
Abstract
Variants in KMT2A, encoding the histone methyltransferase KMT2A, are a growing cause of intellectual disability (ID). Up to now, the majority of KMT2A variants are non-sense and frameshift variants causing a typical form of Wiedemann–Steiner syndrome. We studied KMT2A gene in a cohort of 200 patients with unexplained syndromic and non-syndromic ID and identified four novel variants, one splice and three missense variants, possibly deleterious. We used primary cells from the patients and molecular approaches to determine the deleterious effects of those variants on KMT2A expression and function. For the putative splice variant c.11322-1G>A, we showed that it led to only one nucleotide deletion and loss of the C-terminal part of the protein. For two studied KMT2A missense variants, c.3460C>T (p.(Arg1154Trp)) and c.8558T>G (p.(Met2853Arg)), located at the cysteine-rich CXXC domain and the transactivation domain of the protein, respectively, we found altered KMT2A target genes expression in patient’s fibroblasts compared to controls. Furthermore, we found a disturbed subcellular distribution of KMT2A for the c.3460C>T mutant. Taken together, our results demonstrated the deleterious impact of the splice variant and of the missense variants located at two different functional domains and suggested reduction of KMT2A function as the disease-causing mechanism.
Similar content being viewed by others


Introduction
Intellectual disability (ID) is a common clinically and genetically heterogeneous disorder characterized by limitations in intellectual functioning and adaptive behavior [1]. ID affects 1–3% of the population, but the pathophysiological background remains poorly understood [2]. High-throughput sequencing studies performed in ID patients enabled identification of variants in numerous genes, including several chromatin-regulating factors [3,4,5]. Chromatin consists of the DNA helix spooled around octamers of histones H2A, H2B, H3, and H4. Histones are subject to a plethora of post-translational modifications, which are able to influence a variety of nuclear processes [6]. Variants in genes encoding histone methyltransferases, such as MLL (MLL-1 or KMT2A), MLL2 (KMT2D) and SETD5, and in genes encoding histone demethylases, such as KDM6A, PHF8 (KDM7B) and KDM5C, have been identified in ID patients, suggesting that disruption of histone methylation dynamics may contribute to the disease development [7,8,9,10,11,12].
KMT2A, a histone methyltransferase of the lysine 4 from the histone 3 (H3K4) regulates chromatin-mediated transcription of multiple genes including the Hox and Wnt family factors [13, 14]. KMT2A is a 3972 aminoacids (aa) (NP_001184033.1), multidomain protein comprising three DNA-binding AT-hooks at the N terminus, a cysteine-rich CXXC domain, a plant homeodomain (PHD) finger motif, a bromodomain, a transactivation (TAD) domain, a FYRN domain, a WDR5 interaction (Win) motif, and a C-terminal SET domain (Fig. 1a). The SET domain is involved in the histone monomethylation, dimethylation, or trimethylation activity of the protein [13, 14]. KMT2A is cleaved at two independent sites (CS1, aa 2669–2670 and CS2, aa 2721–2722), and the resulting C-terminal and N-terminal fragments form a stable complex that localized to a subnuclear compartment [15].

a Schematic of KMT2A protein structure (NP_001184033.1), with domains (and motifs) and the location of the identified de novo missense and splice variants. Domains were predicted by SMART program (http://smart.embl-heidelberg.de/). DNA binding AT hooks 1–3 (aa 169–180; aa 217–227; aa 301–309), CXXC domain (aa 1150–1198), zinc finger PHD-type 1–3 (aa 1431–1482; 1479–1533; 1566–1630), TAD domain (aa 2850–2858), bromodomain (aa 1703–1748), FYR N-terminal domain (aa 2021–2077), FYR-C terminal domain (aa 3666–3747), WDR5 interacting motif (aa 3765–3773), SET domain (aa 3832–3948), and post-SET domain (aa 3956–3972). b Sequence analysis of PCR products from the patient’s and parent’s (mother and father) DNA showing the different de novo variants. Asteriks indicate the position of the variants
In 2012, Jones and colleagues identified de novo heterozygous KMT2A variants in five of six individuals with hypertrichosis cubiti associated with short stature, ID, and a distinctive facial appearance, consistent with a diagnosis of Wiedemann–Steiner syndrome (WSS) [7]. In these five patients the KMT2A variants were predicted to result in premature termination of translation and to a nonsense-mediated decay (NMD) of the corresponding transcripts [7]. Since this first description additional WSS patients, including a pair of monozygotic twins, have been reported, especially by using exome analysis approaches, bringing to more than 25 the total number of patients carrying KMT2A variants leading to a premature stop codon [7, 16,17,18,19,20,21,22,23,24,25,26]. KMT2A missense variants have been reported in five patients with WSS, one of them having a severe form of the disease and congenital immunodeficiency [16]. While the pathophysiological mechanism of the non-sense variants is haploinsufficiency, the mechanism for missense variants is unknown.
Here, we studied KMT2A gene in a cohort of 200 patients with unexplained syndromic and non-syndromic ID and identified 42 KMT2A variants in 41 patients. We selected the four novel possibly deleterious KTM2A variants, that occurred de novo and were absent from variant databases, to investigate the cellular and the molecular consequences in patients’ skin fibroblasts.
Materials and methods
Patients
The study was carried out on 200 patients from our cohort of non-syndromic or syndromic ID of unknown cause. Array CGH and fragile X testing were normal in all ID patients. All human subjects research was carried out in accordance with approved ethics guidelines at all locations. KMT2A variants have been submitted to the specific gene variant database LOVD (http://databases.lovd.nl/shared/variants/). All potential sequence variations without discrimination between novel and confirmatory (KMT2A) data have been sent to the LOVD gene variant database. The ID patients 1, 2, 3, and 4 received, respectively, the ID0000105338, ID0000105339, ID0000105340, and ID 0000105341 numbers. The other ID individuals received the ID0000105412–ID0000105423 numbers.
Cell culture and fibroblast synchronization
Fibroblasts were cultured at 37 °C in 5% CO2 in DMEM glutaMax (1 g/L glucose, pyruvate+; ThermoFisher Scientific, Waltham, Massachusetts, USA) supplemented with 10% (v/v) fetal bovine serum (ThermoFisher Scientific), 100 U/mL penicillin, 10 µg/mL streptomycin (ThermoFisher Scientific) and 10 µg/mL ciprofloxacine (Sigma, Saint Louis, Missouri, USA). For COS7 cell and human embryonic kidney (HEK) 293 cell amplification, DMEM glutaMax (1 g/L glucose, pyruvate+) was substituted by DMEM glutaMax high glucose (4.5 g/L glucose, pyruvate−; ThermoFisher Scientific). At cell confluence, cells were cultured with media supplemented with 50% fetal bovine serum for 2 h followed by 22 h with normal media (supplemented with antibiotics) culture condition. Then, the cells were trypsinized, washed in PBS, splited and kept at −80 °C for future RNA and protein extractions.
Antibodies
Rabbit monoclonal KMT2A antibody (clone D6G8N, #14197, Cell Signaling, Danvers, MA, USA) was used for immunofluorescence and western blotting. This antibody recognizes an antigenic motif located at the C-terminal p180 (C180) KMT2A fragment localized before SET domain (manufacturer information). Polyclonal anti-H3K4Me3 (Cell Signaling, #9727) and monoclonal anti-SC35 (Sigma, S4045) were used for immunofluorescence, while monoclonal anti-GAPDH (ThermoFisher Scientific clone 6C5, AM4300) was used for western blotting.
Western blotting
Whole cell lysates were made using RIPA like buffer supplemented with PMSF (Cell Signaling, Danvers, USA) and 1X protease and phosphatase inhibitor cocktail (Roche, Meylan, France). The quantity of protein was evaluated by Bradford method (Bio-Rad protein assay 500-0006, Hercules, CA, USA). Lysates were added with Laemmli sample buffer followed by heat denaturation before run on 4–15% (Bio-Rad, #4561084) or 4% SDS–PAGE. Gels (Bio-Rad mini-protean 3) were transferred to nitrocellulose membranes (GE Healthcare Life Sciences). Membranes were blocked (1 h), incubated overnight with primary antibody, washed and incubated (45 min) with HRP-conjugated secondary antibodies (Santa Cruz, Dallas, Texas, USA), all in 0.05% Tween/PBS with 5% non-fat dry milk or 5% BSA (Sigma). HRP-conjugated antibodies were detected with ECL western blotting detection reagents (Thermo Scientific) according to the manufacturer’s instructions.
qRT-PCR
Total RNA was isolated using RNeasy reagent (Qiagen, Hilden, Germany) with a DNase step, according to the manufacturer’s instructions. Total RNA was retrotranscribed with Maxima reverse transcriptase (Thermo Scientific). Real-time PCR were performed using Sybergreen kit and Roche system. Relative expression was calculated using the ΔΔCt method and normalized to the GAPDH gene. HOXA9, MEIS1, SIX2, CDKN2C, KMT2A and CDKN1B primer sequences are available on request.
Plasmid and transfection
We used KMT2A (NM_005933) Human ORF Clone (Origene, Rockville, USA) (pCMV6-AC-GFP backbone) for generate c.3460C>T (p.(Arg1154Trp)) and c.8558T>G (p.(Met2853Arg)) mutant using QuikChange II XL Site-Directed Mutagenesis Kit (Agilent, Les Ulis, France) and subcloning approaches. To prepare an enzyme-inactive KMT2A, the coding sequence of SET domain was deleted from the entire coding sequence of KMT2A (KMT2A-ΔSET; p.Lys3775Serfs*32). We used lipofectamine 2000 (ThermoFisher Scientific) to transfect COS7 or HEK 293 cells (24-well plates 50×103 cells/well) with 500 ng of WT or mutants KMT2A constructs according to the manufacturer’s instructions.
Immunofluorescence
Human fibroblasts or COS7 cells 48 h post transfection were fixed with 4% paraformaldehyde for 15 min, and then with 100% ice-cold methanol for 10 min. Cells were permeabilized and blocked with PBS supplemented with 0.1% triton X-100 and 0.2% normal goat serum (Dako, Les Ulis, France) (1 h). Coverslips were incubated overnight at 4 °C with the appropriate primary antibodies, washed and incubated with the corresponding secondary antibodies for 45 min at room temperature. Cy3-conjugated donkey-anti-rabbit, Cy3-conjugated donkey-anti-mouse, and FITC-conjugated goat-anti-mouse secondary antibodies (Jackson ImmunoResearch, Suffolk, UK) were used at 1:300. Coverslips were then washed, mounted with Fluoromount-G (eBioscience, Paris, France) containing DAPI (Sigma) and analyzed by a fluorescence microscopy (LEICA DM6000 B) using a ×63 objective.
Results
Study of the KMT2A gene in a cohort of 200 ID patients
Using next-generation sequencing, we studied KMT2A (NM_001197104) in a cohort of 200 patients with unexplained syndromic and non-syndromic ID and identified 42 KMT2A heterozygous variants (among which 31 different) in 41 unrelated patients (one patient carrying two variants) (Table S1). Thirty were missense variants and one was a splice variant. Thirteen variants (12 missense and one splice) were absent in the ExAC database. Among the 12 missense variants, five variants were conserved across species from Xenopus to Human. Three of them were located in functional domains of KMT2A: The c.3460C>T (p.(Arg1154Trp)) and c.3581G>A (p.(Cys1194Tyr)) variants were located at the cysteine-rich CXXC domain and the c.8558T>G (p.(Met2853Arg)) variant at the TAD domain of KMT2A (Fig. 1a). De novo occurrence was observed in the three patients carrying these latter missense variants and in the individual carrying the putative splice variant c.11322-1G>A (Fig. 1b).
Phenotype of ID patients carrying a de novo KMT2A variant
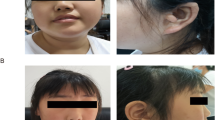
The phenotypic traits of four new independent patients (one female and three males), each carrying a de novo KMT2A variant are listed in the supplementary data on line (Table S2). Distinctive facial features consisted of flat face, short palpebral fissures, hypertelorism or epicantus, low-set ears, wide nasal bridge, thin upper lip, and everted lower lip were present in all patients. We also noted thick eyebrows or synophrys (3/4), strabismus (3/4), long eyelashes (2/3), bulbous nose (2/3), and narrow external auditory canals (2/4) (Fig. 2). Oligodontia was present in one patient. Hands and feeds were small in three patients but largest and swollen in the last one. Clinodactyly was observed in three patients and 2–3 toe syndactyly in two. Ocular features were hyperopia (2/4) and myopia (1/4). Hypertrichosis was present in all patients.

Facial characteristics of the ID patients carrying a KMT2A variant. Patient 2 (P2) carries the c.8558T>G (p.(Met2853Arg)) variant, Patient 3 (P3) carries the c.3581G>A (p.(Cys1194Tyr)) variant and Patient 4 (P4) carries the c.11322–1G>A variant
Digestive features consisted with feeding difficulties and gastro-esophageal reflux were observed in three patients. Uro-genital abnormalities were unilateral cryptorchidism, scrotal hypoplasia, and sacrococcygeal lipoma with neurogenic bladder. Congenital heart defect was present in one patient. Skeletal abnormalities consisted of atlas occipital, C2–C3 fusion, and basilar impression.
All patients had hypotonia and mild to severe developmental delays. Two patients had severe language impairment and one had orofacial dyspraxia. A patient did not walk at 4 years of age and two others had learned to walk between 19 and 30 months of age. Stereotypies and sleep disturbances were noted in three patients. Hyperventilation and generalized seizures were observed in one patient. Two patients present with Chiari type I malformation visible in cerebral magnetic resonance imaging. Interestingly, the patient carrying the c.8558T>G (p.(Met2853Arg)) had a less severe phenotype with normal growth development, no gross retardation, malformation and seizures, mild morphological features, slight hypertrichosis, and mild developmental delay with walking at 19 months (Table S2).
KMT2A mRNA and protein expression in patients’ fibroblasts
To determine the consequences of KMT2A variants on gene and protein expressions we compared the mRNA and the protein levels in fibroblasts from the three patients, bearing either the c.3460C>T (p.(Arg1154Trp)) variant, the c.8558T>G (p.(Met2853Arg)) variant and the intronic variant c.11322-1G>A to four controls(one female and three males) (Fig. 3). Fibroblasts from the patient carrying the c.3581G>A ((p.Cys1194Tyr)) were not available.

a Sequence analysis of RT-PCR products from the patients (c.3460C>T (p.(Arg1154Trp)), c.8558T>G (p.(Met2853Arg)), c.11322–1G>A). In all cases, the two alleles were detected. b KMT2A mRNA expression in controls (WT) and mutated fibroblasts (c.3460C>T (p.(Arg1154Trp)), c.8558T>G (p.(Met2853Arg)), c.11322–1G>A). Y: KMT2A relative expression. Normalization factor was based on the GAPDH reference gene. Each chart represents one KMT2A relative quantification of a stimulated fibroblast culture. Errors bars represent SEM. c Western blot analysis of KMT2A protein of human control (WT1, WT2, WT3, and WT4; WT: all controls) and mutated fibroblasts (c.3460C>T (p.(Arg1154Trp)), c.8558T>G (p.(Met2853Arg)), c.11322–1G>A). GAPDH was used as loading control. Densitometry of western blotting was performed using Image J software. Data in arbitrary units (a.u.) represent the mean ± SEM of four separate experiments. *p < 0.05, **p < 0.01, ***p < 0.001 between control and KMT2A mutated fibroblasts. d Concerning the patient bearing the potential splice variant, RT-PCR using primers in exon 32 and 35 on cDNA from the patient and controls only showed one distinct band. Sequencing of the RT products showed two transcripts, the wild-type transcript and the mutated transcript with a deletion of the first nucleotide G of exon 33. This variant results in a premature stop-codon and is predicted to produce a truncated protein deleted of its C-terminal SET domain (p.Lys3775Serfs*32). This data suggests a change in the acceptor site of intron 32 which uses the mutated nucleotide A and the first G of the exon 33 to reconstitute the acceptor site
We used quantitative reverse transcriptase PCR(RT-PCR) to measure KMT2A levels in the mRNA products (Fig. 3b). The KMT2A mRNA level from the patient carrying the c.8558T>G (p.(Met2853Arg)) variant was not different compared to the controls. In contrast, the KMT2A mRNA level from the patient carrying the c.3460C>T (p.(Arg1154Trp)) variant was significantly increased to ~100% compared to the controls.
To assess the extent to which the observed increase in KMT2A mRNA levels affected the amount of protein, we quantified by western blotting KMT2A protein in fibroblasts from the patients carrying the c.3460C>T (p.(Arg1154Trp)) and the c.8558T>G (p.(Met2853Arg)) variants and from the controls. These variants, involving a single amino acid substitution, were predicted to give rise to full-length KMT2A protein. Using an antibody raised against the C-terminus of protein we detected KMT2A at the expected molecular weight of 180 kDa and at the similar level than in controls (Fig. 3c).
To investigate the molecular consequences of the intronic variant which disrupts the 3’ splice acceptor site c.11322-1G>A (NG_027813.1), we explored the mRNA splicing processing in fibroblasts obtained from the patient. Electrophoresis of the RT-PCR product spanning exon 32 to exon 35 detected a single band at the same molecular weight than the control (data not shown). Sequencing of the RT-PCR products harboring the c.11322-1G>A variant revealed the deletion of the first nucleotide of exon33 (c.11322delG) leading to a frameshift p.(Lys3775Serfs*32) (Fig. 3a–d). Quantitative RT-PCR showed a slightly (~15%) reduced level of KMT2A mRNA in this patient compared to controls (Fig. 3b). This data suggested a partial degradation of the KMT2A mRNA carrying the c.11322delG variant by the NMD mechanisms.
To test whether the KMT2A protein carrying the frameshift variant p.(Lys3775Serfs*32) was expressed, we performed western-blot using an antibody raised to the C-terminal part before the truncation. No truncated KMT2A protein (expected size ~166 kDa) was detected by western blotting (Fig. 3c; Fig. 4S) and we observed ~50% reduction of KMT2A expression in patient’s fibroblasts compared to controls (Fig. 3c).

Mapping nuclear targeting signals of wild-type (WT) and mutated KMT2A. Wild type or mutated KMT2A constructs (c.3460C>T (p.(Arg1154Trp)); c.8558T>G (p.(Met2853Arg))) were transiently transfected into COS7 cells and detected by staining with anti-MLL-1 (KMT2A) antibody. a Representative examples of typical patterns; uniform pattern and dot patterns (small dots or bigger patches absent within the nucleoli). b Distribution (% ± SEM) of the different nuclear patterns of cells expressing wild-type or mutated KMT2A constructs. Results were obtained by using data from more than 600 transfected cells of each construct in four independent experiments. The KMT2A c.3460C>T (p.(Arg1154Trp)) mutant abolishes significantly its capability to produce big dots (***χ 2 test with p < 0.0001)
KMT2A mutants have a nuclear expression
To determine the subcellular localization of the endogenous KMT2A protein, we performed immunofluorescent staining in human fibroblasts. In control fibroblasts we detected the KMT2A protein in the nucleus with no significant staining in the cytoplasm. The protein was excluded from the nucleolus and revealed a pattern of small speckles uniformly distributed in the nucleoplasm (Fig. 1S). Similar pattern was previously observed in COS, HeLa and SV80 cells [27]. In patient’s fibroblasts, the pattern was similar than in controls and consisted of small dots (Fig. 1S). The protein was present within both the nucleoli and the nucleoplasm and occasionally at the nucleus periphery. In both cases, missense and splice variants did not affect the endogenous sub-localization of KMT2A. Moreover, the H3K4me3 epigenetic mark and the SC35 protein, an exclusively nuclear splicing factor mark did not show a significant change in their subcellular distribution in patient’s fibroblasts (Fig. 2S).
Overexpression of wild-type and mutated KMT2A in COS7 cells
Taking into account the low expression of KMT2A in fibroblasts (http://www.proteinatlas.org/ENSG00000118058-KMT2A/tissue/skin) and the likely low sensitivity of immunofluorescence in these cells, we generated KMT2A expression constructs encoding the c.3460C>T (p.(Arg1154Trp)) and the c.8558T>G (p.(Met2853Arg)) KMT2A missense variants to study the impact of KMT2A variant overexpression. Overexpression of WT and mutants constructs in COS7 cells followed by anti-KMT2A immunofluorescent staining revealed a nuclear punctuate pattern for all constructs. This pattern, consisted of small dots and bigger patches, appeared in the transfected COS7 cells less uniform than the endogenous KMT2A spreading observed in human fibroblasts. No co-localization between KMT2A and SC35 was noted in these transfected cells (Fig. 3S). However, in the COS7 cells transfected with the c.3460C>T (p.(Arg1154Trp)) construct (which is located in the CXXC-type zinc finger domain), the nuclear spreading was significantly more homogenous than in the cell transfected with the c.8558T>G (p.(Met2853Arg)) (that is located in the TAD domain) and the WT constructs (Fig. 4).
HOXA9, SIX2, and MEIS1 expression are modulated by KMT2A variants
KMT2A has been shown to associate with transcriptionally active genes. In mouse embryonic fibroblasts (MEFs), ∼3% of genes require KMT2A’s methyltransferase activity for their expression. Among these genes, an abundance of transcriptional regulators, cellular signaling pathways, and signal transduction components that play key roles in regulating developmental processes was identified [28]. Those target genes included members of the clustered HOX genes, such as HOXA9, HOX coregulators, such as MEIS1, SIX2, and the cyclin-dependent kinase inhibitors CDKN2C and CDKN1B [29,30,31]. We analyzed the effect of the c.3460C>T (p.(Arg1154Trp)), the c.8558T>G (p.(Met2853Arg)) and the splice site c.11322-1G>A KMT2A variants on the mRNA expression level of five KMT2A known target genes (HOXA9, MEIS1, SIX2, CDKN2C and CDKN1B) in patient’s fibroblasts using real-time RT-PCR (Fig. 5). Mutated fibroblasts showed a significant down-regulation of SIX2 and MEIS1 expressions, an overexpression of HOXA9, but no change in both CDKN2C and CDKN1B expressions (Fig. 5).

Expression of target genes of KMT2A (CDKN2C, CDKN1B, SIX2, HOXA9 and MEIS1) in wild-type fibroblasts (WT) and in human fibroblasts bearing different KMT2A variants (patients, c.3460C>T (p.(Arg1154Trp)); c.8558T>G (p.(Met2853Arg)), c.11322–1G>A).**p < 0.001, ***p < 0.0005 between control and KMT2A mutated fibroblasts
Discussion
The WSS (OMIM 605130) is a rare severe autosomal dominant ID with no independent living, characterized by hypertrichosis cubiti, short stature, ID, and distinctive facial features (long eyeslashes, thick eyebrows, down slanted and vertically narrowed palpebral fissures, wide nasal bridge, and broad nasal tip) [24, 32]. Since the involvement of KMT2A in WSS, more than 25 different KMT2A variants have been reported. The majority of the identified variants were predicted to cause premature termination of the protein product, leading to severe reduction of protein or to a mature protein product, lacking the SET domain and thus, losing the histone-methyltransferase activity. Major role of KMT2A in neural progenitor proliferation, neuronal and glial differentiation and synaptic plasticity has been shown by data obtained in knock-out mice and zebrafish embryos [33, 34]. Until today, only five missense variants located in the N-terminal region between the two AT hooks, in the CXXC zinc finger domains and the PHD domains, have been identified: (c.838C>A, (p.(Pro280Thr)); c.3481T>G, (p.(Cys1161Gly)); c.3504G>A, (p.(Gly1168Asp)); c.3566G>A, (p.(Cys1189Tyr)), and c.4342T>C, (p.(Cys1448Arg)) [17, 20, 23, 24]. However, no functional tests were performed to assess the deleterious impact of these missense variants.
Here, we studied the KMT2A gene within a large cohort of patients with undiagnosed ID. We identified four novel de novo variants, three missense variants and one putative splice site variant. Since typical WSS were not included in this study, we do not identify KMT2A non-sense and frameshift variants. To assess the deleterious character of the newly identified KMT2A variants we studied the molecular and the cellular issues using patient’s fibroblasts.
The c.3460C>T (p.(Arg1154Trp)) and c.8558T>G (p.(Cys1194Tyr)) variants are located in the CXXC domain of KMT2A. This domain selectively binds to target genes containing unmethylated-CpG stretches. This is essential for target gene recognition, TAD and myeloid transformation in KMT2A fusion proteins. It could play a role in targeting KMT2A to active genes. This structure presents a positively charged surface groove containing a number of essential residues for the DNA binding capacity of KMT2A to CpGs [32]. Previous experiments using chemical shift perturbation analysis, cross-saturation transfer, and site-directed mutagenesis, have shown significant decrease of DNA binding by the mutant p.(Arg1154Ala) [35]. In our study, KMT2A transcript was found to be overexpressed in the patient bearing the c.3460C>T (p.(Arg1154Trp) variant. One hypothesis is that KMT2A protein may contribute to expression regulation via a possible autoregulatory mechanisms [36]. However, we cannot exclude the contribution of the genetic background of this individual. We also observed a significant reduction of SIX2 and MEIS1 expression in fibroblasts from the patient bearing the c.3460C>T (p.(Arg1154Trp)) variant, consistent with impaired DNA binding. This amino acid change reduces the surface of the groove positively charged and affects the subnuclear localization of the overexpressed mutated protein. Surprisingly, HOXA9 is up-regulated, as previously observed by the MLL-1 fusion protein [29]. Auto-regulated expression of HOXA9 may likely explain this result [37].
The de novo missense c.8558T>G (p.(Met2853Trp)) variant is located in the TAD KMT2A domain (aa2850-2858) of KMT2A. This domain appears to be important for KMT2A-mediated transcriptional activation [13]. We showed that this c.8558T>G (p.(Met2853Trp)) mutant did not affect the level of the KMT2A transcript or the subcellular localization of the protein. However, it significantly affects SIX2 and MEIS1 mRNA expression in mutant fibroblasts, confirming the role of this TAD domain for transcriptional activation. Although we observed a reduction in expression of KMT2A-mutated fibroblasts, we cannot exclude a different effect in brain. Interestingly enough, the tested missense mutants did not affect CDKN2C and CDKN1B mRNA expression in human fibroblasts, previously found to be deregulated in Mll knockout mouse embryonic fibroblasts [31]. KMT2A may not regulate the expression of the same set of genes between human and rodents. Alternatively, the relative importance of the targets depends on the species and the stage of development, since fibroblasts were isolated from either mice embryo or from human adult skin biopsies.
Finally, the c.11322-1G>A de novo variant was a putative splice site variant, which disrupted the 3′ splice acceptor site of intron 32. However, sequencing of the cDNA showed a mutated transcript with a deletion of the first nucleotide G of exon 33. This data suggests a change in the acceptor site of intron 32, which uses the mutated nucleotide A and the first G of the exon 33, to reconstitute a functional acceptor site (Fig. 3d). This variant results in a premature stop-codon. It leads to a slight reduction in the level of total KMT2A mRNA in fibroblasts, suggesting that the mutated transcript is not totally degraded through NMD mechanism and may produce a truncated protein. However, we did not detect any truncated protein in patient’s fibroblasts (Fig. 4S). Reduction of KMT2A expression in fibroblasts bearing this splice variant, suggests that the mutated protein is likely degraded during its maturation [38].
Our four patients bearing a de novo KMT2A variant, share several of the features of KMT2A-associated WSS, including mild to moderate developmental delay, severe to profound ID, distinctive facial features (such as narrow palpebral fissures) and hypertrichosis [7]. The most severe phenotype was observed in the patients bearing the c.3460C>T (p.(Arg1154Trp)) and c.3581G>A (p.(Cys1194Tyr)) missense variants located to the CXXC domain. Interestingly, all patients carrying a missense variant in the CXXC domain, presented a more severe phenotype (profound ID, feeding difficulties, short stature) compared to patients with a missense variant in other domains or a splice variant (Table 3S). Missense mutated KMT2A proteins with reduced DNA-binding capacity appear to be more deleterious than the complete loss of KMT2A activity by other mechanisms. KMT2A mislocalization observed in the CXXC missense variant may induce a dominant negative effect by either altering the interactions with nuclear proteins or affecting the wild-type protein in heterozygous patients. Furthermore, seizures an unfrequented sign in WSS was observed in the patient carrying the c.3460C>T (p.(Arg1154Trp)) variant and in only three previously reported individuals (1/3 carrying a CXXC variant and 2/24 carrying another KMT2A variant) [16, 21, 25]. This patient presented also with cardiac abnormalities as previously observed in five WSS patients and abnormal dentition [7, 18,19,20]. Previous reports showed abnormal dentitions including hypodontia in five WSS patients (2/3 carrying a CXXC variant and 3/24 carrying another KMT2A variant) [19,20,21,22,23]. In contrast, the mildest phenotype is associated with the c.8558T>G (p.(Met2853Arg)) variant. This patient presented severe ID, slight hypertrichosis, mild developmental delay with walking at 19 months, and orofacial dyspraxia. Finally, although short stature was considered as an invariant feature of WSS, one of our patients carrying the splice site variant, presented normal height and weight at the age of 10.
In this study, we showed that both variants located at the CXXC domain or the TAD domain affect the expression of some KMT2A target genes, such as HOXA9, SIX2, and MEIS1, but with different pathophysiological mechanisms (Table 4S). In the case of CXXC domain variants the DNA-binding capacities are likely reduced and the subcellular distribution of the nuclear KMT2A proteins is altered, whereas the variant in the TAD domain does not disrupt the nuclear localization, but the “transactivation” activity. Finally, the c.11322-1G>A splice variant affects slightly the KMT2A mRNA expression but significantly HOXA9, SIX2, and MEIS1 expression. This variant may produce a truncated protein rapidly and severely degraded during its maturation. As previously suggested by the identification of truncating variants, our data concerning missense variants in functional domains, confirm that loss of function is the disease-causing mechanism of KMT2A-related disorders.
In conclusion, we firstly demonstrated that the splice site variant results in a frameshift variant, leading to an instable C-terminally truncated protein. Secondly, we showed that the missense variant located in the CXXC domain, affects the subcellular distribution of the nuclear protein. Finally, we observed that all de novo variants affect transcription of several MLL-1 target genes.
References
American Psychiatric Association, American Psychiatric Association. Task force on DSM-IV and American Psychiatric Association. Task force on nomenclature and statistics. Diagnostic and statistical manual of mental disorders: DSM-IV. Washington, DC: American Psychiatric Association; American Psychiatric Press or American Psychiatric Publishing, 1994.
Srivastava AK, Schwartz CE. Intellectual disability and autism spectrum disorders: causal genes and molecular mechanisms. Neurosci Biobehav Rev 2014;46:161–74.
Brookes E, Shid Y. Diverse epigenetic mechanisms of human disease. Annu Rev Genet 2014;48:237–68.
Iwased S, Shid Y, Histone and DNA modifications in mental retardation. Prog Drug Res. 2011;67:147–73
Krumm N, O’Roak BJ, Shendure J, Eichler EE. A de novo convergence of autism genetics and molecular neuroscience. Trends Neurosci 2014;37:95–105.
Kouzarides T. Chromatin modifications and their function. Cell 2007;128:693–705.
Jones WD, Dafou D, McEntagart M, et al.. De novo mutations in MLL cause Wiedemann-Steiner syndrome. Am J Hum Genet 2012;91:358–64.
Ng SB, Bigham AW, Buckingham KJ, et al.. Exome sequencing identifies MLL2 mutations as a cause of Kabuki syndrome. Nat Genet 2010;42:790–3.
Grozeva D, Carss K, Spasic-Boskovic O, et al.. De novo loss-of-function mutations in SETD5, encoding a methyltransferase in a 3p25 microdeletion syndrome critical region, cause intellectual disability. Am J Hum Genet 2014;94:618–24.
Lederer D, Grisart B, Digilio MC, et al. Deletion of KDM6A, a histone demethylaseinteracting with MLL2, in three patients with Kabuki syndrome. Am J Hum Genet 2012;90:119–24.
Laumonnier F, Holbert S, Ronce N, et al.. Mutations in PHF8 are associated with X linked mental retardation and cleft lip/cleft palate. J Med Genet 2005;42:780–6.
Jensen LR, Amende M, Gurok U, et al.. Mutations in the JARID1C gene, which is involved in transcriptional regulation and chromatin remodeling, cause X-linked mental retardation. Am J Hum Genet 2005;76:227–36.
Cosgrove MS, Patel A. Mixed lineage leukemia: a structure-function perspective of the MLL1 protein. FEBS J 2010;277:1832–42.
Zhang P, Bergamin E, Couture JF. The many facets of MLL1 regulation. Biopolymers 2013;99:136–45.
Hsieth JJ, Ernst R, Erdjument-Bromage H, Tempst P, Korsmeyer SJ, Proteolytic cleavage of MLL generates a complex of N- and C-terminal fragments that confers protein stability and subnuclear localization. Mol Cell Biol. 2003;23:186–94
Stellacci E, Onesimo R, Bruselles A, et al.. Congenital immunodeficiency in an individual with Wiedemann-Steiner syndrome due to a novel missense mutation in KMT2A. Am J Med Genet A 2016;170:2389–93.
Calvel P, Kusz-Zamelczyk K, Makrythanasis P, et al.. A case of Wiedemann-Steiner syndrome associated with a 46,XY disorder of sexual development and gonadal dysgenesis. Sex Dev 2015;9:289–95.
Dunkerton S, Field M, Cho V, et al.. A de novo mutation in KMT2A (MLL) in monozygotic twins with Wiedemann-Steiner syndrome. Am J Med Genet A 2015;167A:2182–7.
Miyake N, Tsurusaki Y, Koshimizu E, et al.. Delineation of clinical features in Wiedemann-Steiner syndrome caused by KMT2A mutations. Clin Genet 2016;89:115–9.
Mendelsohn BA, Pronold M, Long R, Smaoui N, Slavotinek AM, Advanced bone age in a girl with Wiedemann-Steiner syndrome and an exonic deletion in KMT2A (MLL). Am J Med Genet A. 2014;164A:2079–83
Martínez F, Caro-Llopis A, Roselló M, et al. High diagnostic yield of syndromic intellectual disability by targeted next-generation sequencing. J Med Genet 2017; doi: https://doi.org/10.1136/jmedgenet-2016-103964. PubMed PMID: 27620904.
Strom SP, Lozano R, Lee H, et al.. De Novo variants in the KMT2A (MLL) gene causing atypical Wiedemann-Steiner syndrome in two unrelated individuals identified by clinical exome sequencing. BMC Med Genet 2014;15:49.
Ko JM, Cho JS, Yoo Y, Seo J, Choi M, Chae JH, Lee HR, Cho TJ, Wiedemann-Steiner syndrome with 2 novel KMT2A mutations. J Child Neurol. 2017;32:237–42
Sun Y, Hu G, Liu H, et al.. Further delineation of the phenotype of truncating KMT2A mutations: the extended Wiedemann-Steiner syndrome. Am J Med Genet A 2017;173:510–4.
Helbig KL, Farwell Hagman KD, Shinde DN, et al.. Diagnostic exome sequencing provides a molecular diagnosis for a significant proportion of patients with epilepsy. Genet Med 2016;18:898–905.
Aggarwal A, Rodriguez-Buritica DF, Northrup H. Wiedemann-Steiner syndrome: novel pathogenic variant and review of literature. Eur J Med Genet 2017;60:285–8.
Yano T, Nakamura T, Blechman J, et al.. Nuclear punctate distribution of MLL-1 is conferred by distinct elements at the N terminus of the protein. Proc Natl Acad Sci USA 1997;94:7286–91.
Wang P, Lin C, Smith ER, et al.. Global analysis of H3K4 methylation defines MLL family member targets and points to a role for MLL1-mediated H3K4 methylation in the regulation of transcriptional initiation by RNA polymerase II. Mol Cell Biol 2009;29:6074–85.
Zeisig BB, Milne T, García-Cuéllar MP, et al.. Hoxa9 and Meis1 are key targets for MLL-ENL-mediated cellular immortalization. Mol Cell Biol 2004;24:617–28.
Bina M, Wyss P, Novorolsky E, et al.. Discovery of MLL1 binding units, their localization to CpG Islands, and their potential function in mitotic chromatin. BMC Genom 2013;28:927.
Milne TA, Hughes CM, Lloyd R, et al.. Menin and MLL cooperatively regulate expression of cyclin-dependent kinase inhibitors. Proc Natl Acad Sci USA 2005;18:749–54.
Koenig R, Meinecke P, Kuechler A, Schäfer D, Müller D. Wiedemann-Steiner syndrome: three further cases. Am J Med Genet A 2010;152A:2372–5.
Jakovcevski M, Ruan H, Shen EY, et al.. Neuronal Kmt2a/Mll1 histone methyltransferase is essential for prefrontal synaptic plasticity and working memory. J Neurosci 2015;35:5097–108.
Huang YC, Shih HY, Lin SJ, et al.. The epigenetic factor Kmt2a/Mll1 regulates neural progenitor proliferation and neuronal and glial differentiation. Dev Neurobiol 2015;75:452–62.
Allen MD, Grummitt CG, Hilcenko C, et al.. Solution structure of the nonmethyl-CpG-binding CXXC domain of the leukaemia-associated MLL histone methyltransferase. EMBO J 2006;25:4503–12.
Whitman SP, Liu S, Vukosavljevic T, et al.. The MLL partial tandem duplication: evidence for recessive gain-of-function in acute myeloid leukemia identifies a novel patient subgroup for molecular-targeted therapy. Blood 2005;106:345–52.
Trivedi CM, Patel RC, Patel CV, Differential regulation of HOXA9 expression by nuclear factor kappa B (NF-kappaB) and HOXA9. Gene. 2008;408:187–95
Inturi R, Wäneskog M, Vlachakis D, et al.. A splice variant of the human phosphohistidine phosphatase 1 (PHPT1) is degraded by the proteasome. Int J Biochem Cell Biol 2014;5:69–75.
Acknowledgements
The authors thank the families for their participation to the study. They want also to thank Thierry Gaillon from the Henri Mondor Hospital molecular diagnostic lab for the technical support, and Patrick Nusbaum and Arnaud Hubas for providing primary cultures of fibroblasts. I.G. received support from La Fondation Jérôme Lejeune and from GIS - Institut des Maladies Rares.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Rights and permissions
About this article

Cite this article
Lebrun, N., Giurgea, I., Goldenberg, A. et al. Molecular and cellular issues of KMT2A variants involved in Wiedemann-Steiner syndrome. Eur J Hum Genet 26, 107–116 (2018). https://doi.org/10.1038/s41431-017-0033-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41431-017-0033-y
This article is cited by
-
Single-cell analysis reveals the spatial-temporal expression of genes associated with esophageal malformations
Scientific Reports (2024)
-
Transcriptional co-activators: emerging roles in signaling pathways and potential therapeutic targets for diseases
Signal Transduction and Targeted Therapy (2023)
-
Expanding the phenotype associated to KMT2A variants: overlapping clinical signs between Wiedemann–Steiner and Rubinstein–Taybi syndromes
European Journal of Human Genetics (2021)
-
A novel deletion mutation in KMT2A identified in a child with ID/DD and blood eosinophilia
BMC Medical Genetics (2019)
-
Histone H3 lysine K4 methylation and its role in learning and memory
Epigenetics & Chromatin (2019)
