
Abstract
Over the past years, several studies have demonstrated that defects in DNA damage response and repair (DDR) genes are present in a significant proportion of patients with prostate cancer. These alterations, particularly mutations in BRCA2, are known to be associated with an increased risk of developing prostate cancer and more aggressive forms of the disease. There is growing evidence that certain DDR gene aberrations confer sensitivity to poly-(ADP ribose) polymerase inhibitors and/or platinum chemotherapy, while other defects might identify cases that are more likely to benefit from immune checkpoint inhibition. The potential prognostic impact and relevance for treatment selection together with the decreasing costs and broader accessibility to next-generation sequencing have already resulted in the increased frequency of genetic profiling of prostate tumours. Remarkably, almost half of all DDR genetic defects can occur in the germline, and prostate cancer patients identified as mutation carriers, as well as their families, will require appropriate genetic counselling. In this review, we summarise the current knowledge regarding the biology and clinical implications of DDR defects in prostate cancer, and outline how this evidence is prompting a change in the treatment landscape of the disease.
Similar content being viewed by others
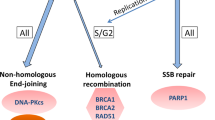

Background
The therapeutic landscape of metastatic castration-resistant prostate cancer (mCRPC) has rapidly evolved over the past 10 years as several agents have been shown to improve the overall survival (OS) of these patients.1,2,3,4,5,6,7 However, as no biomarker has yet been identified for treatment selection, prostate cancer has so far remained a disease treated with a “one-size-fits-all” approach.
The initiation of prostate cancer and the progression of the disease are driven by androgen receptor (AR) signalling, and androgen deprivation therapy (ADT) constitutes the backbone of systemic therapy for patients with advanced disease. However, insights into the biology of prostate cancer have shown that up to 60% of patients with advanced disease have clinically actionable molecular alterations in non-AR-related pathways.8 In particular, mutations in the genes encoding components of the DNA damage response (DDR; Box 1), such as BRCA1 and BRCA2, are common in prostate cancer8,9,10,11,12,13 Such mutations reduce the ability to effectively repair single- and double-strand DNA damage, and thus compromise genomic integrity. This knowledge has resulted in a growing interest in biomarker-driven clinical trials. For example, patients harbouring BRCA1/2 mutations have been shown to be more vulnerable to the action of poly-(ADP ribose) polymerase (PARP) inhibitors (PARPi), agents that prevent cells from repairing DNA damage. There are still several caveats regarding which DDR alterations may sensitise cells to PARP inhibition and when in the treatment sequence is the right time to use PARPi. However, with the recent approval of two of these compounds (olaparib and rucaparib) to treat advanced disease stages, prostate cancer has finally met precision oncology.
Here, we review the prevalence of DDR defects and the clinical implications of these alterations in both localised and advanced prostate cancer, before discussing data from the numerous clinical trials addressing the potential benefit of drugs targeting the DDR pathway in prostate cancer and the challenges physicians may face to incorporate PARPi to daily clinical practice.

a DNA damage response is coordinated by different proteins which functions may be categorised in a simplified way as DNA damage sensors, transducers and effectors. DNA damage can be detected by γ-H2AX and MRN complex (sensors), which activate ATM and ATR. ATM and ATR are key signal transducers of downstream DDR pathways. They activate cell-cycle regulators CHK1 and CHK2 (encoded by CHEK1 and CHEK2, respectively), which in turn signal downstream checkpoints that finally induce the activation of p53. This tumour suppressor determines if the cell initiates cell cycle, DNA repair mechanisms or undergoes apoptosis, depending on the DNA damage and/or DNA repair efficacy. b DNA-damaging agents may cause a range of different DNA lesions, which are repaired by a specific mechanism. ATR, ATM and CHK1/2 are kinases involved in the response to several types of DNA damage. Therefore, the inhibition of these kinases may be effective in certain tumours with DDR alterations or increase the activity or other agents causing DNA damage (i.e. platin salts). Similarly, DNA-PKc is a kinase involved in the NHEJ pathway, which inhibition by targeted drugs is also being tested in multiple tumours including prostate cancer. Platin salts (i.e. cisplatin or carboplatin) induce intra- and interstrand DNA crosslinks. These alterations require the coordinated action of HR, NHEJ and NER pathways to be repaired. When the function of genes involved in DNA repair pathway are altered (either in the somatic or in the germline), the cytotoxic effect of platinum chemotherapy could not be repaired, resulting in apoptotic cell death. PARP inhibitors interact with PARP, inhibiting its function. This result in the accumulation of single-strand breaks, which in turn leads to double-strand breaks (DSBs) formation. “Normal” cells have the ability to repair DSBs; however, these cannot be repaired in the presence of alterations in the function of genes involved in HR pathway (BRCA1/2, PALB2, FA family) (synthetic lethality). MMR deficiency leads to the accumulation of somatic mutations, and hence, more potential neoantigens. Higher somatic mutations and neoantigens have been correlated with better responses to immunotherapy.
Prevalence of alterations in DDR genes in prostate cancer
The high prevalence of genomic alterations that involve DDR genes in prostate cancer has been recognised over the past 5 years.8,9,10,11,12,13,14 In 2015, The Cancer Genome Atlas (TCGA) published the molecular analysis of 333 primary prostate tumours, revealing that 19% of them harboured alterations in different DDR genes, including BRCA2, BRCA1, ATM, CDK12, FANCD2 or RAD51C.12 Even though BRCA2 was reported as the most commonly altered gene, all six cases with germline BRCA2 mutations presented with the same variant, p.K3326* (the pathogenic significance of which is unclear). At the same time, a report of the International Stand Up to Cancer/American Association for Cancer Research Prostate Cancer/Prostate Cancer Foundation Team (SU2C-PCF) identified genomic alterations affecting DDR genes in 23% of the 150 metastatic biopsy samples analysed. BRCA2 was altered in 13% of samples, followed by ATM (7.3%), MSH2 (2%) and BRCA1, FANCA, MLH1, RAD51B and RAD51C (all with a prevalence of 0.3%).8 A larger series that included samples from the aforementioned studies identified DDR defects in 10% and 27% of the primary and metastatic samples, respectively,14 in line with the observation that DDR alterations are associated with high histology grade and metastatic prostate cancer.15,16,17,18 Besides, the prevalence of DDR alterations across different studies may vary depending on the number of genes analysed, the technique used and the clinico-pathological features of the tumours included. The larger series of prostate cancer samples screened for DDR defects has been provided by the PROfound study, a Phase 3 study addressing the benefit of the PARPi olaparib in mCRPC, that successfully screened 2792 biopsies for aberrations in 15 DDR genes involved in the homologous recombination repair (HRR) pathway.19 Such alterations were identified in 28% of the samples analysed,20 with a similar frequency in primary tumours (27%) and in biopsy samples from metastatic sites (32%), suggesting that HRR alterations are probably early events in the evolution of aggressive prostate tumours. BRCA2 (8.7%), CDK12 (6.3%), ATM (5.9%), CHEK2 (1.2%) and BRCA1 (1%) were the most commonly altered genes. Co-occurring aberrations in two or more DDR genes were found in 2.2% of cases, although it is unclear whether this is associated to increased sensitivity to PARPi. The prevalence of mismatch repair (MMR) defects in prostate cancer was established in 2018 in a large series with 1033 patients. In this study, Abida et al.,21 identified a MMR deficiency in 3.1% of cases, and confirmed MSH2 as the most commonly altered MMR gene in prostate cancer (Table 1).
A remarkable finding from the SU2C-PCF study was that 8% of the mCRPC patients harboured a germline DDR mutation—almost half of all the reported DDR aberrations.8 This study was the first to suggest that germline variants in DDR genes associated with an increased risk of cancer were present in metastatic prostate cancer at a prevalence higher than previously acknowledged. A first confirmation of these findings was provided by a retrospective multicentre study that pooled data on germline DNA variants from 692 patients with metastatic prostate cancer from different series (including the aforementioned SU2C-PCF study). In this analysis, 11.8% of the patients were identified as carriers of a deleterious germline mutation in at least one of the 20 DDR genes associated with cancer-predisposition syndromes that were analysed.9 A slightly lower prevalence (7.4%) of mutations in the same genes was reported in PROREPAIR-B, a prospective study13 that screened 419 unselected patients with mCRPC from Spain. This variation in prevalence is likely to be due to the different genetic background of both populations as the series reported by Pritchard et al.9 included a higher prevalence of the Ashkenazi founder mutations BRCA1 c.5266dupC and BRCA2 c.5946delT and the Eastern European founder mutation CHEK2 p.1100del than the prospective Spanish study. Nonetheless, BRCA2 was the most commonly mutated gene in both series (5.3%9 and 3.3%,13 respectively).
It is noteworthy that most of the studies analysing the presence of DDR defects in tumours do not distinguish the germline or somatic origin of the variants identified. A 2019 analysis of over 17,000 tumours, including 1042 prostate cancers, identified BRCA2 aberrations in 92 cases. Unlike for the other cancer types included, the proportion of prostate tumours with germline and somatic BRCA2 mutations was similar and approximately half of the BRCA2 alterations identified through tumour profiling are already present in the germline. Furthermore, the authors describe prostate tumours as the only BRCA2-mutated prostate cancers with a similar rate of biallelic loss (70%) in cases with germline and somatic alterations.22 A lower ratio of germline:somatic alterations have been reported for components of the MMR pathway in prostate cancer, as less than a quarter (22%) of the tumours identified by Abida et al.21 as MMR deficient harboured a germline mutation. This is in line with previous reports that found germline mutations significantly less prevalent in the prostate than in other cancer types with MMR defects such as the colorectal or urothelial ones.23
Clinical implications of DDR gene alterations in prostate cancer
Due to the high prevalence of germline mutations in DDR genes, the National Comprehensive Cancer Network (NCCN) has recommended germline testing for all men with high-risk localised prostate cancer and in those with metastatic disease.24 The development of PARPi to treat prostate cancer has made genomic testing appealing in this setting. Therefore, the number of prostate cancer patients with tumours classified as DDR deficient is likely to increase in the near future. However, the clinical relevance of germline and somatic defects in DDR genes remains largely unclear at present—the exception being germline BRCA2 mutations, which have been shown to be an independent prognostic factor for prostate cancer outcomes in different settings.13,15,25,26
For non-metastatic prostate cancer patients
A range of management options, including active surveillance, radical prostatectomy and/or radiotherapy with or without hormonal deprivation, is currently available to treat patients with localised prostate cancer. Data regarding the implications of germline and somatic DDR defects in early prostate cancer come from retrospective analyses, most of them focused on germline BRCA mutation carriers.
A report on the outcomes of 1211 men undergoing active surveillance, including 11 BRCA1, 11 BRCA2 and 5 ATM germline carriers, has shown that BRCA2 carriers are more likely to undergo a tumour grade re-classification in subsequent biopsies. In this series, the incidence of tumour staging upgrades at 2, 5 and 10 years was 27%, 50% and 78% in BRCA2 carriers compared with 10%, 22% and 40% in non-carriers (P = 0.001).26 These results indicate that active surveillance might not be an appropriate management for germline BRCA2 carriers with tumours classified as “low risk”.
Likewise, no conclusive data are available regarding the potential use of alterations in BRCA2 or other DDR genes for selecting between curative treatment options (radical prostatectomy or radiotherapy). The only existing evidence comes from a retrospective study that analysed the outcomes of 1302 patients, including 18 BRCA1 and 49 BRCA2 carriers, with localised disease. In this series, BRCA1/2 carriers developed metastasis significantly earlier than non-carriers after radical treatment. Metastasis-free survival rates for those surgically treated were 89% and 67% in BRCA1/2 carriers compared with 97% and 91% in non-carriers at 5 and 10 years, respectively (P = 0.024). The difference was even greater for patients treated with radiotherapy, as only 57% and 39% of BRCA1/2 carriers were free from metastasis at 5 and 10 years, respectively, compared with 91% and 80% of non-carriers (P < 0.001).25 A direct comparison of the two groups could not be performed as patients treated with radiotherapy (both carriers and non-carriers) presented with more advanced disease than those who were surgically treated. Therefore, it is not possible to conclude from this study which of the treatment options—surgery or radiotherapy—would be best for BRCA carriers.
For patients with metastatic hormone-sensitive prostate cancer and CRPC
The clinical implications of DDR defects in advanced prostate cancer remain unclear as the available evidence is somehow conflicting. To date, data on the prognosis of advanced prostate cancer patients with DDR gene aberrations and the response to therapies are limited. For metastatic hormone-sensitive prostate cancer (mHSPC), at least two studies in mCRPC patients have provided retrospective data on time-to-castration resistance (TTCR) from initiation of continuous ADT. Annala et al.27 reported that patients with germline mutations in HRR genes presented a significantly shorter TTCR than non-carriers (11.8 vs. 19.0 months, P = 0.031). Similarly, Castro et al.13 showed that germline BRCA2 mutation carriers achieve mCRPC status earlier than non-carriers (13.2 vs. 28 months, P = 0.05). The only prospective data available to date has been reported by Vandekerkhove et al.28 This group analysed the outcomes of 53 patients with de novo mHSPC, and reported a significantly shorter TTCR in the 11 cases with somatic and/or germline DDR alterations compared with those without such aberrations (7.3 months vs. not reached, P = 0.01). However, this difference did not remain significant in the multivariable analysis.28
Three retrospective studies assessed the role of germline DDR mutations in the outcomes of patients with mCRPC treated with AR signalling inhibitors (ARSi) and taxanes. In the first report, Annala et al.29 analysed 176 mCRPC patients, including 22 germline carriers (16 BRCA2). They found that carriers treated with ARSi as first-line therapy had shorter progression-free survival (PFS) than non-carriers (3.3 vs. 6.2 months, P = 0.01), despite apparent prolonged responses to ARSi in some carriers. Importantly, almost all carriers in this series had a circulating tumour DNA (ctDNA) fraction relative to total cell-free DNA of >30%, which correlates strongly with disease burden, poor responses and shorter OS.27 Therefore, it is questionable whether the poor clinical outcomes of these patients are related solely to their germline status, or potentially also affected by other confounding factors, such as tumour burden and a high ctDNA. In a second report, the clinical outcomes of 172 mCRPC patients, including 22 germline carriers (five BRCA2) who were treated with ARSi, were analysed by Antonarakis et al.30 In this study, carriers showed a trend to longer PFS than non-carriers (15 vs. 10.8 months, P = 0.090). Interestingly, those patients who received chemotherapy before ARSi had shorter PFS and cause-specific survival (CSS), suggesting that treatment sequence could be important in this setting. The third report31 analysed the outcomes of 390 mCRPC patients included in the series published previously by Pritchard et al.9 describing the prevalence of germline DDR alterations. This study included 60 germline DDR mutation carriers (37 BRCA2). No significant differences in the responses to the administered therapies or in OS were observed between carriers and non-carriers. However, it should be noted that, unlike in the other studies, up to 47% of carriers in this series had received platinum salts and/or PARPi, which could have a confounding effect on the outcomes.
PROREPAIR-B13 was the first prospective study designed to evaluate the impact of germline DDR mutations on the outcome(s) of mCRPC patients. Out of a total of 419 patients analysed, 68 were found to carry a germline mutation (14 BRCA2). Remarkably, in this cohort, in which none of the carriers received PARPi or platinum salts, CSS was halved in BRCA2 carriers compared with non-carriers (17.4 vs. 33.2 months, P = 0.027). The multivariate analyses identified germline BRCA2 mutations as an independent prognostic factor for CSS in this setting (HR [hazard ratio] 2.11; 95% CI [confidence interval] 1.06–4.18). The differences in CSS were not significant when the outcomes of patients with BRCA2 mutations were analysed together with those harbouring germline ATM or BRCA1 mutations (23.3 vs. 33.2 months, P = 0.264); neither were they significant when carriers of any germline mutation in the 107 DDR genes studied in PROREPAIR-B were compared with non-carriers (28.6 vs. 33.1 months, P = 0.646). Germline mutations in BRCA2 have clearly a deleterious impact on the prognosis of mCRPC patients, while germline mutations in other DDR genes may not affect patients’ survival. Further studies are needed to establish the clinical significance of inherited mutations in less frequently altered genes.
Exploratory analyses in PROREPAIR-B13 showed that germline BRCA2 carriers had worse CSS than non-carriers when they received first-line taxane therapy for mCRPC (10.7 vs. 28.4 months, P < 0.001). By contrast, there were no significant differences in CSS between germline BRCA2 carriers and non-carriers treated initially with ARSi for mCRPC (24.0 vs. 31.2 months, P = 0.901). CSS outcomes were similar for non-carriers treated with either a first-line ARSi or taxane. Despite further validation of these results is required, germline BRCA2 status may be relevant for tailoring the treatment sequence in mCPRC, and mHSPC.
In the past year, relevant data on the clinical implication of CDK12 and MMR alterations have been reported. CDK12 defects are present in 6% of mCRPC19 patients and are often biallelic.32 CDK12 is a kinase involved in HRR but is also key to maintain genomic stability.32 Four retrospective series have demonstrated the clinical aggressiveness of CDK12-inactivated prostate cancer tumours, which often present Gleason grade groups 4 and 5 and metastases at diagnosis. These patients had a rapid progression to castration resistance from ADT initiation and responses to abiraterone and enzalutamide were poor.16,17,18,33 Importantly, one of these studies has suggested that CDK12 alterations may confer poorer outcomes than somatic defects in BRCA1/2, ATM or TP53.16 MMR have also been associated with aggressive features and more advanced disease at diagnosis.34,35 However, conflicting results have been reported on the clinical outcomes of MMR-deficient prostate tumours.34 While a study suggests favourable responses to ADT, others have found that MMR-deficient patients develop castration-resistant disease earlier than the MMR-proficient ones.35,36 These differences in outcomes between studies may be related to the limited number of patients included and the different assays used to detect these alterations (targeted sequencing vs. immunohistochemistry). Further studies are needed to completely elucidate the clinical implications of these and other DDR alterations in prostate cancer.
Targeting DDR genes in prostate cancer
The relatively high prevalence of alterations in DDR genes in patients with advanced prostate cancer provides a unique opportunity to take advantage of these defects by using different therapeutic strategies, including synthetic lethality.
Platinum-based chemotherapy
Platinum-based chemotherapy causes DNA crosslinks that cannot be easily repaired when the HRR pathway is impaired, leading to cell death (Box 1). This strategy has proven successful in treating breast37,38 and ovarian39 cancers with pathogenic mutations in BRCA1 or BRCA2. Although platinum salts are not a standard of care option in prostate cancer patients, their use is recommended in cases of neuroendocrine differentiation.40,41 In the DDR context, several retrospective studies suggest that BRCA2-mutated prostate cancer patients might benefit from this treatment approach.42,43,44,45 The largest of these analyses included 141 men with mCRPC treated with carboplatin and docetaxel at the Dana Farber Cancer Institute between 2001 and 2015, and reported a benefit from this combination for patients with germline BRCA2 mutations.43 Six out of the eight BRCA2 carriers (75%) showed a >50% decline in the levels of prostate-specific antigen (PSA) at 12 weeks, compared with 23 of 133 of non-carriers (17%) (P = 0.001). A >50% PSA decline was associated with longer survival (18.9 months in BRCA2 carriers vs. 9.5 months in non-carriers). A second study also evaluated the efficacy of platinum-based chemotherapy after progression to taxanes in 109 mCRPC patients.45 A PSA decline ≥50% was more frequent in patients with DDR alterations compared with DDR-proficient patients (50% vs. 13%, P = 0.006). This analysis included a subset of patients with DDR aberrations who received platinum chemotherapy after progression on PARPi (n = 8) with clinical benefit observed in a third of them. None of the two patients with ATM mutations responded to a platinum, regardless of prior treatment with PARPi. Several studies are ongoing to validate these results and to establish the role of platinum-based chemotherapy for DDR-deficient prostate tumours in different scenarios (Table 2).
PARP inhibitors and synthetic lethality
Another strategy to treat DDR-deficient tumours is to exploit the synthetic lethal interaction between the inhibition of PARP and the impairment of HRR already present in some tumours. This interaction is thought to result from the PARPi-induced increase in the number of double-strand break (DSBs) or collapsed replication forks, which are lethal in HRR-deficient cells46 (Box 1). Several PARPi, differing in their ability to bind PARP and to trap PARP–DNA complexes,47 are at various stages of clinical development (Table 2). Olaparib was the first-in-class drug to be granted approval in 2014 for the treatment of BRCA-deficient ovarian cancer.48 The first-in-man clinical trial of olaparib in a population of patients with advanced solid tumours enriched for germline BRCA1 and BRCA2 mutations included three mCRPC patients, one of whom benefited from treatment with the drug for over 2 years.49,50 Small numbers of mCRPC patients with germline BRCA mutations were also enrolled in Phase 1 trials of other PARPi, such as talazoparib51 or niraparib as single agents,52 showing promising signs of antitumour activity in these patients with a good safety profile.
TOPARP-A and TOPARP-B
In the TOPARP-A Phase 2 trial,53 50 men with heavily pretreated mCRPC received olaparib 400 mg twice a day. Fourteen out of 16 patients (88%) who harboured a DDR defect (somatic or germline) achieved clinical benefit (including radiological responses, a decrease in PSA levels and/or a decrease in circulating tumour cell count) and durable responses. All seven patients with BRCA2 defects, but also some with BRCA1, ATM, PALB2 and FANCA defects, among others, responded. Subsequently, the TOPARP-B Phase 2 trial54 enrolled 98 mCRPC patients with DDR alterations. Half of them received 300 mg twice daily and the other half received 400 mg twice daily. Radiographic and PSA responses were observed in 39% and 16% of patients in the 300 mg twice daily cohort and in 54% and 24% of patients in the 400 mg twice daily cohort, respectively. Despite the increased responses and prolonged benefit in the 400 mg arm, 37% of these patients required a dose reduction to 300 mg twice daily due to toxicity. The study included 30 men with BRCA1/2 mutations, of whom 52% and 77% achieved a radiographic or PSA response, respectively, as compared with 5% and 11.3% of men with other DDR defects.
PROfound
PROfound19 is the first randomised Phase 3 biomarker-driven clinical trial in mCRPC patients. In this trial, DDR-deficient mCRPC patients who progressed on prior ARSi were randomised 2:1 to receive olaparib 300 mg twice daily or the alternative ARSi according to physician’s choice. Patients with a DDR alteration were stratified into two cohorts based on the previous evidence of activity: cohort A with BRCA1, BRCA2 and ATM alterations; and cohort B, which included alterations in BARD1, BRIP1, CDK12, CHEK1, CHEK2, FANCL, PALB2, PPP2R2A, RAD51B, RAD51C, RAD51D and RAD54L. The primary endpoint was radiographic PFS (rPFS) benefit in cohort A. Crossover to olaparib was allowed upon progression. A total of 245 and 142 patients were included in cohorts A and B, respectively (65.6% had previously received taxane). The study met its primary endpoint and showed a statistically significant benefit in rPFS in patients included in cohort A, with a median rPFS of 7.4 months in men treated with olaparib vs. 3.5 months in those who received abiraterone or enzalutamide (P < 0.001; HR 0.34, 95% CI 0.25–0.47). In an exploratory analysis that looked at the gene-by-gene effect on rPFS, patients with BRCA2 aberrations seemed to benefit the most from olaparib. The final analysis of overall survival has been already reported,55 showing a clear benefit in patients with BRCA1, BRCA2 and ATM mutations (cohort A), with a median OS of 19.1 months in the olaparib arm compared with 14.7 months with ARSi (HR 0.69, p = 0.02), despite 66% of patients in the control arm crossing over to olaparib at radiographic progression. Median OS in the overall population (cohorts A and B) was 17.3 vs 14.0 months (HR 0.79) with olaparib and hormonal treatment, respectively.
TRITON2 and GALAHAD
The preliminary results of two Phase 2 trials (TRITON2 and GALAHAD) evaluating the efficacy of other two PARPi (rucaparib and niraparib, respectively) in heavily pretreated mCRPC patients have also been reported.56,57 Both studies have enrolled patients with DDR defects, although the assays and the gene panels used to screen patients and decide their eligibility are different (Table 3). Patients with alterations in ATM, BARD1, BRCA1, BRCA2, BRIP1, CDK12, CHEK2, FANCA, NBN, PALB2, RAD51, RAD51B, RAD51C, RAD51D or RAD54L detected in tissue, or ATM, BRCA1 or BRCA2 in plasma, are eligible for the TRITON2 study, while GALAHAD is only enrolling men with biallelic tumour alterations in ATM, BRCA1, BRCA2, BRIP1, CHEK2, FANCA, HDAC2 or PALB2 detected in circulating free DNA. Forty-four per cent of men with BRCA1/2 alterations and measurable disease in the TRITON2 study achieved radiographic responses, which, in 60% of cases, lasted for >6 months. Fifty-two per cent also had a ≥50% decrease in PSA levels. In this study, no differences in response have been observed between patients with germline and somatic BRCA2 alterations. In GALAHAD, the reported overall response rate for men with biallelic BRCA1/2 alterations was 41% with a median duration of 5.5 months, and the PSA response rate was 50%. TRITON2 included patients with mono- and biallelic BRCA1/2 alterations, while only patients with biallelic defects were eligible for GALAHAD, but in view of the reported response rates, the impact of zygosity on the response to PARPi is unclear, at least for mCRPC.
These findings are consistent with a report by Jonsson et al.,22 which showed that the clinical benefit of PARPi in BRCA-associated cancer types, including those with increased heritable cancer risk, such as prostate, breast, ovarian and pancreatic cancer, was similar regardless of somatic, germline and mono- or biallelic inactivation.
DDR alterations confer different sensitivity to PARP inhibition
The benefit of PARPi in BRCA2 carriers are well established across all the above-mentioned studies; however, it is unclear whether other DDR alterations beyond BRCA2 may predict response to PARPi. In the TRITON study, only 15% and 10% of patients with non-BRCA DDR defects achieved radiographic and PSA responses, respectively.58 Similar response rates have been observed in GALAHAD.57 Despite early preclinical data suggesting that patients with ATM alterations would respond to PARPi,47 clinical evidence has proven otherwise. First, in a retrospective analysis of 23 mCRPC patients treated with olaparib, none of those with ATM (n = 6) achieved PSA responses compared with 76% of patients with BRCA1/2 alterations (n = 17). Disease progression occurred significantly earlier on ATM-mutated patients.59 These findings have later been confirmed in clinical trials as ATM-deficient tumours have consistently shown limited response to different PARPi.19,54,57,58 Similar lack of response has been seen in patients with CDK12 inactivation.19,54,57,58 On the contrary, promising results have been observed for patients with alterations in PALB2, BRIP1, FANCA or RAD51B.58 However, due to the low prevalence of these aberrations, further data are needed to fully understand their value as predictors of response to PARPi.
PARPi and ARSi
Crosstalk between the AR and DNA repair pathways has been extensively described.60,61,62 First, PARP is involved in androgen-dependent transcription and PARPi, therefore, impair this process.63 Second, the AR pathway regulates the transcription of DNA repair genes; androgen depletion, therefore, impairs HRR,60 which might render the tumour susceptible to PARPi, regardless of HRR mutation status. A Phase 2 randomised trial assessed the efficacy and tolerability of olaparib in combination with abiraterone compared with abiraterone and placebo in mCRPC patients pretreated with docetaxel, irrespective of their DDR status. Eleven out of 71 (15%) men in the olaparib arm and ten out of 71 (14%) patients in the control arm had mutations in HRR genes. However, 61% of patients had only partially characterised HRR status, as the results of germline and plasma testing could not be confirmed by tumour analysis. Time-to-radiographic progression was significantly prolonged in the olaparib plus abiraterone group compared with the abiraterone alone group (13.8 vs. 8.2 months, P = 0.034), regardless of HRR status. No differences in radiological response rates or in PSA responses were observed between the two arms. Importantly, 54% of patients in the olaparib plus abiraterone arm presented severe adverse events compared with 28% in the abiraterone only group, including seven (10%) patients with a serious cardiovascular event.64 Supported by this early data, several currently ongoing Phase 3 trials aim to address the potential synergy between PARPi and ARSi in all mCRPC patients, irrespective of DDR status (Table 2).
Other strategies to treat DDR-deficient tumours
Activation of the DNA damage-sensing proteins ATM and ATR stimulates different downstream effectors, such as checkpoint kinases 1 and 2 (Chk1 and Chk2, encoded by CHEK1 and CHEK2 genes, respectively) and Wee1, all of which are involved in the maintenance of genomic integrity65 (Box 1). Although previous data have suggested that ATM-deficient tumours show an increased sensitivity to PARPi,47 preclinical findings contradict these reports.66 Clinical trials conducted in mCRPC patients have shown very limited benefit of PARPi for patients with ATM alterations.19,54,56,57,58,59 Thus, other strategies should be explored in these patients, and several ATR, ATM and Chk inhibitors either alone or in combination with other agents are at different stages of preclinical or clinical development67,68 (Table 2).
While PROfound19 demonstrated some potential time-to-progression benefit for olaparib compared with an alternative second ARSi in CDK12-altered mCRPC patients, no similar benefit was observed in TOPARP-B54 or in TRITON2.58 CDK12 has been linked to the HRR pathway, but is also a key player in maintaining genomic stability, and CDK12 inactivation delineates a distinct subgroup of prostate cancers that are characterised by marked genomic instability with focal tandem duplications, increased levels of T cell infiltration and neoantigens.32,69 Thus, CDK12-mutated tumours might constitute a different subgroup of prostate cancer that could benefit from immunotherapy.32,69,70 In that respect, the largest cohort to date of CDK12-inactivated prostate cancer patients treated with immunotherapy has been provided by two independent retrospective multicentre series that have, together, described the outcomes of 112 CDK12-mutated tumours.17,18 Of these, 28 received diverse immunotherapy regimens showing favourable responses even in some heavily pretreated cases. Another group of tumours that may benefit from immune checkpoint inhibitors are those MMR-deficient or microsatellite instability-high (MSI-H)71 (Box 1). In 2017, the US Food and Drug Administration approved pembrolizumab for the treatment of solid tumours based on biomarker status (MMR-deficient or MSI-H tumours) rather than the primary site/tumour type. However, exploratory analysis of data from KEYNOTE-199, a multicohort Phase 2 trial of pembrolizumab in mCRPC patients, failed to show a clear correlation between responses to pembrolizumab and DDR defects or MMR deficiency in tumours.72 Further studies assessing the role of immunotherapy in prostate cancer are ongoing (NCT04104893, NCT04019964 and NCT03570619).
Germline and somatic DDR testing in prostate cancer
As discussed above, the identification of DDR defects in prostate cancer could have prognostic and therapeutic implications. Following the approval of PARPi for the treatment of prostate cancer, genomic testing should be considered in most mCRPC patients. Some DDR alterations may occur early in the evolution of aggressive prostate tumours and could be detected in the diagnostic biopsy or prostatectomy. Some other genomic events may be acquired during the progression of the disease and metastatic tumour biopsy has been the preferred approach to collect information on advanced cancer features. However, biopsies of metastatic lesions can be challenging or not feasible, and at the same time, a single biopsy may not capture heterogeneity across metastases. Besides, the PROfound study has revealed that 30% of such samples might not be of sufficient quality for next-generation sequencing to be carried out.20 The analysis of circulating free DNA is a promising approach as it might overcome the difficulty in obtaining tissue in some cases, but it is still early to conclude that these assays can be used reliably. Currently, constitutive DNA testing might be more straightforward, as germline variants and mutations can be easily and reliably evaluated from peripheral blood or saliva, and accepted standards for reporting variants exist. However, somatic and germline DDR mutations may have a similar prevalence, so by screening only for the germline variants, approximately half of the patients who might benefit from PARPi would not be identified.
The high prevalence of germline DDR alterations in prostate cancer and the fact that 30% of patients harbouring a germline DDR mutation do not have a relative affected by cancer9,13 have led to the recommendation of germline screening for all patients with high-risk localised prostate cancer and metastatic disease.73,74 Unfortunately, most guidelines of clinical practice have not implemented this recommendation, and most patients with advanced disease are not offered germline screening. According to the current guidelines, however, families of prostate cancer patients found to carry a germline BRCA2 mutation would not fulfil the criteria for genetic testing until a median of two further breast and/or ovarian cancer cases per family occur.75 The identification of an inherited mutation in a prostate cancer patient would not only have implications for the patient, but should also be followed by genetic testing in all related family members, providing the opportunity for early cancer-specific screening and risk reduction strategies in those found to be carriers. Unaffected men >40 years found to carry a BRCA2 mutation are recommended to have annual PSA-based prostate cancer screening.24 Recommendations for male carriers of germline mutations in non-BRCA genes are less clear. Protocols for prevention and early detection of other tumour types are well established for individuals carrying mutations in certain DDR genes, but this is beyond the scope of this review.
Conclusions and future directions
A sizeable number of patients with prostate cancer have defects in genes that are involved in the DDR pathway. Such alterations are significantly more prevalent in metastatic than in localised prostate cancer. While some DDR alterations may be acquired with time or in response to therapies, others are probably early events in the evolution of aggressive prostate tumours. A significant proportion of the alterations detected in the tumour are already present in the germline. Identification of these inherited alterations is relevant as it should prompt cascade testing in the relatives. Germline mutation carriers are at risk of different cancer types (i.e. breast, ovarian, prostate, colorectal, etc.) and may benefit from screening and early detection programmes. BRCA2 is the most frequently altered DDR gene, both in the germline and somatic. Germline BRCA2 mutations have consistently been associated with poor outcomes in prostate cancer at different disease stages, but the clinical impact of somatic BRCA2 alterations is unclear. CDK12 inactivation also seems to correlate with aggressive disease, but further research is needed to understand the clinical implications of most somatic and germline DDR alterations and the most appropriate management of these patients.
Some alterations in DDR genes, particularly in those involved in HRR, are predictors of response to PARP inhibition. Somatic, germline and bi- and monoallellic alterations in BRCA1 and BRCA2 have been associated with response to PARPi in clinical trials. Whether there is a differential response based on the alteration type is yet to be clarified. Innate resistance in BRCA1/2-altered patients without previous exposition to platinum-based chemotherapy or PARPi has also been noted. Patients with ATM and CDK12 alterations seem to get little advantage from the treatment with PARPi in monotherapy, although several trials are ongoing to address other therapeutic strategies involving ATR inhibitors and immunotherapy regimens among others. It is unclear whether less prevalent genomic events predict response to PARPi, although promising antitumor activity has been seen in patients with PALB2, BRIP1 or FANCA aberrations. Furthermore, responses to PARPi have been reported in biomarker-negative patients, suggesting that there may be DDR dysfunctions undetected by next-generation sequencing that reflect the multiple elements of this complex pathway.76,77,78 Further clinical trials implementing functional assays would be needed to optimise patient selection for PARPi treatments.
To date, clinical trials with PARPi have focused in the mCRPC setting; however, these drugs may also result in a clinical benefit if used in HPSC stages as addressed by several ongoing or planned studies. Usually, these trials involve the combination of a PARPi with continuous ADT or ADT plus ARSi, but an attractive hypothesis tested in the NCT03047135 study is that PARPi in monotherapy (without any antiandrogen therapies) may be active and safe in selected patients with biochemical relapse after prostatectomy preventing these men from the side effects related to androgen depletion. Another important area of research is the synergy between AR and DDR pathways and the potential benefit of combining ARSi and PARPi to treat mCRPC or mHPSC patients unselected for DDR alterations.
PARPi are the first targeted therapy approved for men with advanced prostate cancer, but other targeted therapies have shown promising results and may also be available in a short time. This is a paradigm shift in the management of prostate cancer that has finally entered the era of precision medicine. However, implementing these therapies in daily clinical practice does not come without challenges. First, it may require a level of knowledge about genomics and genetics that may exceed what most physicians received during training. Scientific societies should provide educational support to maximise the benefits of these therapies to patients. Second, acquisition of tissue for the identification of DDR defects and other targets may in some cases be a major difficulty. As previously discussed, there are molecular events that can be identified in the diagnostic biopsy, but this may have been taken years before the patient develops mCRPC, and therefore untraceable. Metastatic tumour biopsies are the preferred approach to obtain information regarding mCRPC tumour features, but these are not always feasible. Besides, a third of samples analysed in the context of clinical trials were inadequate for sequencing using some of the currently available commercial tests. Germline samples are easily collected and analysed, but limiting DDR testing just to germline alterations, about half of the patients with DDR defects (somatic) would not be detected. The development of a liquid biopsy approach may overcome these challenges providing non-invasive tumour samples that could also be used to monitor the emergence of secondary mutations that may restore the function of the gene previously altered.79,80
Despite the limitations of the studies reported to date, the evidence gaps and the difficulties to identify all patients that may benefit from these therapies, PARPi have finally brought precision oncology to prostate cancer, opening the door to new and exciting times.
References
Tannock, I. F., de Wit, R., Berry, W. R., Horti, J., Pluzanska, A., Chi, K. N. et al. Docetaxel plus prednisone or mitoxantrone plus prednisone for advanced prostate cancer. N. Engl. J. Med. 351, 1502–1512 (2004).
de Bono, J. S., Oudard, S., Ozguroglu, M., Hansen, S., Machiels, J. P., Kocak, I. et al. Prednisone plus cabazitaxel or mitoxantrone for metastatic castration-resistant prostate cancer progressing after docetaxel treatment: a randomised open-label trial. Lancet 376, 1147–1154 (2010).
de Bono, J. S., Logothetis, C. J., Molina, A., Fizazi, K., North, S., Chu, L. et al. Abiraterone and increased survival in metastatic prostate cancer. N. Engl. J. Med. 364, 1995–2005 (2011).
Ryan, C. J., Smith, M. R., de Bono, J. S., Molina, A., Logothetis, C. J., de Souza, P. et al. Abiraterone in metastatic prostate cancer without previous chemotherapy. N. Engl. J. Med. 368, 138–148 (2013).
Scher, H. I., Fizazi, K., Saad, F., Taplin, M. E., Sternberg, C. N., Miller, K. et al. Increased survival with enzalutamide in prostate cancer after chemotherapy. N. Engl. J. Med. 367, 1187–1197 (2012).
Beer, T. M., Armstrong, A. J., Rathkopf, D. E., Loriot, Y., Sternberg, C. N., Higano, C. S. et al. Enzalutamide in metastatic prostate cancer before chemotherapy. N. Engl. J. Med. 371, 424–433 (2014).
Parker, C., Nilsson, S., Heinrich, D., Helle, S. I., O’Sullivan, J. M., Fossa, S. D. et al. Alpha emitter radium-223 and survival in metastatic prostate cancer. N. Engl. J. Med. 369, 213–223 (2013).
Robinson, D., Van Allen, E. M., Wu, Y. M., Schultz, N., Lonigro, R. J., Mosquera, J. M. et al. Integrative clinical genomics of advanced prostate cancer. Cell 161, 1215–1228 (2015).
Pritchard, C. C., Mateo, J., Walsh, M. F., De Sarkar, N., Abida, W., Beltran, H. et al. Inherited DNA-repair gene mutations in men with metastatic prostate cancer. N. Engl. J. Med. 375, 443–453 (2016).
Abida, W., Armenia, J., Gopalan, A., Brennan, R., Walsh, M., Barron, D. et al. Prospective genomic profiling of prostate cancer across disease states reveals germline and somatic alterations that may affect clinical decision making. JCO Precis. Oncol. 2017, https://doi.org/10.1200/PO.17.00029 (2017).
Abida, W., Cyrta, J., Heller, G., Prandi, D., Armenia, J., Coleman, I. et al. Genomic correlates of clinical outcome in advanced prostate cancer. Proc. Natl Acad. Sci. USA 116, 11428–11436 (2019).
Cancer Genome Atlas Research Network. The molecular taxonomy of primary prostate cancer. Cell 163, 1011–1025 (2015).
Castro, E., Romero-Laorden, N., Del Pozo, A., Lozano, R., Medina, A., Puente, J. et al. PROREPAIR-B: a prospective cohort study of the impact of germline DNA repair mutations on the outcomes of patients with metastatic castration-resistant prostate cancer. J. Clin. Oncol. 37, 490–503 (2019).
Armenia, J., Wankowicz, S. A. M., Liu, D., Gao, J., Kundra, R., Reznik, E. et al. The long tail of oncogenic drivers in prostate cancer. Nat. Genet. 50, 645–651 (2018).
Castro, E., Goh, C., Olmos, D., Saunders, E., Leongamornlert, D., Tymrakiewicz, M. et al. Germline BRCA mutations are associated with higher risk of nodal involvement, distant metastasis, and poor survival outcomes in prostate cancer. J. Clin. Oncol. 31, 1748–1757 (2013).
Reimers, M. A., Yip, S. M., Zhang, L., Cieslik, M., Dhawan, M., Montgomery, B. et al. Clinical outcomes in cyclin-dependent kinase 12 mutant advanced prostate cancer. Eur. Urol. 77, 333–341 (2020).
Antonarakis, E. S., Velho, P. I., Fu, W., Wang, H., Agarwal, N., Santos, V. S. et al. CDK12-altered prostate cancer: clinical features and therapeutic outcomes to standard systemic therapies, poly (ADP-ribose) polymerase inhibitors, and PD-1 inhibitors. JCO Precis. Oncol. 4, 370–381 (2020).
Schweizer, M. T., Ha, G., Gulati, R., Brown, L. C., McKay, R. R., Dorff, T. et al. CDK12-mutated prostate cancer: clinical outcomes with standard therapies and immune checkpoint blockade. JCO Precis. Oncol. 4, 382–392 (2020).
de Bono, J., Mateo, J., Fizazi, K., Saad, F., Shore, N., Sandhu, S. et al. Olaparib for metastatic castration-resistant prostate cancer. N. Engl. J. Med. 383, 891 (2020).
de Bono, J. S., Fizazi, K., Saad, F., Shore, N., Sandhu, S. K., Mehra, N. et al. 847PDCentral, prospective detection of homologous recombination repair gene mutations (HRRm) in tumour tissue from >4000 men with metastatic castration-resistant prostate cancer (mCRPC) screened for the PROfound study. Ann. Oncol. 30, v325–v355 (2019).
Abida, W., Cheng, M. L., Armenia, J., Middha, S., Autio, K. A., Vargas, H. A. et al. Analysis of the prevalence of microsatellite instability in prostate cancer and response to immune checkpoint blockade. JAMA Oncol. 5, 471–478 (2019).
Jonsson, P., Bandlamudi, C., Cheng, M. L., Srinivasan, P., Chavan, S. S., Friedman, N. D. et al. Tumour lineage shapes BRCA-mediated phenotypes. Nature 571, 576–579 (2019).
Latham, A., Srinivasan, P., Kemel, Y., Shia, J., Bandlamudi, C., Mandelker, D. et al. Microsatellite instability is associated with the presence of Lynch syndrome pan-cancer. J. Clin. Oncol. 37, 286–295 (2019).
Mohler, J. L., Antonarakis, E. S., Armstrong, A. J., D’Amico, A. V., Davis, B. J., Dorff, T. et al. Prostate cancer, version 2.2019, NCCN clinical practice guidelines in oncology. J Natl Compr Canc Netw. 17, 479–505 (2019).
Castro, E., Goh, C., Leongamornlert, D., Saunders, E., Tymrakiewicz, M., Dadaev, T. et al. Effect of BRCA mutations on metastatic relapse and cause-specific survival after radical treatment for localised prostate cancer. Eur. Urol. 68, 186–193 (2015).
Carter, H. B., Helfand, B., Mamawala, M., Wu, Y., Landis, P., Yu, H. et al. Germline mutations in ATM and BRCA1/2 are associated with grade reclassification in men on active surveillance for prostate cancer. Eur. Urol. 75, 743–749 (2019).
Annala, M., Vandekerkhove, G., Khalaf, D., Taavitsainen, S., Beja, K., Warner, E. W. et al. Circulating tumor DNA genomics correlate with resistance to abiraterone and enzalutamide in prostate cancer. Cancer Discov. 8, 444–457 (2018).
Vandekerkhove, G., Struss, W. J., Annala, M., Kallio, H. M. L., Khalaf, D., Warner, E. W. et al. Circulating tumor DNA abundance and potential utility in de novo metastatic prostate cancer. Eur. Urol. 75, 667–675 (2019).
Annala, M., Struss, W. J., Warner, E. W., Beja, K., Vandekerkhove, G., Wong, A. et al. Treatment outcomes and tumor loss of heterozygosity in germline DNA repair-deficient prostate cancer. Eur. Urol. 72, 34–42 (2017).
Antonarakis, E. S., Lu, C., Luber, B., Liang, C., Wang, H., Chen, Y. et al. Germline DNA-repair gene mutations and outcomes in men with metastatic castration-resistant prostate cancer receiving first-line abiraterone and enzalutamide. Eur. Urol. 74, 218–225 (2018).
Mateo, J., Cheng, H. H., Beltran, H., Dolling, D., Xu, W., Pritchard, C. C. et al. Clinical outcome of prostate cancer patients with germline DNA repair mutations: retrospective analysis from an international study. Eur. Urol. 73, 687–693 (2018).
Wu, Y. M., Cieslik, M., Lonigro, R. J., Vats, P., Reimers, M. A., Cao, X. et al. Inactivation of CDK12 delineates a distinct immunogenic class of advanced prostate. Cancer Cell 173, 1770–1782 (2018). e14.
Nguyen, B., Mota, J. M., Nandakumar, S., Stopsack, K. H., Weg, E., Rathkopf, D. et al. Pan-cancer analysis of CDK12 alterations identifies a subset of prostate cancers with distinct genomic and clinical characteristics. Eur. Urol. https://doi.org/10.1016/j.eururo.2020.03.024 (2020).
Antonarakis, E. S., Shaukat, F., Isaacsson Velho, P., Kaur, H., Shenderov, E., Pardoll, D. M. et al. Clinical features and therapeutic outcomes in men with advanced prostate cancer and dna mismatch repair gene mutations. Eur. Urol. 75, 378–382 (2019).
Ritch, E., Fu, S. Y. F., Herberts, C., Wang, G., Warner, E. W., Schonlau, E. et al. Identification of hypermutation and defective mismatch repair in ctDNA from metastatic prostate cancer. Clin. Cancer Res. 26, 1114–1125 (2020).
Rodrigues, D. N., Rescigno, P., Liu, D., Yuan, W., Carreira, S., Lambros, M. B. et al. Immunogenomic analyses associate immunological alterations with mismatch repair defects in prostate cancer. J. Clin. Invest. 128, 5185 (2018).
Byrski, T., Gronwald, J., Huzarski, T., Grzybowska, E., Budryk, M., Stawicka, M. et al. Pathologic complete response rates in young women with BRCA1-positive breast cancers after neoadjuvant chemotherapy. J. Clin. Oncol. 28, 375–379 (2010).
von Minckwitz, G., Schneeweiss, A., Loibl, S., Salat, C., Denkert, C., Rezai, M. et al. Neoadjuvant carboplatin in patients with triple-negative and HER2-positive early breast cancer (GeparSixto; GBG 66): a randomised phase 2 trial. Lancet Oncol. 15, 747–756 (2014).
Yang, D., Khan, S., Sun, Y., Hess, K., Shmulevich, I., Sood, A. K. et al. Association of BRCA1 and BRCA2 mutations with survival, chemotherapy sensitivity, and gene mutator phenotype in patients with ovarian cancer. JAMA 306, 1557–1565 (2011).
Sternberg, C. N., Petrylak, D. P., Sartor, O., Witjes, J. A., Demkow, T., Ferrero, J. M. et al. Multinational, double-blind, phase III study of prednisone and either satraplatin or placebo in patients with castrate-refractory prostate cancer progressing after prior chemotherapy: the SPARC trial. J. Clin. Oncol. 27, 5431–5438 (2009).
Hager, S., Ackermann, C. J., Joerger, M., Gillessen, S. & Omlin, A. Anti-tumour activity of platinum compounds in advanced prostate cancer-a systematic literature review. Ann. Oncol. 27, 975–984 (2016).
Cheng, H. H., Pritchard, C. C., Boyd, T., Nelson, P. S. & Montgomery, B. Biallelic inactivation of BRCA2 in platinum-sensitive metastatic castration-resistant prostate cancer. Eur. Urol. 69, 992–995 (2016).
Pomerantz, M. M., Spisak, S., Jia, L., Cronin, A. M., Csabai, I., Ledet, E. et al. The association between germline BRCA2 variants and sensitivity to platinum-based chemotherapy among men with metastatic prostate cancer. Cancer 123, 3532–3539 (2017).
Zafeiriou, Z., Bianchini, D., Chandler, R., Rescigno, P., Yuan, W., Carreira, S. et al. Genomic analysis of three metastatic prostate cancer patients with exceptional responses to carboplatin indicating different types of DNA repair deficiency. Eur. Urol. 75, 184–192 (2019).
Mota, J. M., Barnett, E., Nauseef, J. T., Nguyen, B., Stopsack, K. H., Wibmer, A. et al. Platinum-based chemotherapy in metastatic prostate cancer with DNA repair gene alterations. JCO Precis. Oncol. 4, 355–366 (2020).
Ashworth, A. & Lord, C. J. Synthetic lethal therapies for cancer: what’s next after PARP inhibitors? Nat. Rev. Clin. Oncol. 15, 564–576 (2018).
Murai, J., Huang, S. Y., Das, B. B., Renaud, A., Zhang, Y., Doroshow, J. H. et al. Trapping of PARP1 and PARP2 by clinical PARP inhibitors. Cancer Res. 72, 5588–5599 (2012).
Ledermann, J., Harter, P., Gourley, C., Friedlander, M., Vergote, I., Rustin, G. et al. Olaparib maintenance therapy in patients with platinum-sensitive relapsed serous ovarian cancer: a preplanned retrospective analysis of outcomes by BRCA status in a randomised phase 2 trial. Lancet Oncol. 15, 852–861 (2014).
Fong, P. C., Boss, D. S., Yap, T. A., Tutt, A., Wu, P., Mergui-Roelvink, M. et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N. Engl. J. Med. 361, 123–134 (2009).
Sandhu, S. K., Omlin, A., Hylands, L., Miranda, S., Barber, L. J., Riisnaes, R. et al. Poly (ADP-ribose) polymerase (PARP) inhibitors for the treatment of advanced germline BRCA2 mutant prostate cancer. Ann. Oncol. 24, 1416–1418 (2013).
de Bono, J., Ramanathan, R. K., Mina, L., Chugh, R., Glaspy, J., Rafii, S. et al. Phase I, dose-escalation, two-part trial of the PARP inhibitor talazoparib in patients with advanced germline BRCA1/2 mutations and selected sporadic cancers. Cancer Discov. 7, 620–629 (2017).
Sandhu, S. K., Schelman, W. R., Wilding, G., Moreno, V., Baird, R. D., Miranda, S. et al. The poly(ADP-ribose) polymerase inhibitor niraparib (MK4827) in BRCA mutation carriers and patients with sporadic cancer: a phase 1 dose-escalation trial. Lancet Oncol. 14, 882–892 (2013).
Mateo, J., Carreira, S., Sandhu, S., Miranda, S., Mossop, H., Perez-Lopez, R. et al. DNA-repair defects and olaparib in metastatic prostate cancer. N. Engl. J. Med. 373, 1697–1708 (2015).
Mateo, J., Porta, N., Bianchini, D., McGovern, U., Elliott, T., Jones, R. et al. Olaparib in patients with metastatic castration-resistant prostate cancer with DNA repair gene aberrations (TOPARP-B): a multicentre, open-label, randomised, phase 2 trial. Lancet Oncol. 21, 162–174 (2020).
Hussain, M., Mateo, J., Fizazi, K., Saad, F., Shore, N., Sandhu, S. et al. for the PROfound Trial Investigators. Survival with Olaparib in Metastatic Castration-Resistant Prostate Cancer. N Engl J Med. https://doi.org/10.1056/NEJMoa2022485 2020. Online ahead of print.
Abida, W., Campbell, D., Patnaik, A., Shapiro, J. D., Sautois, B., Vogelzang, N. J. et al. Rucaparib in men with metastatic castration-resistant prostate cancer harboring a BRCA1 or BRCA2 gene alteration. J Clin Oncol. https://doi.org/10.1200/JCO.20.01035, JCO2001035 (2020).
Smith, M. R., Sandhu, S. K., Kelly, W. K., Scher, H. I., Efstathiou, E., Lara, P. N. et al. LBA50Pre-specified interim analysis of GALAHAD: a phase II study of niraparib in patients (pts) with metastatic castration-resistant prostate cancer (mCRPC) and biallelic DNA-repair gene defects (DRD). Ann. Oncol. 30, v851–v934 (2019).
Abida, W., Campbell, D., Patnaik, A., Shapiro, J. D., Sautois, B., Vogelzang, N. J. et al. Non-BRCA DNA damage repair gene alterations and response to the PARP inhibitor rucaparib in metastatic castration-resistant prostate cancer: analysis from the phase 2 TRITON2 study. Clin Cancer Res. 26, 2487–2496 (2020).
Marshall, C. H., Sokolova, A. O., McNatty, A. L., Cheng, H. H., Eisenberger, M. A., Bryce, A. H. et al. Differential response to olaparib treatment among men with metastatic castration-resistant prostate cancer harboring BRCA1 or BRCA2 versus ATM mutations. Eur. Urol. 76, 452–458 (2019).
Polkinghorn, W. R., Parker, J. S., Lee, M. X., Kass, E. M., Spratt, D. E., Iaquinta, P. J. et al. Androgen receptor signaling regulates DNA repair in prostate cancers. Cancer Discov. 3, 1245–1253 (2013).
Goodwin, J. F., Schiewer, M. J., Dean, J. L., Schrecengost, R. S., de Leeuw, R., Han, S. et al. A hormone-DNA repair circuit governs the response to genotoxic insult. Cancer Discov. 3, 1254–1271 (2013).
Asim, M., Tarish, F., Zecchini, H. I., Sanjiv, K., Gelali, E., Massie, C. E. et al. Synthetic lethality between androgen receptor signalling and the PARP pathway in prostate cancer. Nat. Commun. 8, 374 (2017).
Schiewer, M. J., Goodwin, J. F., Han, S., Brenner, J. C., Augello, M. A., Dean, J. L. et al. Dual roles of PARP-1 promote cancer growth and progression. Cancer Discov. 2, 1134–1149 (2012).
Clarke, N., Wiechno, P., Alekseev, B., Sala, N., Jones, R., Kocak, I. et al. Olaparib combined with abiraterone in patients with metastatic castration-resistant prostate cancer: a randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Oncol. 19, 975–986 (2018).
Harper, J. W. & Elledge, S. J. The DNA damage response: ten years after. Mol. Cell 28, 739–745 (2007).
Rafiei, S., Fitzpatrick, K., Liu, D., Cai, M. Y., Elmarakeby, H. A., Park, J. et al. ATM loss confers greater sensitivity to ATR inhibition than PARP inhibition in prostate cancer. Cancer Res. 80, 2094–2100 (2020).
Jin, M. H. & Oh, D. Y. ATM in DNA repair in cancer. Pharm. Ther. 203, 107391 (2019).
Mei, L., Zhang, J., He, K. & Zhang, J. Ataxia telangiectasia and Rad3-related inhibitors and cancer therapy: where we stand. J. Hematol. Oncol. 12, 43 (2019).
Sokol, E. S., Pavlick, D., Frampton, G. M., Ross, J. S., Miller, V. A., Ali, S. M. et al. Pan-cancer analysis of CDK12 loss-of-function alterations and their association with the focal tandem-duplicator phenotype. Oncologist 24, 1526–1533 (2019).
Antonarakis, E. S. Cyclin-dependent Kinase 12, immunity, and prostate cancer. N. Engl. J. Med. 379, 1087–1089 (2018).
Le, D. T., Durham, J. N., Smith, K. N., Wang, H., Bartlett, B. R., Aulakh, L. K. et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 357, 409–413 (2017).
Antonarakis, E. S., Piulats, J. M., Gross-Goupil, M., Goh, J., Ojamaa, K., Hoimes, C. J. et al. Pembrolizumab for treatment-refractory metastatic castration-resistant prostate cancer: multicohort, open-label phase II KEYNOTE-199 Study. J. Clin. Oncol. JCO1901638 (2019).
Mohler, J. L., Antonarakis, E. S., Armstrong, A. J., D’Amico, A. V., Davis, B. J., Dorff, T. et al. Prostate cancer, version 2.2019, NCCN clinical practice guidelines in oncology. J. Natl Compr. Canc Netw. 17, 479–505 (2019).
Gillessen, S., Attard, G., Beer, T. M., Beltran, H., Bjartell, A., Bossi, A. et al. Management of patients with advanced prostate cancer: report of the advanced prostate cancer consensus conference 2019. Eur. Urol. 77, 508–547 (2020).
Oliva, L., Lozano, R., Llacer, C., Aragon, I., Pajares, B. I., Saez, M. I. et al. Risk prediction tools available for germline BRCA1/2 mutations underperform in prostate cancer patients. Eur. Urol. Oncol. https://doi.org/10.1016/j.euo.2019.06.019 (2019).
Hoppe, M. M., Sundar, R., Tan, D. S. P. & Jeyasekharan, A. D. Biomarkers for homologous recombination deficiency in cancer. J. Natl Cancer Inst. 110, 704–713 (2018).
Redon, C. E., Nakamura, A. J., Zhang, Y. W., Ji, J. J., Bonner, W. M., Kinders, R. J. et al. Histone gammaH2AX and poly(ADP-ribose) as clinical pharmacodynamic biomarkers. Clin. Cancer Res. 16, 4532–4542 (2010).
Castroviejo-Bermejo, M., Cruz, C., Llop-Guevara, A., Gutierrez-Enriquez S., Ducy M., Ibrahim, Y. H. et al. A RAD51 assay feasible in routine tumor samples calls PARP inhibitor response beyond BRCA mutation. EMBO Mol. Med. 10, e9172 (2018).
Quigley, D., Alumkal, J. J., Wyatt, A. W., Kothari, V., Foye, A., Lloyd, P. et al. Analysis of circulating cell-free DNA identifies multiclonal heterogeneity of BRCA2 reversion mutations associated with resistance to PARP inhibitors. Cancer Discov. 7, 999–1005 (2017).
Goodall, J., Mateo, J., Yuan, W., Mossop, H., Porta, N., Miranda, S. et al. Circulating cell-free DNA to guide prostate cancer treatment with PARP ihibition. Cancer Discov. 7, 1006–1017 (2017).
Lindahl, T. Instability and decay of the primary structure of DNA. Nature 362, 709–715 (1993).
Lindahl, T. & Wood, R. D. Quality control by DNA repair. Science 286, 1897–1905 (1999).
Harper, J. W. & Elledge, S. J. The DNA damage response: ten years after. Mol. Cell 28, 739–745 (2007).
Caldecott, K. W. Single-strand break repair and genetic disease. Nat. Rev. Genet. 9, 619–631 (2008).
Hanawalt, P. C. Subpathways of nucleotide excision repair and their regulation. Oncogene 21, 8949–8956 (2002).
Li, G. M. Mechanisms and functions of DNA mismatch repair. Cell Res. 18, 85–98 (2008).
Prakash, R., Zhang, Y., Feng, W. & Jasin, M. Homologous recombination and human health: the roles of BRCA1, BRCA2, and associated proteins. Cold Spring Harb. Perspect. Biol. 7, a016600 (2015).
Ceccaldi, R., Sarangi, P. & D’Andrea, A. D. The Fanconi anaemia pathway: new players and new functions. Nat. Rev. Mol. Cell Biol. 17, 337–349 (2016).
Deriano, L. & Roth, D. B. Modernizing the nonhomologous end-joining repertoire: alternative and classical NHEJ share the stage. Annu. Rev. Genet. 47, 433–455 (2013).
Acknowledgements
None.
Author information
Authors and Affiliations
Contributions
R.L., E.C., C.C. and D.O. designed and wrote the whole manuscript; I.M.A., Y.C. and P.P.L.-C. reviewed and edited the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent to publish
Not applicable.
Data availability
Not applicable.
Competing interests
R.L. declares speaker fees from Roche, Janssen and Sanofi, and travel support from Roche and Janssen. E.C. declares honoraria from Astellas Pharma, AstraZeneca, Bayer, Clovis, Janssen-Cilag, Pfizer and Roche, consulting or Advisory Roles for AstraZeneca, Bayer, Janssen and MSD, research funding from AstraZeneca (Inst), Bayer (Inst) and Janssen (Inst) and travel support from Bayer, Janssen, Roche and Astellas Pharma. C.C. declares travel support from Ipsen and Novartis. D.O. declares honoraria from Bayer, Janssen and Sanofi, consulting or advisory role from AstraZeneca, Bayer, Clovis Oncology, Daiichi-Sankyo Janssen, MSD and Roche, research funding from Astellas (Inst), AstraZeneca (Inst), Bayer (Inst), Genentech (Inst), Janssen (Inst), Medivation (Inst), MSD (Inst), Pfizer (Inst), F. Hoffman-Roche (Inst) and Tokai Pharmaceuticals (Inst) and travel support from Bayer, Ipsen, Janssen and F. Hoffman-Roche. I.M.A., Y.C. and P.P.L.-C. do not have any relationships to disclose.
Funding information
The authors at the Prostate Cancer Clinical Research Unit (CNIO) and the Genitourinary Cancer Translational Research Group (IBIMA) are supported by: grants from “Instituto de Salud Carlos III” (PI16/01565 and PI19/01380 to D.O., JR18/00011 and PI19/01475 to E.C., CM17-00221 to R.L.), a Department of Defense US IMPACT award (W81XWH-18-1-0193), a CRIS Excellence in research award (E19-26 to D.O.), a “Fundación Científica de la AECC” research grant (PROYE19054OLMO to D.O.), a 2017 Prostate Cancer Foundation Young investigator Award to E.C., grants from “Ministerio de Economía, Industria y Competitividad” (RYC-2015-18625 to D.O. and FJCI-2016-28121 to I.M.A), a grant from “Ministerio de Educación, Cultura y Deportes (FPU15/05126 to Y.C.), a 2019 ESMO clinical research fellowship to C.C. and unrestricted grants from “Fundación CRIS contra el Cáncer”.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lozano, R., Castro, E., Aragón, I.M. et al. Genetic aberrations in DNA repair pathways: a cornerstone of precision oncology in prostate cancer. Br J Cancer 124, 552–563 (2021). https://doi.org/10.1038/s41416-020-01114-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41416-020-01114-x
This article is cited by
-
Loss of PDE4D7 expression promotes androgen independence, neuroendocrine differentiation and alterations in DNA repair: implications for therapeutic strategies
British Journal of Cancer (2023)
-
Interstitial pneumonia after regression by olaparib for neuroendocrine prostate cancer with BRCA1 mutation: a case report
International Cancer Conference Journal (2023)
-
Genomic and Phenotypic Biomarkers for Precision Medicine Guidance in Advanced Prostate Cancer
Current Treatment Options in Oncology (2023)
-
The DACH1 gene is frequently deleted in prostate cancer, restrains prostatic intraepithelial neoplasia, decreases DNA damage repair, and predicts therapy responses
Oncogene (2023)
-
Homologous recombination deficiency (HRD) score in aggressive prostatic adenocarcinoma with or without intraductal carcinoma of the prostate (IDC-P)
BMC Medicine (2022)
