
Abstract
The pattern of X-chromosome inactivation (XCI) can affect the clinical severity of X-linked disorders in females. XCI pattern analysis has been conducted mainly by HUMARA assay, a polymerase chain reaction-based assay using a methylation-sensitive restriction enzyme. However, this assay examines the XCI ratio of the androgen receptor gene at the genomic DNA level and does not reflect the ratio of either targeted gene directly or at the mRNA level. Here, we report four females with Dent disease, and we clarified the correlation between XCI and female cases of Dent disease using not only HUMARA assay but also a novel analytical method by RNA sequencing. We constructed genetic analysis for 4 female cases showing high level of urinary low-molecular-weight proteinuria and their parents. Their XCI pattern was analyzed by both HUMARA assay and an ultra-deep targeted RNA sequencing of the CLCN5 gene using genomic DNA and mRNA extracted from both leukocytes and urine sediment. All four cases possessed pathogenic variants of the CLCN5 gene. XCI analysis revealed skewed XCI in only two cases, while the other two showed random XCI. All assay results of HUMARA and targeted RNA sequencing in both leukocytes and urinary sediment were clearly identical in all four cases. We developed a novel XCI analytical assay of ultra-deep targeted RNA sequencing and revealed that skewed XCI explains the mechanism of onset of female Dent disease in only half of such cases.
Similar content being viewed by others

Introduction
Dent disease is a rare, X-linked, recessive, renal proximal tubulopathy. It is divided into Dent-1 caused by CLCN5 variants (MIM #300009), which involves 60% of all cases, and Dent-2 caused by OCRL variants (MIM #300555), which involves 15% of all cases [1]. All male patients with hemizygous pathogenic variants in either CLCN5 or OCRL show very high levels of low-molecular-weight protein (LMWP) excretion into urine, and some of them also show hypercalciuria (with or without nephrocalcinosis) or progressive kidney disease. Female carriers usually show normal to very mild elevation of urinary LMWP. Specifically, it has been reported that 60% of female carriers show such elevation, with a concentration of urinary LMWP of about 1500 μg/ml, which is 50 times lower than that in male patients [2]. However, a very small number of female cases showing severe phenotypes such as pronounced urine LMWP or chronic kidney disease (CKD) have been reported [3].
X-chromosome inactivation (XCI) is a transcriptional silencing mechanism of either the paternal X chromosome or the maternal one [4]. Which copy of the X chromosome that is inactivated is determined at random, and the proportion of XCI in each allele varies between individuals. However, some females show preferential inactivation of one of the two X chromosomes (called skewed XCI). This skewed XCI affects the onset or severity of diseases among X-linked disease carrier females [4], but no studies have reported results about the correlation between the pattern of XCI and the onset of Dent disease in females.
XCI pattern analysis has generally been conducted by human androgen receptor (HUMARA) assay, a polymerase chain reaction-based X-chromosome inactivation assay using a methylation-sensitive restriction enzyme [5]. However, this assay has some limitations. First, it is an indirect method based on measuring the ratio of methylated alleles of human androgen receptor genes, and it does not reflect the methylation of the pathogenic gene such as CLCN5 gene in Dent disease. Therefore, segregation analysis using familial samples is needed to know which allele has a pathogenic variant. Second, the HUMARA assay involves genomic analysis of DNA methylation, so it does not evaluate the expression of mRNA, which directly reflects the protein expression levels, that is, the clinical severity. In this context, in some reports it has been asserted that the ratio of DNA methylation between alleles did not reflect the ratio of RNA expression of each allele, so it did not reflect the clinical severity [6]. As a potential option to overcome this issue, a new method that uses a high-throughput RNA expression assay using whole RNA sequencing by a next-generation sequencing (NGS) approach has recently been reported [7]. Although this method can overcome the limitations of the HUMARA assay, the complexity of the procedure of whole RNA sequencing and analysis is a major shortcoming.
Here, we report four female patients with pronounced urine LMWP excretion whose heterozygous CLCN5 variants were detected by targeted sequencing. We aimed to clarify the correlation between skewed XCI and the onset of Dent disease. The XCI pattern was analyzed using both HUMARA assay and ultra-deep targeted RNA sequencing with genomic DNA and mRNA extracted from both peripheral leukocytes and urine sediment. Urine sediment typically detaches from the kidney glomerulus or tubules and enters the urine. We targeted CLCN5 transcripts directly by ultra-deep RNA sequencing, which is a novel method for examining XCI patterns in female cases of X-linked diseases.
Materials and methods
Ethics and data collection
All procedures were reviewed and approved by the Institutional Review Board of Kobe University School of Medicine. Informed consent was obtained from all patients or their parents.
Genomic DNA and total RNA isolation from leukocytes and urine sediment
Genomic DNA was extracted from peripheral blood leukocytes and urine sediment of patients using the Quick Gene Mini 80 system (Wako Pure Chemical Industries, Ltd., Tokyo, Japan) and Quick-DNA Urine kit (Zymo Research Corporation, Irvine, CA, USA). A RiboPure Blood Kit (Invitrogen, Carlsbad, CA, USA) and RNA stabilization agent (RNAlater; Invitrogen) were used for total RNA extraction from blood leukocytes, and ZR Urine RNA Isolation Kit (Zymo Research Corporation) was used for urine sediment. The obtained genomic DNA was used for targeted sequencing, Sanger sequencing, and HUMARA assay. Transcript analysis and ultra-deep targeted RNA sequencing were performed using the total RNA.
Targeted genomic DNA sequencing
Because our patients were female and they showed non specific manifestations to Dent disease or Lowe syndrome except for extreme elevation of urinary LMWP, it was difficult to guess the pathogenic genes. Therefore, comprehensive analysis was conducted on genes (Supplemental Table 1) associated with Dent disease, Lowe syndrome, and other renal tubular diseases by NGS using Miseq (Illumina, San Diego, CA, USA) for all patients. The sample library for NGS analysis was prepared using HaloPlex Target Enrichment kit 500 kb for Illumina (Agilent Technologies, Santa Clara, CA, USA), in accordance with the manufacturer’s workflow. Briefly, 225 ng of genomic DNA was used for a restricted reaction and hybridized at 54 °C for 16 h with NGS probes. All indexed DNA samples were amplified by polymerase chain reaction and sequenced using the Miseq platform. The results were analyzed using SureCall 3.0 (Agilent Technologies).
Sanger sequencing and transcript analysis
The results of mutational analyses of CLCN5 obtained by targeted sequencing were confirmed using Sanger sequencing. Mutational sites of CLCN5 in each patient were amplified by PCR. PCR-amplified products were purified and subjected to direct sequencing using a dye terminator cycle sequencing kit (Amersham Biosciences, Piscataway, NJ, USA) and an automatic DNA sequencer (ABI Prism 3130; Perkin-Elmer, Applied Biosystems, Foster City, CA, USA). For variant descriptions, NM_000084 was used as a reference sequence. Total RNA was reverse-transcribed to cDNA using Ecodry Premix (Double Primed; Clontech Laboratories Inc., Mountain View, CA, USA). cDNA was amplified by RT-PCR using the following pair of CLCN5 primers to detect abnormal splicing in Case 1:
Forward primer: 5′-CCTGGTGTAGGGACCTATG-3′
Reverse primer: 5′-TGTGAGTGATGCTTTTTCCG-3′.
After 40 cycles of amplification, PCR products were separated on 1.5% agarose gel and the purified products were sequenced using a dye terminator cycle sequencing kit and an automatic DNA sequencer as mentioned above.
HUMARA method
The HUMARA assay was performed as described by Allen et al. [5]. Briefly, 200 ng of genomic DNA from blood leukocytes and urine sediment was digested by a methylation-sensitive enzyme (Hpa2) and the same volume of undigested DNA was amplified with FAM-labeled forward primer and reverse primer specific for regions either side of polymorphic CAG repeats (Supplemental Table 2). The PCR product was mixed with internal size standard (GeneScan 500 LIZ dye Size Standard; Perkin Elmer, Applied Biosystems) in deionized formamide and subjected to automatic DNA sequencing (ABI Prism 3100; Perkin Elmer, Applied Biosystems). Quantification of data and their visualization in graphs were performed using GeneScan software. The XCI pattern was defined as random (from 50:50 to 80:20), skewed (from 80:20 to 90:10), or extremely skewed (more than 90:10).
Ultra-deep targeted RNA sequencing using NGS
For RNA expression analysis of alleles of CLCN5 variants, reverse transcription of total RNA to cDNA was performed using Ecodry Premix (Double Primed; Clontech Laboratories Inc.). cDNA synthesis was amplified by PCR using the following pair of Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) specific primer to check the genome DNA contamination. The primer pair was designed to amplify both genome DNA and cDNA to detect genome DNA contamination in cDNA: Forward primer: 5′-CCCTTCATTGACCTCAAC-3′, reverse primer: 5′-TTCACACCCATGACGAAC-3′. The PCR products were subjected to Agilent 2100 bioanalyzer (Agilent Technologies). After that, 40 cycles of RT-PCR for cDNA were performed using forward and reverse primers designed to target the variants (Supplemental Table 3). After 150-bp PCR products were purified on 1.5% agarose gel, the ends of these cDNA fragments were repaired and adenyl nucleotides were added using TruSeq Nano DNA Library Preparation kit (Illumina), following the TruSeq workflow. Templates were ligated to adapters and these libraries were amplified by PCR. After the quantification of template libraries had been performed using Agilent High Sensitivity DNA kit (Agilent Technologies), these samples were sequenced using the Miseq platform. SureCall 3.0 software was used for reading the data. We analyzed the ratio of alleles with and without a variant for estimation of the XCI ratio.
Results
Patient history
In all four cases, urinary proteinuria was found by a urinary screening test at one or three years old and pronounced urinary LMWP was detected by detailed examination. None of these cases had subjective symptoms or hypercalciuria, calcinosis in the kidney, CKD, or other extrarenal symptoms. Pronounced urinary LMWP had been identified in the fathers of Cases 1, 3, and 4. The mother in Case 2 had never shown proteinuria and did not have high urinary LMWP. Renal biopsy was performed in Cases 1 and 3, with minor glomerular abnormality being found in both of them (Table 1).
Detection of pathogenic variants of CLCN5
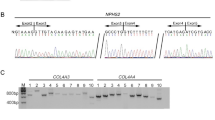
Targeted sequencing by NGS and Sanger sequencing revealed that all four cases had heterozygous variants of CLCN5 (Fig. 1, Table 1), but no other pathogenic variants. Cases 1 and 4 possessed a novel splicing site variant (c.337 + 5 G > C) and a novel missense variant (c.800 A > T, p.Glu267Val), respectively. Case 2 and her mother, and Case 3 had a known missense variant (c.608 C > T, p.Ser203Leu) and a known nonsense variant (c.2152 C > T, p.Arg718*), respectively. The variant in Case 4 was estimated to be pathogenic by in silico analysis using three different software programs for pathogenicity prediction (SIFT, PolyPhen-2, Mutation Taster). The splicing site variant c.337 + 5 G > C resulted in a 23-bp insertion at the transcript level and was revealed to be pathogenic (Fig. 1b,c).

Sanger sequencing results of CLCN5. a A novel splicing variant (c.337 + 5 G > C), a known missense variant (c.608 C > T, p.Ser203Leu), a known nonsense variant (c.2152 C > T, p.Arg718*), and a novel missense variant (c.800 A > T, p.Glu267Val) were detected in Cases 1–4, respectively. b The chromatogram of the transcript in Case 1. A 23-bp insertion was found between exon 2 and exon 3 in Case 1. c A schema of the splicing pattern found in Case 1. The splice site variant (c.337 + 5 G > C) disrupted the original acceptor site (gt in blue) and resulted in the generation of an aberrant acceptor site (gt in red) that led to a 23-bp insertion
X-chromosome inactivation analysis
In all cases and the mother of Case 2, HUMARA assay was performed using genomic DNA derived from blood leukocytes and urine sediment. This assay revealed that Case 1 had an extremely skewed XCI pattern, with a ratio of 100:0 (i.e., the rate of activation of the X chromosome including the pathogenic variant to that of the other chromosome) in both leukocytes and urine sediment. Case 3 also showed skewed XCI patterns of 87:13 in leukocytes and 94:6 in urine sediment. Skewed XCI patterns were not observed in Case 2 and her mother, or in Case 4, in either leukocytes or urine sediment (Fig. 2 and Table 2).

XCI analysis by HUMARA assay in peripheral leukocytes and urine sediment. The peaks colored black indicate the alleles with CLCN5 variants and the other peaks indicate the alleles without such variants. Section (a) corresponds to PCR products of undigested DNA in peripheral leukocytes in patients and their parents. Section (b) shows the PCR products in leukocytes and urine sediment after digestion with a methylation-sensitive enzyme (Hpa2)
We conducted targeted RNA sequencing for all cases and the mother of Case 2 (Supplemental Fig. 1 and Table 2). The depth of sequencing in the patients ranged from 0.3 to 2.3 million in the NGS analysis, representing ultra-deep sequencing. RNA sequencing did not recognize any genome DNA sequences of CLCN5 in all cases (Data not shown). In addition, PCR amplification of GAPDH showed that there was no contamination of genomic DNA in cDNA in each case (Supplemental Fig. 2). All assay results were completely identical, indicating the reliability of all methods (Table 2).
Discussion
We report four females with Dent disease possessing heterozygous CLCN5 pathogenic variants. All cases showed severe urinary LMWP excretion. For these four cases and an asymptomatic one (the mother of Case 2), we performed XCI analysis using HUMARA assay and ultra-deep targeted RNA sequencing to clarify the correlation between skewed XCI and the onset of Dent disease in females. To the best of our knowledge, this is the first report describing the XCI patterns in female cases of Dent disease. We applied DNA methylation and RNA sequencing analysis to both leukocytes and urine sediment extracted from the subjects.
Correlations between XCI ratio and clinical manifestations have been reported in many X-linked diseases, such as Fabry disease [8] and Duchenne/Becker muscular dystrophy [9, 10]. For Dent disease, the performance of an XCI assay has been reported in only a single case [11]. In this case report, the mother of a boy with severe Dent disease was described as being asymptomatic and XCI using HUMARA assay for genomic DNA extracted from leukocytes showed 100% inactivation of the allele with the pathogenic variant, which led to her being asymptomatic. It is believed that the XCI pattern differs among tissues, so genomic DNA extracted from renal tissue may be needed for XCI analysis of kidney diseases [11]. In addition, HUMARA assay shows the methylation of the androgen receptor gene at the genomic DNA level [5]. This means that, when using the HUMARA assay, neither the targeted gene nor the mRNA expression that affects phenotypes is evaluated directly. In addition, this assay needs segregation analysis at the same time and in cases with de novo variants or homozygous CAG repeat numbers, we could not determine the XCI proportion. That is also the limitation of the HUMARA assay. In view of these limitations, in the current study, we used genomic DNA and mRNA derived from leukocytes and urinary sediment originating from the kidney [3, 12], and also conducted not only HUMARA assays but also RNA expression analyses.
The results of our study show that two of the four cases with severe urinary LMWP had skewed XCI. The incidence of skewed XCI in healthy young females was reported to be 7–34% [13,14,15], so this finding (of an association in 50% of cases) may support the association between skewed XCI and female Dent disease. However, a skewed XCI pattern was not found in the other two cases. In Case 2, although this individual and her mother had the same genotype and XCI pattern, they showed significantly different levels of urinary LMWP. This indicated that there are other mechanisms besides skewed XCI that contribute to the onset of Dent disease in females. One possible mechanism enhancing female phenotypes is digenic mutations in modifier genes associated with tubular functions. Although we did not find any pathogenic variants in genes associated with renal tubular diseases including OCRL (Supplemental Table 1), some of the variants with high allele frequency in healthy controls can affect the severity of the Dent disease. Furthermore, the variants in other known genes such as INPP5B or unknown genes can also affect the phenotypes [16].
The data obtained in this study indicate that the XCI patterns in blood leukocytes and renal tissue were almost the same. Some previous reports have described XCI patterns in different tissues and also indicated significant similarities among different tissues including blood and kidney [13, 17, 18]. It was also reported that the differences of XCI patterns between tissues tend to be smaller at younger ages than at older ones [13]. However, some individuals showed extreme variation of XCI patterns between tissues [13]. In our study, the patterns between leukocytes and urine sediment were almost the same in all five studied individuals.
We have developed a novel XCI analytical method of ultra-deep targeted RNA sequencing. With this assay, we measured the ratio of activation of variant and normal alleles directly for the CLCN5 gene at the transcript level. Physiologically, this method much more strongly reflects the influence of XCI on the clinical manifestations. However, our findings revealed that the results obtained by the HUMARA assay and the ultra-deep RNA sequencing assay were identical in all five individuals. Although the RNA expression assay overcomes the limitations of the HUMARA assay as described above, we revealed that both assays are actually reliable.
The current study has some limitations. First, the number of cases examined here was very small because female Dent disease cases are very rare. Second, we applied nested PCR for the RNA sequencing of cDNA extracted from urinary sediments because the total amount of mRNA was low. However, the bias caused by this seemed to be small because the results from our method were concordant with those obtained using leukocytes and the HUMARA assay.
In conclusion, we developed a novel XCI analytical assay of targeted ultra-deep RNA sequencing and revealed that skewed XCI explains the mechanism of onset of female Dent disease in only two out of the four studied cases. We also revealed that the assay results from using genomic DNA and mRNA, and leukocytes and urinary sediment matched perfectly. Both genomic DNA and mRNA assays and leukocytes and urinary sediment were shown to be useful tools and materials for determining the patterns of XCI in females.
References
Blanchard A, Curis E, Guyon-Roger T, Kahila D, Treard C, Baudouin V, et al. Observations of a large Dent disease cohort. Kidney Int. 2016;90:430–9.
Mansour-Hendili L, Blanchard A, Le Pottier N, Roncelin I, Lourdel S, Treard C, et al. Mutation Update of the CLCN5 Gene Responsible for Dent Disease 1. Hum Mutat. 2015;36:743–52.
Igarashi T, Inatomi J, Ohara T, Kuwahara T, Shimadzu M, Thakker RV. Clinical and genetic studies of CLCN5 mutations in Japanese families with Dent’s disease. Kidney Int. 2000;58:520–7.
Orstavik KH. X chromosome inactivation in clinical practice. Hum Genet. 2009;126:363–73.
Allen RCutler, Zoghbi HudaY, Moseley AnnemarieB, R. HM, Belmont JohnW. Methylation of Hpall and Hhal Sites Near the Polymorphic CAG Repeat in the Human Androgen-Receptor Gene Correlates with X Chromosome Inactivation. Am J Hum Genet. 1992;51:1229–39.
Sabina I, Swierczek LP, Jelinek Jaroslav, Agarwal Neeraj, Hammoud Sue, Wilson Andrew, Hickman Kimberly, Parker CharlesJ, Cairns BradleyR, Prchal JosefT. Methylation of AR locus does not always reflect X chromosome inactivation state. Blood. 2011;119:e100–9.
Szelinger S, Malenica I, Corneveaux JJ, Siniard AL, Kurdoglu AA, Ramsey KM, et al. Characterization of X chromosome inactivation using integrated analysis of whole-exome and mRNA sequencing. PLoS ONE. 2014;9:e113036.
Echevarria L, Benistan K, Toussaint A, Dubourg O, Hagege AA, Eladari D, et al. X-chromosome inactivation in female patients with Fabry disease. Clin Genet. 2016;89:44–54.
Viggiano E, Ergoli M, Picillo E, Politano L. Determining the role of skewed X-chromosome inactivation in developing muscle symptoms in carriers of Duchenne muscular dystrophy. Hum Genet. 2016;135:685–98.
Viggiano E, Picillo E, Ergoli M, Cirillo A, Del Gaudio S, Politano L Skewed X-chromosome inactivation plays a crucial role in the onset of symptoms in carriers of Becker muscular dystrophy. J Gene Med. 2017;19.
Addis M, Meloni C, Tosetto E, Ceol M, Cristofaro R, Melis MA, et al. An atypical Dent’s disease phenotype caused by co-inheritance of mutations at CLCN5 and OCRL genes. Eur J Hum Genet. 2013;21:687–90.
Kaito H, Nozu K, Fu XJ, Kamioka I, Fujita T, Kanda K, et al. Detection of a transcript abnormality in mRNA of the SLC12A3 gene extracted from urinary sediment cells of a patient with Gitelman’s syndrome. Pediatr Res. 2007;61:502–5.
Sharp A, Robinson D, Jacobs P. Age- and tissue-specific variation of X chromosome inactivation ratios in normal women. Human Genet. 2000;107:343–9.
Hatakeyama C, Anderson CL, Beever CL, Penaherrera MS, Brown CJ, Robinson WP. The dynamics of X-inactivation skewing as women age. Clin Genet. 2004;66:327–32.
Bolduc V, Chagnon P, Provost S, Dube MP, Belisle C, Gingras M, et al. No evidence that skewing of X chromosome inactivation patterns is transmitted to offspring in humans. J Clin Invest. 2008;118:333–41.
Duran D, Jin SC, DeSpenza T Jr., Nelson-Williams C, Cogal AG, Abrash EW, et al. Digenic mutations of human OCRL paralogs in Dent’s disease type 2 associated with Chiari I malformation. Hum Genome Var. 2016;3:16042.
de Hoon B, Monkhorst K, Riegman P, Laven JS, Gribnau J. Buccal swab as a reliable predictor for X inactivation ratio in inaccessible tissues. J Med Genet. 2015;52:784–90.
Bittel DC, Theodoro MF, Kibiryeva N, Fischer W, Talebizadeh Z, Butler MG. Comparison of X-chromosome inactivation patterns in multiple tissues from human females. J Med Genet. 2008;45:309–13.
Acknowledgements
The authors gratefully acknowledge the cooperation of the attending patients and physicians in this study. The authors also thank Edanz Group (www.edanzediting.com/ac) for editing a draft of this manuscript.
Funding
All phases of this study were supported by a grant from the Ministry of Health, Labour and Welfare (Japan) for Research on Rare Intractable Diseases in Kidney and Urinary Tract [H24-nanchitou (nan)-ippan-041 to Kazumoto Iijima] in the Research on Measures for Intractable Diseases Project and a Grant-in-Aid from the Ministry of Culture, Sports, Science and Technology [KAKENHI] (Subject ID: 15K09691 to Kandai Nozu, 26293203 to Kazumoto Iijima and 17K16087 to Shogo Minamikawa).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Minamikawa, S., Nozu, K., Nozu, Y. et al. Development of ultra-deep targeted RNA sequencing for analyzing X-chromosome inactivation in female Dent disease. J Hum Genet 63, 589–595 (2018). https://doi.org/10.1038/s10038-018-0415-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s10038-018-0415-1
This article is cited by
-
Structural basis of pH-dependent activation in a CLC transporter
Nature Structural & Molecular Biology (2024)
-
A novel quantitative targeted analysis of X-chromosome inactivation (XCI) using nanopore sequencing
Scientific Reports (2023)
-
Prenatal diagnosis and genetic counseling of an inherited Xq24q25 deletion associated with normal phenotype
Molecular Cytogenetics (2022)
-
X-chromosome inactivation patterns in females with Fabry disease examined by both ultra-deep RNA sequencing and methylation-dependent assay
Clinical and Experimental Nephrology (2021)
-
Case report: a Chinese girl with dent disease 1 and turner syndrome due to a hemizygous CLCN5 gene mutation and Isochromosome (Xq)
BMC Nephrology (2020)
