
Abstract
Wolfram syndrome 1 (WS1) is a rare autosomal recessive neurodegenerative disease characterized by diabetes insipidus, diabetes mellitus, optic atrophy, deafness, and other abnormalities. WS1 usually results in death before the age of 50 years. The pathogenesis of WS1 is ascribed to mutations of human WFS1 gene on chromosome 4p encoding a transmembrane protein called wolframin, which has physiological functions in membrane trafficking, secretion, processing, and/or regulation of ER calcium homeostasis. Different types of WFS1 mutations have been identified, and some of these have been associated with a dominant, severe type of WS. Mutations of CISD2 gene cause autosomal recessive Wolfram syndrome 2 (WS2) characterized by the absence of diabetes insipidus and psychiatric disorders, and by bleeding upper intestinal ulcer and defective platelet aggregation. Other WFS1-related disorders such as DFNA6/14/38 nonsyndromic low-frequency sensorineural hearing loss and Wolfram syndrome-like disease with autosomal dominant transmission have been described. WS1 is a devastating disease for the patients and their families. Thus, early diagnosis is imperative to enable proper prognostication, prevent complications, and reduce the transmission to further progeny. Although there is currently no effective therapy, potential new drugs have been introduced, attempting to improve the progression of this fatal disease.
Similar content being viewed by others

Main
Mutations of CISD2 gene cause autosomal recessive Wolfram syndrome 2 (WS2) characterized by the absence of diabetes insipidus and psychiatric disorders, and by bleeding upper intestinal ulcer and defective platelet aggregation.
Other WFS1-related disorders such as DFNA6/14/38 nonsyndromic low-frequency sensorineural hearing loss and Wolfram-syndrome-like disease with autosomal dominant transmission have been described.
WS1 is a devastating disease for the patients and their families. Thus, early diagnosis is imperative to enable proper prognostication, prevent complications, and reduce the transmission to further progeny. Although there is currently no effective therapy, potential new drugs have been introduced, attempting to improve the progression of this fatal disease.
Wolfram syndrome 1
Wolfram syndrome 1 (WS1; MIM 222300) was described for the first time in 1938 by Wolfram and Wagener (1) and is characterized by diabetes insipidus, diabetes mellitus, optic atrophy, and deafness (DI DM OA D). This rare, neurodegenerative, progressive disease is inherited as an autosomal recessive disorder (2, 3, 4). The concurrence of early-onset nonautoimmune insulin-dependent DM and bilateral OA are the minimal criteria for the diagnosis of WS1 (1).
Epidemiology of WS1
The syndrome is very rare, with a prevalence of 1 in 770,000 (5) and 1 in 500,000 in children (6) in the United Kingdom; 1 in 100,000 in North America (4); 1 in 710,000 in the Japanese population (7), and 1 in 68,000 in the Lebanese population (8). The highest prevalence, of 1 in 54,478, is in a small district in a Sicilian population (Italy) (9). The high prevalence of WS1 in Lebanese and Sicilian populations could be due to the high rates of consanguinity in these populations (8, 9). In the United Kingdom, the carrier frequency is 1/354 (5).
Non-autoimmune insulin-dependent DM is the first manifestation of WS1. From studies performed with insulin-dependent DM patients of different ethnic origins, it has been found that many patients could be affected by WS1. The prevalence of this syndrome in DM patients is variable, and it has been estimated to be 0.57% in the United Kingdom as against 4.8% in the Lebanese population (5, 8). In pediatric insulin-dependent DM patients, Zmyslowska et al. have found that WS1 was diagnosed with a delay of at least 7 years as all WS1 patients were primarily misdiagnosed as having insulin-dependent type-1 DM (10).
Lombardo et al. found a relative prevalence of WS1 of 1 in 22.3 in a Sicilian population (Italy) with juvenile-onset, insulin-dependent DM aged <30 years. The prevalence was 1 in 51.2 in WS1 patients from nonconsanguineous parents (9).
Genetics of Wolfram syndromes
The human WS1 (WFS1) nuclear gene was identified in 1998 (11, 12), maps to chromosome 4p16, and is composed of eight exons spanning 33.4 kb of genomic DNA. WFS1 encodes wolframin, a transmembraneous protein in the endoplasmic reticulum (ER) with 890 amino acid (aa) residues and an apparent molecular mass of about 100 kDa (11). Wolframin, a hydrophobic, tetrameric protein, is composed of nine transmembrane segments and large hydrophilic regions at both termini (13). Wolframin is highly expressed in brain tissue such as the hippocampus, amygdala, allocortex, and olfactory bulb (14), in pancreatic β-cells, and in the heart (15).
Mutations of CISD2 gene cause a different recessive type of WS, named “WS2” (16). Patients with WS2 exhibit bleeding upper intestinal ulcer and defective platelet aggregation, without DI and psychiatric disorders (17).
CISD2 maps to chromosome 4q22-q24 and is expressed in numerous tissues, including the brain and pancreas (18, 19). It encodes ERIS (ER intermembrane small protein), a small protein of 135 aa, which is located in the mitochondria-associated ER membranes. It distributes dynamically between the ER and mitochondrial outer membrane (20). ERIS is important for the regulation of glucose homeostasis, and insulin sensitivity (21), for calcium homeostasis (18), and in autophagy (22).
Recently, a novel CISD2 mutation has been found in a Moroccan patient with a classical phenotype of WS1, but without mutations of WFS1. Thus, WS1 and WS2 could be a continuous clinical spectrum with overlapping phenotype as WFS1 and CISD2 reside in the same pathway, although CISD2 does interact directly with wolframin (23).
Molecular biology, physiology, and pathophysiology of WS1
The importance of wolframin is due to its localization in the ER. ER is involved in posttranslational modification, folding, and assembly of newly synthesized proteins such as insulin, calcium storage, redox regulation, steroid synthesis, and apoptosis (15, 24, 25, 26). Alterations in ER function cause an accumulation of misfolded proteins and the activation of unfolded protein response (UPR), a state called “ER stress”. UPR counteracts ER stress and restores ER homeostasis. WFS1 mutations cause an increase in ER stress levels, pancreatic β-cell alterations, and stress-induced apoptosis (25).
WFS1 plays a key role in ER stress in beta-cells. It negatively regulates transcription factor 6α (ATF6α), a key transcription factor involved in ER stress, attenuates activation of the ER stress response element (ERSE) promoter by ATF6α, and stabilizes E3 ubiquitin ligase HRD1, thus suppressing ER stress signaling (15).
ER stress is involved not only in WS1, but also in other neurodegenerative diseases (Alzheimer’s disease, Parkinson’s disease, and amyotrophic lateral sclerosis) (27). Thus, WS1 could be a useful model for the evaluation of the impact of ER stress in these severe disorders.
Mutations of WFS1 lead to cell apoptosis by alterations of ER stress regulation. In the ER, the C-terminal domain of WFS1, which is situated in the ER lumen, interacts with the C-terminal domain (aa 652–890) of Na+/K+ ATPase beta-1 subunit (ATP1B1). Thus, alterations of WFS1 could cause a decrease in Na-pump activity (28).
Cellular apoptosis is also influenced by calcium release and uptake in the ER. It has been shown that wolframin is a calmodulin (CaM), which interacts with many cellular proteins and that it regulates several Ca2+ signal transduction processes (29). Therefore, WFS1 controls the storage of cellular ER calcium levels and consequently cell apoptosis (26). Mutations of WFS1 gene could alter the regulation of cytosolic Ca2+ homeostasis, which, in turn, causes perturbation of mitochondrial dynamics. A deficiency of WFS1 gene causes an ER stress cascade, which impairs the function of the inositol 1,4,5-triphosphate receptor (IP3R) calcium channel. Thus, cytosolic Ca2+ homeostasis is perturbed and, in turn, also mitochondrial dynamics (inhibited mitochondrial fusions, altered mitochondrial trafficking, and augmented mitophagy) (30). Mitochondrial alterations result in lower levels of ATP which alter neuronal development. Therefore, mitochondrial involvement could explain the clinical symptoms of WS1. Moreover, a thorough understanding of this mechanism could allow the identification of potential therapeutic targets in WS1, as well as in neuropsychiatric diseases (30).
Neuropathological postmortem case studies have shown abnormalities in multiple regions of the brain of WS1 patients (31). By in vivo neuroimaging, alterations of brain structures, such as generalized brain atrophy, especially in the cerebellum, medulla, and pons, the absence of a signal from the posterior pituitary, and a reduced signal from the optic nerve have been detected early in patients (32).
Experimental studies have shown high expression of mice wolframin (Wfs1) mRNA and protein in the brain from birth to early adulthood (14, 33). A relatively constant Wfs1 mRNA expression in the supraoptic nucleus and the magnocellular nucleus of mice during development has been found, but it weakens in postnatal life (34).
Thus, it is important to define WFS1 expression patterns for better knowledge of the progression and evolution of WS1.
Recently, Tekko et al. have described the initiation pattern of Wfs1 expression in the mouse forebrain by mRNA in situ hybridization. As a marker of neuronal differentiation and synaptic contacts, expression of the synaptophysin (Syp1) gene, which encodes a synaptic vesicle protein, has been evaluated (34). It has been shown that in the brain-development phases, expression of Syp1 starts earlier than that of Wfs1 as the expression of Wfs1 begins only when the different brain structures have already been completed, reaching the adult pattern after 3 postnatal weeks. Indeed, Wfs1 expression was absent or very weak in the early stages of brain development. Thus, it has been hypothesized that WFS1 might be regulated independently during the postnatal period and its role might be to protect the brain from neurodegeneration. The silencing of WFS1 in HEK cells causes upregulation of a complex molecular network called “protein trafficking, cell morphology, cellular function, and maintenance network”. In particular, some genes (ADAM 19, TOMM20, and EPAS1) and proteins (heat shock protein 8 and transthyretin) might be involved in neurodegeneration via mitochondrial dysfunction or UPR dysfunction (35, 36).
Finally, Hershey et al. have shown abnormalities in different brain regions, as well as increased intracranial volume already in young patients (37).
The pathogenetic mechanisms that underlie the neurodegeneration of WS1 are very complex as brain abnormalities may be the product of two separate pathological processes, both neurodegenerative and neurodevelopmental.
Wolframin is highly expressed in pancreatic islet β-cells and it may help to fold a protein precursor of insulin (called proinsulin) into a mature hormone that controls blood glucose levels (38, 39). WFS1 seems to regulate the activation and secretion of insulin, as well as other peptides (40).
WFS1 regulates β cellular survival as suggested by an increase of apoptosis caused by an increase of insulin demand in Wfs1-deficient mice (41). These mice exhibit alterations of insulin pathways as suggested by lower insulin levels and higher proinsulin concentrations (35).
Wolframin is also localized to the secretory granules of pancreas (42) and is involved in prohormone processing (43). A wolframin deficiency causes progressive loss of β-cells, impaired glucose tolerance and cell cycle progression, and activation of ER stress/UPR. Probably, wolframin also plays an important role during embryogenesis, especially in pancreatic development. Indeed, Xu et al. have found that the Wfs1 protein is localized to the mesenchyme in the rat pancreas (44).
Ivask et al. have found a lower number of pancreatic islets in Wfs1-deficient (Wfs1KO) mice than those in heterozygous (Wfs1HZ) or wild-type (WT) mice. Therefore, Wfs1KO pancreatic islets secrete less insulin compared with WT and Wfs1HZ islets. This Wfs1 deficiency alters the pathways related to tissue morphology, endocrine system development, and function (45).
Recently, microRNA (miRNA) deregulation has been emerging as a contributor to the neurodegeneration of WS1, as well as of neurodegenerative diseases.
MiRNAs are mainly expressed in the human brain where they exercise regulatory action on synaptogenesis and other neuronal activities through interaction with target genes. Human miRNA regulates the functions of nearly 50% of all genes implicated in the expression of the proteins (46).
A putative miRNA-binding site has been identified in the WFS1 gene, called rs1046322. It is located in the 3′ UTR of the WFS1 gene. The mutated minor A allele of rs1046322 (a G>A substitution) weakens the binding of a specific miRNA (miR-668) that regulates the wolframin protein synthesis. Mutated homozygotes AA for rs1046322 of WFS1 have higher levels of aggression, impulsiveness, depression, and anxiety than the normal GG homozygotes and AG heterozygotes. Thus, rs1046322 SNP could be a key regulator of WFS1 expression (47).
Genotypes and genotype–phenotype correlations
More than 170 different mutations have been identified in WFS1 (https://lovd.euro-wabb.org//home.php?select_db=WFS1). They are situated principally in exon 8 and are often inactivating (nonsense or frameshift) mutations.
WFS1 mutations may be dominantly or recessively inherited and the onset of the clinical picture is highly variable in both appearance and degree of severity (48).
Moreover, it is difficult to establish genotype–phenotype correlations because of the molecular complexity of WS1, the different clinical characteristics, and the small size of patient cohorts (30–60 patients). Some studies have found that a compound heterozygosity for two missense mutations may cause a relatively mild phenotype (49), whereas the coexistence of two inactivating mutations may predispose to an earlier onset of DM and OA (50). It has been shown that the age of onset of DM in WS1 patients carrying complete loss-of-function mutations was lower than those carrying partial or minor loss-of-function mutations (7, 51).
A study by Chaussenot et al. has shown no association between the genotype and neurological symptoms in WS1 patients (52).
de Heredia et al. have performed a review of clinical and genetic data of a wide group of 412 patients affected by WFS1-related disorders, recruited from 49 studies which have been published since 1998.
In total, 178 different mutations in the entire WFS1 were found that are principally located in transmembrane domains, N-end of the wolframin, and the last 100 amino acids. These mutations have shown a different effect on WFS1 expression and, thus, they have been classified as in Tables 1 and 2 (53).
Recently, novel dominant heterozygous mutations of WFS1 have been associated with severe phenotypes of WS.
Bonnycastle et al. have found a novel nonsynonymous variant (p. Trp314Arg) in WFS1 that segregates completely with dominantly inherited nonsyndromic adult-onset diabetes mellitus in a large four-generations family. Other than diabetes, the p. Trp314Arg carriers of the family had none of the common clinical signs of WS1 (54).
Morikawa et al. have found a de novo heterozygous 4 aa in-frame deletion (p.N325_I328del) in an 11-month-old Japanese female WS patient with insulin-dependent DM, congenital cataract, and severe bilateral hearing loss (55). Although cataract is not typical of WS1, other cases associated with WFS1 heterozygous mutations have been described (56).
Three heterozygous missense mutations in WFS1 (4/5 confirmed de novo) have been found in five patients with diabetes diagnosed before 12 months (5/5), sensorineural deafness diagnosed soon after birth (5/5), congenital cataracts (4/5), and hypotonia (4/5) (56).
Therefore, specific dominant heterozygous mutations of WFS1 cause a severe phenotype of WS by an activation of a pathway of severe constitutive ER stress. This type of WS differs genetically and clinically from recessive WS1.
Natural history, symptoms, and treatment options in WS1
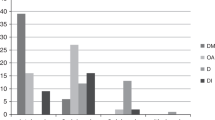
Some studies have shown the order of onset of the different clinical symptoms of WS1 (52, 57, 58). de Heredia et al. have found that from 412 patients with age specified for any clinical symptoms, 98.21% had DM; 82.14% had OA; 48.21% had D; 37.76% had DI; 19.39% had urological manifestations; and 17.09% had neurological symptoms (53). Most patients die of respiratory failure. WS1 has high morbidity and mortality, and the median age of death is around 30 (range 25–49) years (6, 11). However, two peaks of higher frequency might be observed—one at 24 and the other at 45 years of age (53).
Here we describe the main clinical features of WS1 as the following:
Nonautoimmune Insulin-dependent DM
WS1 almost always starts with nonautoimmune and non-HLA-linked insulin-dependent DM, at an average age of 6 years (range 3 weeks to 16 years). Microvascular complications are rare and they do not progress as quickly as in the more frequent insulin-dependent DM (58, 59).
Almost all patients require insulin treatment (60). It seems that in WS1 patients, insulin-dependent DM has different clinical characteristics from common type-1 diabetes: earlier diagnosis, lower prevalence of ketoacidosis, autoantibodies that are less often positive, longer duration of remission, and higher frequency of severe hypoglycemia. Severe hypoglycemia is a result of neurologic dysfunction caused by a perturbation of ER function. Microvascular complications also including microvascular retinopathy are less frequent than in type-1 DM, probably because hyperglycemia occurs when there are high levels of ER stress (51).
There are few reliable data regarding the degree of metabolic control (based on HbA1c level) and insulin requirement in comparison with insulin-dependent DM (T1DM) patients.
WS1 nonautoimmune insulin-dependent DM is characterized by a daily insulin requirement and a mean HbA1c lower than in type-1 diabetes (60).
In WS1, a persistence of residual insulin secretion causes a progression of nonautoimmune insulin-dependent DM toward total insulin deficiency which is not as quick as in type-1 diabetes mellitus.
In a group of seven pediatric WS1 patients with nonautoimmune insulin-dependent DM, Zmyslowska et al. have obtained similar results, showing that the glycemic variability was smaller than in subjects with T1DM (61).
Nonautoimmune insulin-dependent DM of WS1 begins in preschool age without ketoacidosis, it is antibody-negative, and has surprisingly long periods of remission. Therefore, WS1 must be suspected in these cases.
Optic Atrophy
Optic atrophy is a criterion required for the diagnosis of WS1 (5). In WS1 patients, OA occurs in the first decade; it is progressive and often leads to blindness. It begins at an average age of 11 years (6 weeks to 19 years) with reduced visual acuity and loss of color vision. Other less-frequent ocular abnormalities reported are cataract (29.6–66.6%), abnormal papillary light reflexes, nystagmus, pigmentary maculopathy, and glaucoma (62). Pigmentary retinopathy has also been described in 30% of 15 Jordanian WS1 patients from four inbred families. This ocular complication is very rare in WS1 patients and these data are surprising. Indeed, few cases of WS1 associated with any type of retinal pigmentary changes have been described (63, 64, 65, 66, 67). Pigmentary retinopathy can occur commonly in mitochondrial disorders such as Kearns–Sayre syndrome and the neuropathy, ataxia, and retinitis pigmentosa (NARP) syndrome. Recent observations have shown that in WS1, there are dramatic alterations in mitochondrial dynamics (30) and it could explain the detection of pigmentary retinopathy in WS1 patients, albeit rare.
Recently, microspherophakia in two WS1 siblings with congenital cataract, glaucoma, and OA has also been described (68).
In 18 WS1 children at relatively early stages, multiple ophthalmic markers, such as retinal nerve fiber layer thickness, have been identified. It would appear that these alterations correlate with the overall disease severity (62).
The average retinal thickness is significantly lower in pediatric/adolescent patients with WS1 compared with both patients with insulin-dependent, antibody-negative DM and control subjects. No differences have been found between children/adolescents with insulin-dependent, antibody-negative DM and control subjects or between adult WFS1 mutation carriers and control subjects. Thus, retinal thinning is a clinical feature of WS1 and may be a potential marker of disease progression in WS1 patients (69).
In WS1 patients, an annual eye examination is essential, including visual acuity, color-vision testing, fundoscopy, visual field, and optical coherence tomography scan. The monitoring of the efficacy of potential therapy can be performed by visually evoked potentials.
Unfortunately, there are no medical drugs for the treatment of OA. Some authors have proposed the utility of idebenone or docosahexaenoic acid to delay the progression of OA. However, there are few studies that confirm the efficacy of this therapy (70, 71).
Diabetes Insipidus
Clinically, at an average age of 14 years (3 months to 40 years), 73–75% of WS1 patients present partial cranial DI. The range in the age of onset is broad, probably because of delays in establishing the correct diagnosis (5). The test of the concentrating ability of urine is important in evaluating for DI and it must be performed in subjects with T1DM, color-vision defects, deafness, neurological abnormalities, and so on. Therapy of central DI follows routine practices and WS1 patients respond well to intranasal or oral desmopressin.
Sensorineural Deafness
D presents at an average age of 12.5 years (range 5–39 years) (5) and is a feature seen in 62% of WS1 patients (6). D in WS1 patients affects high frequencies first and progresses relatively slowly (5). Pennings et al. have found that in WS1 females, hearing loss (HL) was more altered than in WS1 males (72). However, in other studies, no gender differences in the degree of D have been found (3, 6). With increasing age, D in WS1 patients is more pronounced than in other patients with hypoacusia, probably as a consequence of progressive central nervous system deterioration (72).
The follow-up of D in WS1 patients includes an audiometry test every year or every 2 years, and an auditory brainstem response audiometry to evaluate the clinical course and efficacy of any therapy.
Hearings aids and cochlear implants are the main therapeutic tools.
Neurological and Psychiatric Symptoms
Neurological complications and psychiatric disorders are frequent in WS1 patients.
Neurological abnormalities are found in 62% of WS1 patients, developing at an average age of 16 years (mean age 30 years, range 5–44 years). However, the onset of neurological symptoms is often earlier (48) than those previously reported (5). de Heredia et al. have found that neurological symptoms appear at 10–30 years of age with a median of 23 years and two peaks—one at 13 and the other at 30 years of age (53).
The most common symptom is cerebellar ataxia of the trunk that should be evaluated yearly or twice a year by a neurologist. Dysarthria and dysphagia are frequent. As dysphagia could cause aspiration pneumonia, a swallowing treatment with a speech-language pathologist is important. Some patients must undergo surgical procedures.
Polysomnography and overnight oximetry test are needed as brainstem atrophy often causes central apnea that results in death (5). In some cases, a tracheostomy is required.
Respiratory failure or dysphagia due to brainstem involvement are the common causes of mortality (5).
Other frequent neurological signs include loss of gag reflex, loss of olfaction, myoclonus, epilepsy, and nystagmus. In WS1 patients, autonomic neuropathy is frequent and is characterized by orthostatic hypotension, anhidrosis, hypoidrosis or hyperidrosis, constipation, gastroparesis, hypothermia, or hyperpyrexia (57). Therefore, a thorough medical history is required. Headache has also been reported in WS1 patients (53).
Frequently, WS1 patients (60%) are affected by episodes of severe depression, psychosis, smell and sleep abnormalities, or organic brain symptoms, as well as impulsive verbal and physical aggression, whereas WFS1 heterozygotes may be predisposed to psychiatric illness. Frequently, cognitive and psychiatric symptoms begin in later stages of the disease (52, 73).
Smell- and sleep-related symptoms could be useful to monitor WS1 patients longitudinally (73).
When there are psychiatric manifestations, psychiatric counseling may be helpful. Generally, cognitive performance is preserved in WS1 patients. However, Chaussenot et al. (52) have found that in a group of 59 WS1 patients, a cognitive impairment was ranked third (32%), after cerebellar ataxia and peripheral neuropathy.
Reproductive Biology and Endocrinology
In WS1 patients, anterior pituitary dysfunctions, and in male patients, primary gonadal atrophy and hypergonadotropic hypogonadism have been described. In women, there are abnormalities in menstrual cycles, but ovarian function is normal with a regular ability to become pregnant. Short stature and growth hormone (GH) deficiency have been described in WS1 patients (5, 8).
As deficient GH secretion with deficient corticotropin secretion is one of the most common abnormalities, WS1 patients could be monitored for a possible severe growth retardation which could respond to GH administration. Moreover, these patients require steroid supplementation during periods of stress, such as severe infection (8).
Additional Anomalies
Hydroureteronephrosis, urinary incontinence, and recurrent infections are the common signs of a neurogenic bladder. The median age of the onset of urological manifestations is 20 years, although numerous patients develop the symptoms at 10–20 years of age. Three peaks have been observed: one at 13, the second at 21, and the third at 33 years of age (53).
However, urinary tract infections have been described as the first manifestation of WS1 at a very early age (74).
Early identification of renal and urological manifestations is recommended by assessment of renal function, measurement of post void residual urine volume by ultrasound, a renal ultrasound, and urodynamic testing. The therapy for bladder dysfunction includes anticholinergic drugs and clean intermittent catheterization.
Gastrointestinal symptoms include bowel dysmotility (24%), symptoms of gastroparesis (29%), and bowel incontinence (48).
In WS1 patients, congenital heart diseases are very rare. However, some cases have been reported (75). In 16.1% of 31 Lebanese patients, valvular heart disease, particularly pulmonary stenosis, has been described (8). In 68 WS1 patients, only 3 cases had cardiac anomalies: tetralogy of Fallot in two, and pulmonary valve stenosis in one (76). Aloi et al. have described a tetralogy of Fallot in a compound heterozygous for WFS1 mutations (77). Thus, although congenital heart diseases in WS1 patients are rarely described, cardiac monitoring could be useful.
Other WFS1-related disorders
DFNA6/14/38 Nonsyndromic Low-frequency Sensorineural Hearing Loss
Mutations in WFS1 may also cause DFNA6/14/38, which is characterized by low-frequency SNHL without profound deafness (75).
DFNA6/14/38 low-frequency sensorineural hearing loss (LFSNHL) is characterized by autosomal dominant transmission, but some recessive or sporadic cases have also been described (78, 79, 80). Most mutations are located in exon 8 and are small noninactivating (missense) mutations, whereas most of the pathogenic mutations in WS1 are inactivating mutations (81). The age at the onset of the HL varies from early childhood to young adulthood (79). HL in DFNA6/14/38 can be either progressive or nonprogressive (79). Frequently, the diagnosis of DFNA6/14/38 is delayed because low-frequency HL may not alter language comprehension. Thus, DFNA6/14/38 becomes manifest when high-frequency HL occurs (75).
WS-like Disease
WS-like disease has been described in two families (82) and by Eiberg et al. (83) in which there was the same pathogenic mutation in exon 8. The patients were affected by LFSNHL, diabetes mellitus, and psychiatric illness. OA was present in the family of Eiberg et al. (83), and absent in the family of Valéro et al. (82). Moreover, an association of OA and SNHL has also been described as a phenotype caused by dominant mutations in WFS1 patients.
Diagnosis of WS1
An accurate, prompt diagnosis of WS1 is important so as to begin follow-up of patients early, and to develop all required therapeutic strategies. History and the observation of OA after a diagnosis of nonautoimmune insulin-dependent DM under the age of 16 years lead to the suspicion of WS1. However, in many subjects, a significant delay occurs between the time of diagnosis of the first signs of the syndrome and the diagnosis of WS1.
Genetic tests are needed to confirm the diagnosis of WS1. The molecular complexity of WS1 complicates the establishment of genotype–phenotype correlations and the diagnosis of WS1 remains essentially clinical.
It is important that the follow-up of diagnosed WS1 patients includes regular neurological, ophthalmologic, and psychiatric consultations, audiometry, as well as endocrine checkups.
Molecular tests have been developed to help clinicians in WS1 diagnosis. It is advisable to perform a screening of the entire WFS1. Recently, exome-sequencing- and genome-sequencing-based diagnostic methods for WS1 have been developed.
New chances for therapies
The role of ER stress in cells has been demonstrated in clinical and genetic studies. Indeed, acquired or inherited ER perturbations may lead to rare genetic syndromes such as WS, as well as several common diseases such as insulin-dependent and non-insulin-dependent DM, atherosclerosis, and neurodegenerative diseases (15, 35, 37).
Thus, medical drugs that can restore the normal ER pathway should be the focus of research, and WS1 represents a good human cell model of ER disease.
Recently, drug repurposing has been hypothesized as being useful for WS1 therapy. With drug repurposing, drugs approved by regulatory agencies are used to treat other diseases.
In the treatment of WS1, the drugs should act on specific areas of ER dysfunction, as modulators of ER stress, regulators of ER calcium homeostasis, and cellular proteostasis (84).
WS1 represents the first human cell model of ER disease. Shang and co-workers have developed an experimental model generating β cells in vitro by induction of pluripotent stem cells (iPSCs) derived from skin cells of patients with WS1. These cells were characterized by high levels of ER stress and decreased insulin content. Interestingly, 4-phenylbutyric acid, a chemical protein-folding and trafficking chaperone, has restored normal insulin synthesis and the ability to upregulate insulin secretion. Thus, the chaperones may be used as drugs to treat WS1, and WS1 iPSCs could be used as an experimental model to evaluate the efficacy of these drugs. Currently, it seems that two chaperones, 4-phenylbutyric acid and tauroursodeoxycholic acid, can improve the function of β cells, prevent ER stress-mediated β-cell death, and neurodegeneration in WS1 patients (38).
Recently, Lu et al. have shown that in WFS1 knockout mice, as well as in neural progenitor cells derived from iPSCs of WS1 patients, the calpain protease is hyperactivated by increasing cytosolic calcium levels. Thus, potential drugs for therapy of WS1 should act on the pathway of calpain, and the hyperactivation of calpain may be a biomarker for WS1 (71).
Some studies have been performed to search for drugs that may stabilize ER calcium levels. It seems that dantrolene, which targets the ryanodine receptor localized to the ER membrane, prevents β-cell death and neurodegeneration (71).
Novel small molecules have been proposed to maintain ER homeostasis. It has been shown that WFS1 binds to SERCA, a sarco/endoplasmic reticulum Ca2+–ATPase, which regulates ER calcium homeostasis. Thus, for the therapy of WS1, a drug that activates SERCA and maintains high levels of ER calcium could be useful. Ryanodine receptors and inositol triphosphate receptors could be other regulator calcium channel drugs (85).
Finally, regenerative therapy and gene therapy have been proposed to treat WS1.
The iPSCs from patients’ skin cells could be useful to correct WFS1 mutations. Moreover, iPSCs could be differentiated into insulin-producing β cells, retinal cells, and neurons for transplantation (86).
Conclusion
WS1 is a rare monogenic disorder characterized by ER stress in cell lines and in animal models (34). It is considered as a prototype of ER disease and its characteristics could make it a useful model for understanding more complex diseases such as DM, OA, D, and other multiple neurodegenerative diseases.
A multidisciplinary approach is important for early detection of WS1 symptoms. Early diagnosis may recognize treatable complications and, hence, reduce morbidity and mortality. Moreover, genetic counseling is mandatory.
Further studies are needed to better understand the pathophysiology of WS1, to provide comprehensive genetic counseling in affected families, and ultimately to improve prevention strategies, as well as treatments, for this devastating disease.
Change history
10 October 2018
The original version of this Article erroneously cropped part of the abstract. The abstract has now been corrected in the PDF and HTML versions of this Article.
References
Wolfram DJ, Wagener HP . Diabetes mellitus and simple optic atrophy among siblings: report of four cases. Mayo Clin Proc 1938;13:715–718.
Page MM, Asmal AC, Edwards CR . Recessive inheritance of diabetes: the syndrome of diabetes insipidus, diabetes mellitus, optic atrophy and deafness. Q J Med 1976;45:505–520.
Cremers CW, Wijdeveld PG, Pinckers AJ . Juvenile diabetes mellitus, optic atrophy, hearing loss, diabetes insipidus, atonia of the urinary tract and bladder, and other abnormalities (Wolfram syndrome). A review of 88 cases from the literature with personal observations on 3 new patients. Acta Paediatr Scand Suppl 1977;264:1–16.
Fraser FC, Gunn T . Diabetes mellitus, diabetes insipidus, and optic atrophy. An autosomal recessive syndrome? J Med Genet 1977;14:190–193.
Barrett TG, Bundey SE, Macleod AF . Neurodegeneration and diabetes: UK nationwide study of Wolfram (DIDMOAD) syndrome. Lancet 1995;346:1458–1463.
Kumar S . Wolfram syndrome: important implications for pediatricians and pediatric endocrinologists. Pediatr Diabetes 2010;11:28–37.
Matsunaga K, Tanabe K, Inoue H et al. Wolfram syndrome in the Japanese population; molecular analysis of WFS1 gene and characterization of clinical features. PLoS ONE 2014;9:e106906.
Medlej R, Wasson J, Baz P et al. Diabetes mellitus and optic atrophy: a study of Wolfram syndrome in the Lebanese population. J Clin Endocrinol Metab 2004;89:1656–1661.
Lombardo F, Salzano G, Di Bella C et al. Phenotypical and genotypical expression of Wolfram syndrome in 12 patients from a Sicilian district where this syndrome might not be so infrequent as generally expected. J Endocrinol Invest 2014;37:195–202.
Zmyslowska A, Borowiec M, Fichna P et al. Delayed recognition of Wolfram syndrome frequently misdiagnosed as type 1diabetes with early chronic complications. Exp Clin Endocrinol Diabetes 2014;122:35–38.
Inoue H, Tanizawa Y, Wasson J et al. A gene encoding a transmembrane protein is mutated in patients with diabetes mellitus and optic atrophy (Wolfram syndrome). Nat Genet 1998;20:143–148.
Strom TM, Hörtnagel K, Hofmann S et al. Diabetes insipidus, diabetes mellitus, optic atrophy and deafness (DIDMOAD) caused by mutations in a novel gene (wolframin) coding for a predicted transmembrane protein. Hum Mol Genet 1998;7:2021–2028.
Hofmann S, Philbrook C, Gerbitz KD et al. Wolfram syndrome: structural and functional analyses of mutant and wild-type wolframin, the WFS1 gene product. Hum Mol Genet 2003;12:2003–2012.
Takeda K, Inoue H, Tanizawa Y et al. WFS1 (Wolfram syndrome 1) gene product: predominant subcellular localization to endoplasmic reticulum in cultured cells and neuronal expression in rat brain. Hum Mol Genet 2001;10:477–484.
Fonseca SG, Ishigaki S, Oslowski CM et al. Wolfram syndrome 1 gene negatively regulates ER stress signaling in rodent and human cells. J Clin Invest 2010;120:744–755.
Amr S, Heisey C, Zhang M et al. A homozygous mutation in a novel zinc-finger protein, ERIS, is responsible for Wolfram syndrome 2. Am J Hum Genet 2007;81:673–683.
al-Sheyyab M, Jarrah N, Younis E et al. Bleeding tendency in Wolfram syndrome: a newly identified feature with phenotype genotype correlation. Eur J Pediatr 2001;160:243–246.
Solovyova N, Veselovsky N, Toescu EC et al. Ca(2+) dynamics in the lumen of the endoplasmic reticulum in sensory neurons: direct visualization of Ca(2+)-induced Ca(2+) release triggered by physiological Ca(2+) entry. EMBO J 2002;21:622–630.
Chen YF, Wu CY, Kirby R et al. A role for the CISD2 gene in lifespan control and human disease. Ann N Y Acad Sci 2010;1201:58–64.
Chen YF, Kao CH, Chen YT et al. Cisd2 deficiency drives premature aging and causes mitochondria-mediated defects in mice. Genes Dev 2009;15:1183–1194.
Wang CH, Tsai TF, Wei YH . Role of mitochondrial dysfunction and dysregulation of Ca(2+) homeostasis in insulin insensitivity of mammalian cells. Ann N Y Acad Sci 2015;1350:66–76.
Chang NC, Nguyen M, Germain M et al. Antagonism of Beclin 1-dependent autophagy by BCL-2 at the endoplasmic reticulum requires NAF-1. EMBO J 2010;3:606–618.
Rouzier C, Moore D, Delorme C et al. A novel CISD2 mutation associated with a classical Wolfram syndrome phenotype alters Ca2+ homeostasis and ER-mitochondria interactions. Hum Mol Genet 2017;1:1599–1611.
Ishihara H, Takeda S, Tamura A et al. Disruption of the WFS1 gene in mice causes progressive beta-cell loss and impaired stimulus-secretion coupling in insulin secretion. Hum Mol Genet 2004;13:1159–1170.
Yamada T, Ishihara H, Tamura A et al. WFS1-deficiency increases endoplasmic reticulum stress, impairs cell cycle progression and triggers the apoptotic pathway specifically in pancreatic beta-cells. Hum Mol Genet 2006;15:1600–1609.
Osman AA, Saito M, Makepeace C et al. Wolframin expression induces novel ion channel activity in endoplasmic reticulum membranes and increases intracellular calcium. J Biol Chem 2003;278:52755–52762.
Kim J, Xu W, Reed JC . Cell death and endoplasmic reticulum stress: disease relevance and therapeutic opportunities. Nat Rev Drug Discov 2008;7:1013–1030.
Sütt S, Altpere A, Reimets R et al. Wfs1-deficient animals have brain-region-specific changes of Na+, K+-ATPase activity and mRNA expression of α1 and β1 subunits. J Neurosci Res 2015;93:530–537.
Yurimoto S, Hatano N, Tsuchiya M et al. Identification and characterization of wolframin, the product of the wolfram syndrome gene (WFS1), as a novel calmodulin-binding protein. Biochemistry 2009;48:3946–3955.
Cagalinec M, Liiv M, Hodurova Z et al. Role of mitochondrial dynamics in neuronal development: mechanism for Wolfram syndrome. PLoS Biol 2016;19:e1002511.
Hilson JB, Merchant SN, Adams JC et al. Wolfram syndrome: a clinicopathologic correlation. Acta Neuropathol 2009;118:415–428.
Ito S, Sakakibara R, Hattori T . Wolfram syndrome presenting marked brain MR imaging abnormalities with few neurologic abnormalities. AJNR Am J Neuroradiol 2007;28:305–306.
Kawano J, Fujinaga R, Yamamoto-Hanada K et al. Wolfram syndrome 1 (Wfs1) mRNA expression in the normal mouse brain during postnatal development. Neurosci Res 2009;64:213–230.
Tekko T, Lilleväli K, Luuk H et al. Initiation and developmental dynamics of Wfs1 expression in the context of neural differentiation and ER stress in mouse forebrain. Int J Dev Neurosci 2014;35:80–88.
Kõks S, Overall RW, Ivask M et al. Silencing of the WFS1 gene in HEK cells induces pathways related to neurodegeneration and mitochondrial damage. Physiol Genomics 2013;45:182–190.
Westermark GT, Westermark P . Transthyretin and amyloid in the islets of Langerhans in type-2 diabetes. Exp Diabetes Res 2008;2008:429274 1-7.
Hershey T, Lugar HM, Shimony JS et al. Early brain vulnerability in Wolfram syndrome. PLoS ONE 2012;7:e40604.
Shang L, Hua H, Foo K et al. β-cell dysfunction due to increased ER stress in a stem cell model of Wolfram syndrome. Diabetes 2014;63:923–933.
Lemaire K, Schuit F . Integrating insulin secretion and ER stress in pancreatic β-cells. Nat Cell Biol 2012;14:979–981.
Fonseca SG, Fukuma M, Lipson KL et al. WFS1 is a novel component of the unfolded protein response and maintains homeostasis of the endoplasmic reticulum in pancreatic beta-cells. J Biol Chem 2005;280:39609–39615.
Akiyama M, Hatanaka M, Ohta Y et al. Increased insulin demand promotes while pioglitazone prevents pancreatic beta cell apoptosis in Wfs1 knockout mice. Diabetologia 2009;52:653–663.
Hatanaka M, Tanabe K, Yanai A et al. Wolfram syndrome 1 gene (WFS1) product localizes to secretory granules and determines granule acidification in pancreatic beta-cells. Hum Mol Genet 2011;20:1274–1284.
Tein K, Kasvandik S, Kõks S et al. Prohormone convertase 2 activity is increased in the hippocampus of Wfs1 knockout mice. Front Mol Neurosci 2015;8:45–50.
Xu R, Xia B, Geng J et al. Expression and localization of Wolfram syndrome 1 gene in the developing rat pancreas. World J Gastroenterol 2009;15:5425–5431.
Ivask M, Hugill A, Kõks S . RNA-sequencing of WFS1-deficient pancreatic islets. Physiol Rep 2016;4:e12750.
Sethupathy P, Collins FS . MicroRNA target site polymorphisms and human disease. Trends Genet 2008;24:489–497.
Kovacs-Nagy R, Elek Z, Szekely A et al. Association of aggression with a novel microRNA binding site polymorphism in the wolframin gene. Am J Med Genet B Neuropsychiatr Genet 2013;162B:404–412.
Chaussenot A, Rouzier C, Quere M et al. Mutation update and uncommon phenotypes in a French cohort of 96 patients with WFS1-related disorders. Clin Genet 2015;87:430–439.
d'Annunzio G, Minuto N, D'Amato E et al. Wolfram syndrome (diabetes insipidus, diabetes, optic atrophy, and deafness): clinical and genetic study. Diabetes Care 2008;31:1743–1745.
Cano A, Rouzier C, Monnot S et al. Identification of novel mutations in WFS1 and genotype-phenotype correlation in Wolfram syndrome. Am J Med Genet A 2007;143A:1605–1612.
Rohayem J, Ehlers C, Wiedemann B et al. Diabetes and neurodegeneration in Wolfram syndrome: a multicenter study of phenotype and genotype. Diabetes Care 2011;34:1503–1510.
Chaussenot A, Bannwarth S, Rouzier C et al. Neurologic features and genotype-phenotype correlation in Wolfram syndrome. Ann Neurol 2011;69:501–508.
deHeredia ML, Clèries R, Nunes V . Genotypic classification of patients with Wolfram syndrome: insights into the natural history of the disease and correlation with phenotype. Genet Med 2013;15:497–506.
Bonnycastle LL, Chines PS, Hara T et al. Autosomal dominant diabetes arising from a Wolfram syndrome 1 mutation. Diabetes 2013;62:3943–3950.
Morikawa S, Tajima T, Nakamura A et al. A novel heterozygous mutation of the WFS1 gene leading to constitutive endoplasmic reticulum stress is the cause of Wolfram syndrome. Pediatr Diabetes 2017;8:1–8.
De Franco E, Flanagan SE, Yagi T et al. Dominant ER stress-inducing wfs1 mutations underlie a genetic syndrome of neonatal/infancy-onset diabetes, congenital sensorineural deafness, and congenital cataracts. Diabetes 2017;66:2044–2053.
Rigoli L, Lombardo F, Di Bella C . Wolfram syndrome and WFS1 gene. Clin Genet 2011;79:103–117.
Rigoli L, Lombardo F, Salzano G et al. Identification of one novel causative mutation in exon 4 of WFS1 gene in two Italian siblings with classical DIDMOAD syndrome phenotype. Gene 2013;526:487–489.
Ganie MA, Bhat D . Current developments in Wolfram syndrome. J Pediatr Endocrinol Metab 2009;22:3–10.
Cano A, Molines L, Valéro R et al. Microvascular diabetes complications in Wolfram syndrome (diabetes insipidus, diabetes mellitus, optic atrophy, and deafness [DIDMOAD]): an age- and duration-matched comparison with common type 1 diabetes. Diabetes Care 2007;30:2327–2330.
Zmyslowska A, Fendler W, Niwald A et al. Retinal thinning as a marker of disease progression in patients with Wolfram syndrome. Diabetes Care 2015;38:e36–e37.
Hoekel J, Chisholm SA, Al-Lozi A et al. Ophthalmologic correlates of disease severity in children and adolescents with Wolfram syndrome. J AAPOS 2014;18:461–465.
Cremers CW, Wijdeveld PG, Pinckers AJ . Juvenile diabetes mellitus, optic atrophy, hearing loss, diabetes insipidus, atonia of the urinary tract and bladder, and other abnormalities (Wolfram syndrome). A review of 88 cases from the literature with personal observations on 3 new patients. Acta Paediatr Scand Suppl. 1977;264:1–16.
Gunn T, Bortolussi R, Little JM et al. Juvenile diabetes mellitus, optic atrophy, sensory nerve deafness, and diabetes insipidus—a syndrome. J Pediatr 1976;89:565–570.
Barrett TG, Bundey SE . Wolfram (DIDMOAD) syndrome. J Med Genet 1997;34:838–841.
Al-Till M, Jarrah NS, Ajlouni KM . Ophthalmologic findings in fifteen patients with Wolfram syndrome. Eur J Ophthalmol 2002;12:84–88.
Dhalla MS, Desai UR, Zuckerbrod DS . Pigmentary maculopathy in a patient with Wolfram syndrome. Can J Ophthalmol 2006;41:38–40.
Chacón-Camacho O, Arce-Gonzalez R, Granillo-Alvarez M et al. Expansion of the clinical ocular spectrum of Wolfram Syndrome in a family carrying a novel WFS1 gene deletion. Ophthalmic Genet 2013;34:243–248.
Zmyslowska A, Fendler W, Szadkowska A et al. Glycemic variability in patients with Wolfram syndrome is lower than in insulin-dependent diabetes mellitus. Acta Diabetol 2015: 29–32.
Bababeygy SR, Wang MY, Khaderi KR et al. Visual improvement with the use of idebenone in the treatment of Wolfram syndrome. J Neuroophthalmol 2012;32:386–389.
Lu S, Kanekura K, Hara T et al. A calcium-dependent protease as a potential therapeutic target for Wolfram syndrome. Proc Natl Acad Sci USA 2014;9:E5292–E5301.
Pennings RJ, Huygen PL, van den Ouweland JM et al. Sex-related hearing impairment in Wolfram syndrome patients identified by inactivating WFS1 mutations. Audiol Neurootol 2004;9:51–62.
Bischoff AN, Reiersen AM, Buttlaire A et al. Selective cognitive and psychiatric manifestations in Wolfram Syndrome. Orphanet J Rare Dis 2015;10:66–69.
Fukuma M, Ariyasu D, Hatano M et al. Early-onset urological disorders due to Wolfram syndrome: A case of neonatal onset. Clin Pediatr Endocrinol 2016;25:67–69.
Tranebjaerg L . WFS1-Related Disorders. GeneReviews 2014. http://www.gentests.org.
Kinsley BT, Swift M, Dumont RH et al. Morbidity and mortality in the Wolfram syndrome. Diabetes Care 1995;18:1566–1570.
Aloi C, Salina A, Pasquali L et al. Wolfram syndrome: new mutations, different phenotype. PLoS ONE 2012;7:e29150.
Cryns K, Sivakumaran TA, Van den Ouweland JM et al. Mutational spectrum of the WFS1 gene in Wolfram syndrome, nonsyndromic hearing impairment, diabetes mellitus, and psychiatric disease. Hum Mutat 2003;22:275–287.
Lesperance MM, Hall JW 3rd, San Agustin TB et al. Mutations in the Wolfram syndrome type 1 gene (WFS1) define a clinical entity of dominant low-frequency sensorineural hearing loss. Arch Otolaryngol Head Neck Surg 2003;129:411–420.
Sun Y, Cheng J, Lu Y et al. Identification of two novel missense WFS1 mutations, H696Y and R703H, in patients with non-syndromic low-frequency sensorineural hearing loss. J Genet Genomics 2011;38:71–76.
Rigoli L, Di Bella C . Wolfram syndrome 1 and Wolfram syndrome 2. Curr Opin Pediatr 2012;24:512–517.
Valéro R, Bannwarth S, Roman S et al. Autosomal dominant transmission of diabetes and congenital hearing impairment secondary to a missense mutation in the WFS1 gene. Diabet Med 2008;25:657–661.
Eiberg H, Hansen L, Kjer B et al. Autosomal dominant optic atrophy associated with hearing impairment and impaired glucose regulation caused by a missense mutation in the WFS1 gene. J Med Genet 2006;43:435–440.
Calamini B, Morimoto RI . Protein homeostasis as a therapeutic target for diseases of protein conformation. Curr Top Med Chem 2012;12:2623–2640.
Zatyka M, Da Silva Xavier G, Bellomo EA et al. Sarco(endo)plasmic reticulum ATPase is a molecular partner of Wolfram syndrome 1 protein, which negatively regulates its expression. Hum Mol Genet. 2015;24:814–827.
Urano F . Wolfram syndrome iPS cells: the first human cell model of endoplasmic reticulum disease. Diabetes 2014;63:844–846.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Additional information
Publisher’s note:
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Rigoli, L., Bramanti, P., Di Bella, C. et al. Genetic and clinical aspects of Wolfram syndrome 1, a severe neurodegenerative disease. Pediatr Res 83, 921–929 (2018). https://doi.org/10.1038/pr.2018.17
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/pr.2018.17
This article is cited by
-
ISR inhibition reverses pancreatic β-cell failure in Wolfram syndrome models
Cell Death & Differentiation (2024)
-
A p.Val412Serfs pathogenic variant associated with Wolfram-like syndrome and leukodystrophy
The Egyptian Journal of Neurology, Psychiatry and Neurosurgery (2023)
-
Next generation sequencing identifies a pathogenic mutation of WFS1 gene in a Moroccan family with Wolfram syndrome: a case report
Journal of Medical Case Reports (2023)
-
WFS1 autosomal dominant variants linked with hearing loss: update on structural analysis and cochlear implant outcome
BMC Medical Genomics (2023)
-
Wfs1E864K knock-in mice illuminate the fundamental role of Wfs1 in endocochlear potential production
Cell Death & Disease (2023)
