Quantitative RT–PCR showing the expression of E2F target genes in HCT116 WT, HCT116 p53−/−, and HCT116 p21−/− cells treated with 0.5 μM doxorubicin (Doxo), 20 μM nutlin-3, or vehicle control for 24 h. Error bars indicate s.d. of representative experiments performed in triplicate.
Negative E2F4 complexes mediate the transcriptional downregulation of p53-repression targets
There are eight E2F genes in humans, encoding both activating and repressing transcription factors that regulate genes involved in cell-cycle progression, DNA replication and repair, and chromatin dynamics.38 We focused on E2F1, E2F4, and E2F7, which represent different classes of E2F proteins. Whereas E2F1 normally binds to RB and mediates gene activation, E2F4 predominantly binds to p107 and p130 and mediates gene repression.39, 40 E2F7 also mediates gene repression but is not dependent on the RB pocket proteins.41 To investigate the role of these E2Fs in the p53-dependent repression of survivin and CDC25C, we located putative E2F binding sites on their promoters,42 and analyzed the relative occupancy of each of these E2Fs on their binding sites in EJp53 cells. While E2F1 showed similar promoter occupancies on the selected p53-repression targets regardless of p53 status, the promoter occupancy of E2F4 on the same targets dramatically increased with p53 induction (from 0.2% to 0.5% input to 2.0–3.2% input) (Figure 6a). Under the same conditions, neither E2F1 nor E2F4 occupied a control sequence located within the acetylcholine receptor (AChR) (Figure 6a). As additional controls, E2F1 and E2F4 also bound to the promoters of well-established E2F targets: CDC2, E2F1, CDC6, and cyclin A (Figure 6a).37 Of note, we observed even a greater increase in E2F4 promoter occupancy on the survivin and CDC25C promoters than on these well-established E2F targets. In contrast, E2F7 did not bind to any of the p53-repression target promoters tested and only bound to a subset (E2F1 and CDC6 but not CDC2 or cyclin A) of established E2F targets (Figure 6a).43 Therefore, based on our chromatin immunoprecipitation (ChIP) analysis, E2F7 was unlikely involved in the downregulation of these p53-repression targets.

E2F4, but not E2F1 or E2F7, is recruited to p53-repression target promoters in response to p53 or p21 induction. ChIP analysis showing E2F1, E2F4, and E2F7 promoter occupancy on p53 repression and E2F target promoters in the presence and absence of (a) p53 in EJp53 and (b) p21 in EJp21 cells. p53 and p21 were induced in their respective cells by the complete removal of tet for 72 h. AChR was used as a negative control. Target sequences were detected by quantitative RT–PCR of eluted DNA. The promoter occupancy is shown as the percent of input DNA. Error bars indicate s.d. of representative experiments performed in triplicate.
We next investigated whether p21 induction in EJp21 cells can also mediate the recruitment of E2F4 to p53-repression target promoters. As shown in Figure 6b, p21 induction in these cells resulted in similar E2F1, E2F4, and E2F7 binding patterns on p53 repression and established E2F target promoters to those observed with p53 induction in EJp53 cells. Of note, the extent of E2F4 binding was lower in EJp21 than in EJp53 cells, which correlated with lower levels of p21 induction in these cells and the lesser degree of downregulation of p53-repression target genes analyzed (Figures 3c and d). All of these results argued that p53 target gene repression was mediated by p21-dependent activation of repressive E2F4 complexes, and their binding to these promoters.
The p53-repression target alternative reading frame (ARF) is not regulated by p21 or E2F4 repressive complexes
Recently, ARF was reported to be a direct p53-repression target.44 Using our same strategy to test whether its repression is dependent on p21 and E2F4, we found that while ARF was repressed by induction of p53, induction of neither p21 nor p16 was able to decrease its expression level in EJ carcinoma cells (Figure 7a). Furthermore, knockdown of p21 in EJp53 cells did not abrogate p53-mediated repression of ARF expression (Figure 7b). We also compared E2F1 and E2F4 occupancy on the ARF promoter in the presence and absence of either p53 or p21 induction. Unlike the other reported p53-repression targets, or well-established E2F target genes, we observed no correlation between E2F4 promoter occupancy and ARF repression in response to p53 or p21 induction (Figures 7c and d). These findings suggested that the mechanism of ARF repression was independent of p21 and E2F4 and thus differed from that of survivin, CDC25C, and CDC25B repression targets.

The p53-repression target ARF is not regulated by p21 or E2F4 repressive complexes. (a) Quantitative RT–PCR showing the expression of ARF in EJp53, EJp21, and EJp16 cells. p53, p21, and p16 were induced in their respective cells by the complete removal of tet for the indicated time points. (b) Quantitative RT–PCR showing the expression of ARF in EJp53 cells stably expressing shGFP or shp21. p53 was induced in by reducing the tet concentration from 1.0 to 0.02 μg/ml. (c, d) ChIP analysis of the indicated genes showing the ratio of E2F4 to E2F1 promoter occupancy in (c) EJp53 cells induced for p53 for 72 h and (d) EJp21 cells induced for p21 for 24 and 48 h. Error bars indicate s.d. of representative experiments performed in triplicate.
E2F4 promoter recruitment by RB pocket proteins is essential for p53-mediated repression
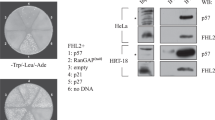
E2F4 mainly interacts with unphosphorylated p107 and p130 pocket proteins, and this interaction is critical for its recruitment to target promoters.40, 45 The HPV E7 oncoprotein disrupts the formation of these complexes, and hence the ability of E2F4 to bind to target promoters and repress gene expression.46 We therefore hypothesized that in the presence of HPV E7, E2F4 would no longer be recruited to p53-repression target promoters. To determine whether this was the case, we infected EJp21 cells with an HPV E7 expressing lentivirus or vector control and induced p21 expression (Figures 8a and b). In the presence of HPV E7, E2F4 recruitment to p21-dependent p53-repression target promoters was significantly impaired (Figure 8c), and p21 failed to downregulate its repression targets (Figure 8d). Moreover, p21 overexpression failed to downregulate the expression of these same target genes in HeLa cells (Supplementary Figures 1a and b), which endogenously express HPV E7.47 These results indicate that proper pocket protein function and E2F4 binding to p53-repression target promoters are required for p21-dependent downregulation of p53-repressed genes.

E2F4 promoter recruitment by RB family members is essential for p53-mediated gene repression. p21 was induced in EJp21 cells expressing either HPV E7 or vector control by the removal of tet. (a, b) Quantitative RT–PCR of p21 (a) and immunoblot of p21 and HPV E7 levels (b). (c) ChIP analysis showing E2F4 promoter occupancy on p53-repression target promoters. AChR was used as a negative control. Target sequences were detected by quantitative RT–PCR of eluted DNA. The promoter occupancy is shown as the percent of input DNA. (d) Quantitative RT–PCR of p53-repression target genes. Error bars indicate s.d. of representative experiments performed in triplicate.
Since E2F4 mainly interacts with p130 and p107, we hypothesized that this repression was independent of RB. To test this possibility, we utilized Saos-2 osteosarcoma cells, which contain a non-functional RB gene due to deletion of exons 21–27, but possess functional p130 and p107 proteins.48 Similarly to EJp21 cells, overexpression of p21 in Saos-2 cells (Supplementary Figure 1c) resulted in the downregulation of survivin, CDC25C, and CDC25B but not ARF (Supplementary Figure 1d). These results demonstrated that functional RB was not required for p21-dependent repression of p53 target genes.
p53-dependent gene repression is globally associated with E2F4 promoter occupancy
To determine whether E2F4 promoter occupancy is universally associated with p53-mediated repression, we utilized bioinformatic tools to compare a ChIP-seq data set for E2F449 with an expression microarray data set of p53-regulated genes.34 We found a strong negative correlation between the E2F4 ChIP-seq peak score, a measure of E2F4’s binding strength on each promoter,49 and the gene expression levels at both 12 and 24 h after p53 induction, with a Pearson correlation of −0.2458 and −0.4215, respectively (P-value<2e−16 for both time points) (Figure 9a). These analyses indicated that the promoters of a large number of p53-repression targets were able to bind E2F4 and, conversely, that those of p53 activation targets generally were not.

Global correlation between p53-mediated gene repression and E2F4 promoter occupancy. (a) Heatmap showing the correlation between the E2F4 ChIP-seq peak score and the p53 microarray expression levels. Row Z-score was performed on the log2-transformed values for each gene. (b) E2F4 ChIP-seq peak scores were divided into low (<4.5), medium (4.5–7.5), high (7.5–20), and very high (>20) binding groups. The percentage of p53 upregulated and downregulated genes, normalized for total gene number, that fall into each of these categories is shown in the form of a bar graph.
To further assess the correlation between p53-dependent gene repression and E2F4 binding, we divided the E2F4 ChIP-seq scores into groups of low (<4.5), medium (4.5–7.5), high (7.5–20), or very high (>20) binding affinities and compared the percentages of p53 upregulated and downregulated genes in each group. Since there were many more downregulated than upregulated genes, we normalized these results to the total number of genes in each group. Strikingly, over 70 and 90% of p53-regulated genes that bound E2F4 with high or very high affinity, respectively, were downregulated, while the majority of genes that had low or no binding were upregulated in response to p53 (Figure 9b). Of note, the p53-repression targets survivin, CDC25C, and CDC25B fit within the high and very high E2F4 binding groups. This data mining approach uncovered a large number of additional genes repressed by p53 that may also be regulated indirectly through p21 and E2F4 (Supplementary Table 1). Furthermore, the p53-repressed genes that displayed very strong E2F4 binding were functionally enriched in cell cycle, DNA replication, and DNA repair KEGG (Kyoto Encyclopedia of Genes and Genomes) pathways (Supplementary Table 2), consistent with the known functions of E2F transcription factors.38, 50 Together, these findings established repression through E2F4 as a general mechanism of p53-mediated repression.
Discussion
Direct versus indirect repression by p53
Previous studies indicated that p53-mediated repression occurred through direct p53 binding to the promoters of genes such as survivin and CDC25C.17, 18 The main evidence involved ChIP and EMSAs indicating p53 binding to specific repression elements.51, 52 By contrast, we demonstrated that the expression of these same genes was downregulated by p21 in cell lines that do not contain p53 and that in the absence of p21, p53 was unable to repress these same target genes. Induction of p21 in the absence of p53 is physiologically relevant since growing evidence suggests that p21 may also be activated independent of p53-mediated transcription.53 These results establish that neither p53 presence nor its association with repression target promoters is required for this repression. Other groups have shown that DNA binding of transcription factors can occur without any effect on the transcription of proximal genes.54, 55 One possibility is that these ‘nonfunctional sites’ protect their transcription factors from degradation and thus may have a role in other aspects of the overall transcriptional response.55 Our present findings are also consistent with more recent evidence that p53-mediated repression can occur indirectly through p21,1, 10 and we have recently shown that p21 is also required for the downregulation of SUV39H1 mRNA expression by p53.56 Thus, we conclude that p21 is both necessary and sufficient for p53-dependent repression of these target genes.
Indirect repression by p53 requires p21 and the recruitment of E2F4 repression complexes
We observed that induction of p16 alone, in addition to either p53 or p21, could equally downregulate the expression of several p53-repression targets (survivin, CDC25C, and CDC25B), implicating the involvement of their common downstream RB-E2F pathway in their repression. In addition, we observed by ChIP analysis that the promoter occupancy of these genes by E2F4 was increased by induction of both p53 and p21, providing novel evidence that E2F4 is recruited to these target promoters. Furthermore, abrogation of E2F4 binding to target promoters prevented the downregulation of these genes, demonstrating that p53-dependent gene repression through p21 is mediated via the recruitment of E2F4 repression complexes.
Our findings that inactivation of RB alone was not enough to eliminate E2F4-mediated repression of p53 targets suggest that the other pocket proteins, p130 and/or p107, cooperate with E2F4 in regulating p53-dependent gene repression through p21. Since p53 activation has been shown to increase p130 protein expression in a p21-dependent manner, while decreasing the levels of RB and p107, p130 is most likely the pocket protein involved.57 E2F4 is known to have a crucial role in cell proliferation,49 accounting for the majority of E2Fs associated with their target promoters during G0 and G1 cell-cycle phases.58 Recently, it was shown to have a major role in the repression of cell-cycle genes through interaction with the larger DREAM complex (which also consists of p130) in G0 and G1 cell-cycle phases.59, 60 These findings further implicate p130 as the pocket protein most likely responsible in concert with E2F4 for mediating repression of p53 target genes through p53 upregulation of p21.
A recent study in which genome-wide E2F4 binding was analyzed by ChIP-seq identified E2F4 occupancy on the promoters of over 7000 genes.49 To determine the extent by which E2F4 mediates p53-dependent repression, we mined these data, and compared it with that of p53-regulated genes identified from our recent p53 microarray.34 Using this approach, we were able to identify a large number of additional p53-repressed genes that also bound E2F4 on their promoters. In addition to the genes we identified as E2F4-mediated p53 indirect targets, several other reported p53 direct repression targets11, 12, 13, 14, 15, 19 showed binding of E2F4 on their promoters including CHK2, cyclin B, CKS1B, RECQL4, and CDC20, all of which were categorized in the very high binding group (see Supplementary Table 1). Thus, p53-mediated, and p21-dependent, transcriptional repression is globally regulated through E2F4.
p21-independent repression of ARF
We observed that ARF, while downregulated by p53, was not regulated by or dependent on p21. Furthermore, E2F4 was not significantly recruited onto the ARF promoter during p53 upregulation. Our data are consistent with another study that could not detect E2F4 on the ARF promoter in quiescent fibroblasts.61 E2F3b has been shown to repress transcription of ARF in quiescent mouse embryonic fibroblasts.62 Moreover, we have observed that E2F3b is upregulated in response to p53 and that it contains putative p53 response elements (our unpublished data). Thus, it is conceivable that ARF repression and possibly repression of other p53 targets occur indirectly via p53-dependent upregulation of E2F3b.
Physiological relevance of p53-dependent, E2F4-mediated gene repression
The mechanism of p53-mediated repression is of particular interest, since we and others have found that many more genes are downregulated than upregulated upon p53 induction in several different cell model systems.34, 63, 64, 65, 66 Furthermore, p53-repressed genes, which were very strongly enriched for E2F4 binding, have proliferation promoting functions including DNA replication, DNA repair, and cell-cycle progression. In response to cellular stress, p53 prevents cell-cycle progression and the replication of damaged DNA, in part by repressing such genes. Thus, cancer cells could lose the ability to repress these genes either by viral inactivation of RB pocket proteins or by loss of p53 function, leading to increased proliferation and the propagation of damaged DNA.
Materials and methods
Cell culture, treatments, and lentiviral expression
EJp53, EJp21, and EJp16 cells, bladder carcinoma cells that express a tetracycline-regulated WT p53, p21, or p16, respectively,35, 67 were grown in Dulbecco’s Modified Eagle’s Medium (Invitrogen, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (Sigma-Aldrich, St Louis, MO, USA), 50 units/ml of penicillin/streptomycin, 750 μg/ml G418, 100 μg/ml hygromycin, and 1.0 μg/ml tetracycline. HCT116 WT, HCT116 p53−/−, and HCT116 p21−/− colon carcinoma cells were cultured in Dulbecco’s Modified Eagle’s Medium supplemented with 10% fetal bovine serum and 50 units/ml of penicillin/streptomycin.31, 32 Saos-2 osteosarcoma and HeLa cervical cancer cells were grown in Dulbecco’s Modified Eagle’s Medium supplemented with 10% fetal bovine serum and 50 units/ml of penicillin/streptomycin. All cells were cultured at 37 °C and 90% humidity in a 5% CO2 incubator. (+/−)-Nutlin-3 (Cayman Chemicals, Ann Arbor, MI, USA; 10004372) was used at a concentration of 20 μM for 24 h. Doxorubicin (Sigma-Aldrich; D1515) was used at 0.5 μM for 24 h. To ectopically express p21 and HPV16 E7, we used NSPI-derived lentiviral vectors.68 Knockdown of p21 was achieved with pLK0.1-derived lentiviral vectors.56 Lentiviral production and infection were carried out as previously described.69
Cell-cycle analysis
Trypsinized cells were stained with propidium iodide using the CycleTEST PLUS DNA reagent kit (BD Biosciences, San Jose, CA, USA), according to the manufacturer’s instructions. In all, 20 000 stained cells were acquired on an FACS Calibur (BD Biosciences), and data were analyzed with the FlowJo 7.6 software (Tree Star, Ashland, OR, USA).
RNA extraction, cDNA synthesis, and real-time PCR analysis
Total RNA was extracted from cells using QIAshredder (Qiagen, Valencia, CA, USA) and RNeasy Mini kit (Qiagen) following the manufacturer’s instructions. First-strand cDNA synthesis was performed using Superscript II reverse transcriptase (Invitrogen) according to the manufacturer’s instructions. Quantitative real-time PCR was carried out on the Mx3005P PCR machine (Agilent Technologies, Palo Alto, CA, USA) using the FastStart SYBR Green Master (Roche Diagnostics, Indianapolis, IN, USA; 04673492001) with gene-specific primers (Supplementary Table 3). PCRs were performed in 96-well plates in 15 μl volumes under the following conditions: 95 °C for 15 min, followed by 40 cycles of 94 °C for 15 s, 61 °C for 30 s, and 72 °C for 30 s. Specificity was verified by a dissociation curve for each set of primers. Data were analyzed with the MxPro software (Agilent Technologies). 18S RNA was used as a normalizer.
Immunoblot analysis
Whole-cell extracts were obtained by solubilizing cells in lysis buffer (50 mM Tris–HCl, pH 7.5, 150 mM NaCl, 1% Triton X-100), supplemented with protease and phosphatase inhibitors (Roche Diagnostics; 04693124001 and 04906845001). Protein samples (20–40 μg) were subjected to SDS–PAGE, transferred onto an Immobilon-FL filter (Millipore, Saint Charles, MO, USA) and probed with the indicated antibodies: p53 (1801; Mount Sinai School of Medicine Hybridoma Center, New York, NY, USA), p21 (BD Biosciences; 556431), MDM2 (Calbiochem, San Diego, CA, USA; OP46), HPV16 E7 (Santa Cruz Biotechnology, Santa Cruz, CA, USA, sc-6981), and β-actin (Sigma-Aldrich; A5441). Detection was carried out with an Odyssey Infrared Imaging System (LI-COR Biosciences, Lincoln, NE, USA) with IR-dye-tagged secondary antibodies: Alexa Fluor 680 or Alexa Fluor 750 (Invitrogen).
Chromatin immunoprecipitation
The ChIP assay was performed following a protocol provided by Millipore with several modifications. Crosslinked and washed cells were resuspended in 1 ml cell lysis buffer (0.5% IGEPAL CA-630, 85 mM KCl, and 20 mM Tris–HCl pH 8.1) supplemented with protease and phosphatase inhibitors, and incubated for 10 min on ice. Cell pellets were then resuspended in 200 μl of nuclei lysis buffer (1% SDS, 10 mM EDTA, and 50 mM Tris–HCl pH 8.1) supplemented with protease and phosphatase inhibitors, and incubated on ice for 10 min. Genomic DNA was sheared by sonication to an average size of 300 bp using the Bioruptor (Diagenode, Denville, NJ, USA) in 1.5 ml tubes on high power with 12 cycles of 30 s on and 30 s off. DNA was precleared with the Protein A/G PLUS-Agarose beads (Santa Cruz Biotechnology; sc-2003) and incubated overnight with E2F1 (Santa Cruz Biotechnology; sc-193), E2F4 (Santa Cruz Biotechnology; sc-866), E2F7 (Santa Cruz Biotechnology; sc-66870) or IgG (Millipore; 12-371) antibodies. Immunoprecipitations were performed using Protein A/G PLUS-Agarose beads. DNA was purified using the QIAquick PCR Purification Kit (Qiagen). Quantitative real-time PCR using promoter-specific primers (Supplementary Table 3) was performed to identify the amount of immunoprecipitated target DNA sequence. 1% of the sample before immunoprecipitation was used as an input control.
ChIP-seq and microarray analysis
A cutoff for the expression microarray of p53-regulated genes using 5 ng of doxycycline to induce p53 was set at 0.2 for upregulated and downregulated genes.34 We then used a perl script to find the E2F4 ChIP-seq peak score of all of these genes from the p53 microarray.49 We made a heatmap using R (R development core, www.r-project.org) of these genes, their binding coefficient and the microarray levels. Correlation with the E2F4 ChIP-seq peak score was computed in R. The P-value was determined using Pearson’s correlation test.
References
Vousden KH, Prives C . Blinded by the light: the growing complexity of p53. Cell 2009; 137: 413–431.
Mandinova A, Lee SW . The p53 pathway as a target in cancer therapeutics: obstacles and promise. Sci Transl Med 2011; 3: 64rv61.
Brooks CL, Gu W . New insights into p53 activation. Cell Res 2010; 20: 614–621.
El-Deiry WS . The role of p53 in chemosensitivity and radiosensitivity. Oncogene 2003; 22: 7486–7495.
Alarcon-Vargas D, Ronai Z . p53-Mdm2–the affair that never ends. Carcinogenesis 2002; 23: 541–547.
Riley T, Sontag E, Chen P, Levine A . Transcriptional control of human p53-regulated genes. Nat Rev Mol Cell Biol 2008; 9: 402–412.
Kannan K, Amariglio N, Rechavi G, Jakob-Hirsch J, Kela I, Kaminski N et al. DNA microarrays identification of primary and secondary target genes regulated by p53. Oncogene 2001; 20: 2225–2234.
Kitayner M, Rozenberg H, Kessler N, Rabinovich D, Shaulov L, Haran TE et al. Structural basis of DNA recognition by p53 tetramers. Mol Cell 2006; 22: 741–753.
Ho J, Benchimol S . Transcriptional repression mediated by the p53 tumour suppressor. Cell Death Differ 2003; 10: 404–408.
Bohlig L, Rother K . One function—multiple mechanisms: the manifold activities of p53 as a transcriptional repressor. J Biomed Biotechnol 2011; 2011: 464916.
Sengupta S, Shimamoto A, Koshiji M, Pedeux R, Rusin M, Spillare EA et al. Tumor suppressor p53 represses transcription of RECQ4 helicase. Oncogene 2005; 24: 1738–1748.
Rother K, Li YY, Tschop K, Kirschner R, Muller GA, Mossner J et al. Expression of cyclin-dependent kinase subunit 1 (Cks1) is regulated during the cell cycle by a CDE/CHR tandem element and is downregulated by p53 but not by p63 or p73. Cell Cycle 2007; 6: 853–862.
Innocente SA, Lee JM . p53 is a NF-Y- and p21-independent, Sp1-dependent repressor of cyclin B1 transcription. FEBS Lett 2005; 579: 1001–1007.
Imbriano C, Gurtner A, Cocchiarella F, Di Agostino S, Basile V, Gostissa M et al. Direct p53 transcriptional repression: in vivo analysis of CCAAT-containing G2/M promoters. Mol Cell Biol 2005; 25: 3737–3751.
Matsui T, Katsuno Y, Inoue T, Fujita F, Joh T, Niida H et al. Negative regulation of Chk2 expression by p53 is dependent on the CCAAT-binding transcription factor NF-Y. J Biol Chem 2004; 279: 25093–25100.
Dalvai M, Mondesert O, Bourdon JC, Ducommun B, Dozier C . Cdc25B is negatively regulated by p53 through Sp1 and NF-Y transcription factors. Oncogene 2011; 30: 2282–2288.
St Clair S, Giono L, Varmeh-Ziaie S, Resnick-Silverman L, Liu WJ, Padi A et al. DNA damage-induced downregulation of Cdc25C is mediated by p53 via two independent mechanisms: one involves direct binding to the cdc25C promoter. Mol Cell 2004; 16: 725–736.
Hoffman WH, Biade S, Zilfou JT, Chen J, Murphy M . Transcriptional repression of the anti-apoptotic survivin gene by wild type p53. J Biol Chem 2002; 277: 3247–3257.
Banerjee T, Nath S, Roychoudhury S . DNA damage induced p53 downregulates Cdc20 by direct binding to its promoter causing chromatin remodeling. Nucleic Acids Res 2009; 37: 2688–2698.
Laptenko O, Prives C . Transcriptional regulation by p53: one protein, many possibilities. Cell Death Differ 2006; 13: 951–961.
Sherr CJ, Roberts JM . CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev 1999; 13: 1501–1512.
Tang X, Milyavsky M, Shats I, Erez N, Goldfinger N, Rotter V . Activated p53 suppresses the histone methyltransferase EZH2 gene. Oncogene 2004; 23: 5759–5769.
Shats I, Milyavsky M, Tang X, Stambolsky P, Erez N, Brosh R et al. p53-dependent down-regulation of telomerase is mediated by p21waf1. J Biol Chem 2004; 279: 50976–50985.
Gottifredi V, Karni-Schmidt O, Shieh SS, Prives C . p53 down-regulates CHK1 through p21 and the retinoblastoma protein. Mol Cell Biol 2001; 21: 1066–1076.
Esteve PO, Chin HG, Pradhan S . Molecular mechanisms of transactivation and doxorubicin-mediated repression of survivin gene in cancer cells. J Biol Chem 2007; 282: 2615–2625.
Lohr K, Moritz C, Contente A, Dobbelstein M . p21/CDKN1A mediates negative regulation of transcription by p53. J Biol Chem 2003; 278: 32507–32516.
Murphy M, Ahn J, Walker KK, Hoffman WH, Evans RM, Levine AJ et al. Transcriptional repression by wild-type p53 utilizes histone deacetylases, mediated by interaction with mSin3a. Genes Dev 1999; 13: 2490–2501.
Spurgers KB, Gold DL, Coombes KR, Bohnenstiehl NL, Mullins B, Meyn RE et al. Identification of cell cycle regulatory genes as principal targets of p53-mediated transcriptional repression. J Biol Chem 2006; 281: 25134–25142.
Roperch JP, Alvaro V, Prieur S, Tuynder M, Nemani M, Lethrosne F et al. Inhibition of presenilin 1 expression is promoted by p53 and p21WAF-1 and results in apoptosis and tumor suppression. Nat Med 1998; 4: 835–838.
Godefroy N, Bouleau S, Gruel G, Renaud F, Rincheval V, Mignotte B et al. Transcriptional repression by p53 promotes a Bcl-2-insensitive and mitochondria-independent pathway of apoptosis. Nucleic Acids Res 2004; 32: 4480–4490.
Bunz F, Dutriaux A, Lengauer C, Waldman T, Zhou S, Brown JP et al. Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science 1998; 282: 1497–1501.
Waldman T, Lengauer C, Kinzler KW, Vogelstein B . Uncoupling of S phase and mitosis induced by anticancer agents in cells lacking p21. Nature 1996; 381: 713–716.
Tewey KM, Rowe TC, Yang L, Halligan BD, Liu LF . Adriamycin-induced DNA damage mediated by mammalian DNA topoisomerase II. Science 1984; 226: 466–468.
Kracikova M, Akiri G, George A, Sachidanandam R, Aaronson SA . A threshold mechanism mediates p53 cell fate decision between growth arrest and apoptosis. Cell Death Differ 20: 576–588.
Sugrue MM, Shin DY, Lee SW, Aaronson SA . Wild-type p53 triggers a rapid senescence program in human tumor cells lacking functional p53. Proc Natl Acad Sci USA 1997; 94: 9648–9653.
Macip S, Igarashi M, Fang L, Chen A, Pan ZQ, Lee SW et al. Inhibition of p21-mediated ROS accumulation can rescue p21-induced senescence. EMBO J 2002; 21: 2180–2188.
Zhu W, Giangrande PH, Nevins JR . E2Fs link the control of G1/S and G2/M transcription. EMBO J 2004; 23: 4615–4626.
Chen HZ, Tsai SY, Leone G . Emerging roles of E2Fs in cancer: an exit from cell cycle control. Nat Rev Cancer 2009; 9: 785–797.
Wells J, Boyd KE, Fry CJ, Bartley SM, Farnham PJ . Target gene specificity of E2F and pocket protein family members in living cells. Mol Cell Biol 2000; 20: 5797–5807.
Crosby ME, Almasan A . Opposing roles of E2Fs in cell proliferation and death. Cancer Biol Ther 2004; 3: 1208–1211.
Lammens T, Li J, Leone G, De Veylder L . Atypical E2Fs: new players in the E2F transcription factor family. Trends Cell Biol 2009; 19: 111–118.
Cartharius K, Frech K, Grote K, Klocke B, Haltmeier M, Klingenhoff A et al. MatInspector and beyond: promoter analysis based on transcription factor binding sites. Bioinformatics 2005; 21: 2933–2942.
Di Stefano L, Jensen MR, Helin K . E2F7, a novel E2F featuring DP-independent repression of a subset of E2F-regulated genes. EMBO J 2003; 22: 6289–6298.
Zeng Y, Kotake Y, Pei XH, Smith MD, Xiong Y . p53 binds to and is required for the repression of Arf tumor suppressor by HDAC and polycomb. Cancer Res 2011; 71: 2781–2792.
Chicas A, Wang X, Zhang C, McCurrach M, Zhao Z, Mert O et al. Dissecting the unique role of the retinoblastoma tumor suppressor during cellular senescence. Cancer Cell 2010; 17: 376–387.
Bindra RS, Glazer PM . Basal repression of BRCA1 by multiple E2Fs and pocket proteins at adjacent E2F sites. Cancer Biol Ther 2006; 5: 1400–1407.
Nor Rashid N, Yusof R, Watson RJ . Disruption of repressive p130-DREAM complexes by human papillomavirus 16 E6/E7 oncoproteins is required for cell-cycle progression in cervical cancer cells. J Gen Virol 2011; 92: 2620–2627.
Tsang NM, Little JB . Effect of restoration of retinoblastoma gene function on the radiosensitivity of cells of human tumor cell lines. Radiat Res 1994; 140: 172–179.
Lee BK, Bhinge AA, Iyer VR . Wide-ranging functions of E2F4 in transcriptional activation and repression revealed by genome-wide analysis. Nucleic Acids Res 2011; 39: 3558–3573.
Huang da W, Sherman BT, Lempicki RA . Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res 2009; 37: 1–13.
Wang B, Xiao Z, Ko HL, Ren EC . The p53 response element and transcriptional repression. Cell Cycle 2010; 9: 870–879.
Li B, Lee MY . Transcriptional regulation of the human DNA polymerase delta catalytic subunit gene POLD1 by p53 tumor suppressor and Sp1. J Biol Chem 2001; 276: 29729–29739.
Jung YS, Qian Y, Chen X . Examination of the expanding pathways for the regulation of p21 expression and activity. Cell Signal 2010; 22: 1003–1012.
Ucar D, Beyer A, Parthasarathy S, Workman CT . Predicting functionality of protein-DNA interactions by integrating diverse evidence. Bioinformatics 2009; 25: i137–i144.
Burger A, Walczak AM, Wolynes PG . Abduction and asylum in the lives of transcription factors. Proc Natl Acad Sci USA 2010; 107: 4016–4021.
Mungamuri SK, Benson EK, Wang S, Gu W, Lee SW, Aaronson SA . p53-mediated heterochromatin reorganization regulates its cell fate decisions. Nat Struct Mol Biol 2012; 19: 478–484.
Helmbold H, Komm N, Deppert W, Bohn W . Rb2/p130 is the dominating pocket protein in the p53-p21 DNA damage response pathway leading to senescence. Oncogene 2009; 28: 3456–3467.
Takahashi Y, Rayman JB, Dynlacht BD . Analysis of promoter binding by the E2F and pRB families in vivo: distinct E2F proteins mediate activation and repression. Genes Dev 2000; 14: 804–816.
Litovchick L, Florens LA, Swanson SK, Washburn MP, DeCaprio JA . DYRK1A protein kinase promotes quiescence and senescence through DREAM complex assembly. Genes Dev 2011; 25: 801–813.
Litovchick L, Sadasivam S, Florens L, Zhu X, Swanson SK, Velmurugan S et al. Evolutionarily conserved multisubunit RBL2/p130 and E2F4 protein complex represses human cell cycle-dependent genes in quiescence. Mol Cell 2007; 26: 539–551.
Komori H, Enomoto M, Nakamura M, Iwanaga R, Ohtani K . Distinct E2F-mediated transcriptional program regulates p14ARF gene expression. EMBO J 2005; 24: 3724–3736.
Aslanian A, Iaquinta PJ, Verona R, Lees JA . Repression of the Arf tumor suppressor by E2F3 is required for normal cell cycle kinetics. Genes Dev 2004; 18: 1413–1422.
Mirza A, Wu Q, Wang L, McClanahan T, Bishop WR, Gheyas F et al. Global transcriptional program of p53 target genes during the process of apoptosis and cell cycle progression. Oncogene 2003; 22: 3645–3654.
Kho PS, Wang Z, Zhuang L, Li Y, Chew JL, Ng HH et al. p53-regulated transcriptional program associated with genotoxic stress-induced apoptosis. J Biol Chem 2004; 279: 21183–21192.
Hammond EM, Mandell DJ, Salim A, Krieg AJ, Johnson TM, Shirazi HA et al. Genome-wide analysis of p53 under hypoxic conditions. Mol Cell Biol 2006; 26: 3492–3504.
Roy S, Jeffrey R, Tenniswood M . Array-based analysis of the effects of trichostatin A and CG-1521 on cell cycle and cell death in LNCaP prostate cancer cells. Mol Cancer Ther 2008; 7: 1931–1939.
Fang L, Igarashi M, Leung J, Sugrue MM, Lee SW, Aaronson SA . p21Waf1/Cip1/Sdi1 induces permanent growth arrest with markers of replicative senescence in human tumor cells lacking functional p53. Oncogene 1999; 18: 2789–2797.
Akiri G, Cherian MM, Vijayakumar S, Liu G, Bafico A, Aaronson SA . Wnt pathway aberrations including autocrine Wnt activation occur at high frequency in human non-small-cell lung carcinoma. Oncogene 2009; 28: 2163–2172.
Benson EK, Lee SW, Aaronson SA . Role of progerin-induced telomere dysfunction in HGPS premature cellular senescence. J Cell Sci 2010; 123: 2605–2612.
Acknowledgements
This research was supported by grants from the Breast Cancer Research Foundation and by Award Number P01CA080058 from the National Cancer Institute. EB was supported by the NCI Training Program in Cancer Biology (T32CA078207). We thank S Lee, Cutaneous Biology Research, MGH Harvard Medical School for helpful discussions.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on the Oncogene website
Rights and permissions
This work is licensed under the Creative Commons Attribution-NonCommercial-No Derivative Works 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Benson, E., Mungamuri, S., Attie, O. et al. p53-dependent gene repression through p21 is mediated by recruitment of E2F4 repression complexes. Oncogene 33, 3959–3969 (2014). https://doi.org/10.1038/onc.2013.378
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/onc.2013.378
Keywords
This article is cited by
-
Unraveling the molecular mechanism of l-menthol against cervical cancer based on network pharmacology, molecular docking and in vitro analysis
Molecular Diversity (2023)
-
Porcine pancreatic ductal epithelial cells transformed with KRASG12D and SV40T are tumorigenic
Scientific Reports (2021)
-
Human tumor necrosis factor alpha-induced protein eight-like 1 exhibited potent anti-tumor effect through modulation of proliferation, survival, migration and invasion of lung cancer cells
Molecular and Cellular Biochemistry (2021)
-
Transcriptome analysis of HPV-induced warts and healthy skin in humans
BMC Medical Genomics (2020)
-
Transcriptional mutagenesis dramatically alters genome-wide p53 transactivation landscape
Scientific Reports (2020)
