
Abstract
Here we report an in-silico approach for identification, characterization and validation of deleterious non-synonymous SNPs (nsSNPs) in the interleukin-8 gene using three steps. In first step, sequence homology-based genetic analysis of a set of 50 coding SNPs associated with 41 rsIDs using SIFT (Sorting Intolerant from Tolerant) and PROVEAN (Protein Variation Effect Analyzer) identified 23 nsSNPs to be putatively damaging/deleterious in at least one of the two tools used. Subsequently, structure-homology based PolyPhen-2 (Polymorphism Phenotyping) analysis predicted 9 of 23 nsSNPs (K4T, E31A, E31K, S41Y, I55N, P59L, P59S, L70P and V88D) to be damaging. According to the conditional hypothesis for the study, only nsSNPs that score damaging/deleterious prediction in both sequence and structural homology-based approach will be considered as ‘high-confidence’ nsSNPs. In step 2, based on conservation of amino acid residues, stability analysis, structural superimposition, RSMD and docking analysis, the possible structural-functional relationship was ascertained for high-confidence nsSNPs. Finally, in a separate analysis (step 3), the IL-8 deregulation has also appeared to be an important prognostic marker for detection of patients with gastric and lung cancer. This study, for the first time, provided in-depth insights on the effects of amino acid substitutions on IL-8 protein structure, function and disease association.
Similar content being viewed by others

Introduction
Single nucleotide polymorphisms (SNPs) represent the most common type of genetic variation in humans1. Identification of single nucleotide polymorphisms having implications in inherited human diseases is among major challenges in human and medical genetics. Genetic variation caused by these SNPs, in particular non-synonymous SNPs (nsSNPs), occurring in protein coding regions alter the encoded amino acid at mutated site and may cause structural and functional changes in the mutated protein. However, not all such structural and functional changes due to nsSNPs are potentially damaging or deleterious. Some nsSNPs affect structural properties, while others show functional consequences. Additionally, some nsSNPs may be associated with a disease condition but others may not be related with any diseased phenotype, and are therefore, considered to be neutral. Functional consequences of any nsSNP, to a large extent, are based on attributes of the polymorphism2. Some attributes depend only on the sequence information, for example the types of residue found at the SNP location2. Therefore, it is very important to use an appropriate computational approaches and empirical rules based on probabilistic and machine learning to facilitate the discrimination of deleterious nsSNPs from neutral ones. In this work, we aimed to predict the structural and functional consequences of nsSNPs mapped in genetic variants of human interleukin-8 (IL8) gene.
The human interleukin-8 (IL-8 or CXCL8) is a pro-inflammatory chemokine that belongs to the supergene family of CXC chemokines and have associated role in inflammatory and infectious diseases3. There are two main families of chemokines; one that includes IL-8 has first two cysteine residues in the protein sequence separated by one amino acid (CXC chemokines), while in other family the first two cysteines in protein sequence are adjacent (CC chemokines)4, 5. In humans, IL-8 is expressed and secreted by various cells such as neutrophils, monocyte, endothelial and epithelial cells. These cells participate in inflammatory response after being challenged with a stimulus or signaling molecule, such as lipopolysaccharides (LPS)6, 7. The role of IL-8 is to activate inflammatory (immune) cells and amplify the inflammatory response. IL-8 accomplishes this by acting as a chemotactic factor (or chemoattractant) and call leucocytes, such as neutrophils, from the peripheral blood to the site of inflammation in tissues8. IL-8 binds to the G-protein coupled receptors, CXCR1 and CXCR2, present on the surface of neutrophils and triggers cell signaling leading to neutrophils activation. Activated neutrophils release their granular content containing lytic enzymes that help in neutralizing the pathogen. In this way, IL-8 plays a key role in neutrophil-mediated innate immune response. For instance, human cells such as epithelial cells and mononuclear cells over-express IL8 gene in response to bacterial infection, Phorbol 12–myristate 13–acetate (PMA), and LPS treatment9, 10. The over-expression results in directed migration of neutrophils to the epithelial cells as a part of innate immune response so as to discourage the pathogen invasion9, 10. Besides this, the IL8 gene has been implicated in a number of chronic inflammatory and genetic diseases, and therefore, the IL8 gene is consistently being focused in the field of human medical genetics11,12,13,14,15.
The genetic diseases caused by polymorphic IL8 genetic variants are basically an outcome of two situations. First being the altered expression of IL8 gene, which results from polymorphisms in the IL8 gene promoter region and results in turn altered binding of transcription factors onto their respective consensus sequence (transcription factor binding sites) in the IL8 gene promoter region. The altered level of expression of IL8 gene controls the level of pro-inflammation response and is linked with several disease phenotypes11,12,13,14,15. The second situation is structural changes in receptor binding sites in IL-8 protein that affect binding of IL-8 to its receptors (CXCR1 and CXCR2) present on the surface of inflammatory cells. This situation arises due to genetic polymorphisms in the IL8 gene coding region that affects IL-8 protein structure, in particular, its receptor binding site. These structural changes in receptor binding sites also influence IL-8 binding to its receptor, and as a consequence, affect IL-8 mediated cell signaling and activation of inflammatory cells. In this work, we focus on mapping and identification of non-synonymous genetic polymorphisms in the IL8 gene coding region that are expected to cause structural and functional alterations in IL-8 protein that may affect IL-8 binding to its cognate receptors present on surface of inflammatory cells.
The human IL8 gene is located on chromosome 4q12-q135. In human, the IL8 gene encodes IL-8 protein as a 99 amino acid long precursor protein, which eventually undergoes processing to form several active isoforms of IL-8 protein16, 17. The expression of IL8 gene is regulated mainly by transcription factor, namely nuclear factor-κB (NF-κB) through tumor necrosis factor (TNF) and TNF receptor associated factor 6 (TRAF6)18, 19; however, consensus sequences for some other factors have also been traced on IL8 gene promoter18. The polymorphisms in the IL8 promoter region influence binding of these transcription factors onto IL8 gene promoter region and thus affect the transcriptional expression of IL8 gene. To date most of the genetic studies focused mainly on polymorphisms identified in IL8 gene promoter region. For instance, a single nucleotide polymorphism (SNP) of T/A at 251 nucleotides position upstream to start codon (−251 T/A; rs4073) in the IL8 gene has been shown to modulate the level of IL-8 after treatment with LPS7, 20. This polymorphism in the IL8 gene has been found associated with several inflammatory diseases such as chronic and aggressive periodontitis; macular degeneration and bronchiolitis; and several type of cancers such as hepatocellular carcinoma, lung cancer, breast cancer, and gastric cancer20,21,22,23,24,25,26. Regulation of IL-8 mediated inflammatory responses against bacterial infection is a well-known etiological factor for gastric cancer27. A meta-analysis study suggested that IL-8–251 allele A > T polymorphism might be a risk factor for gastric cancer27, 28. The −251A allele susceptibility in development of low penetrance cancer was also found in a meta-analysis of 42 case control studies26. In an another case control study it was found that inflammatory bowel disease and colorectal cancer risk might be associated with polymorphism in IL-8 −251 T/A29. A cohort study in north Indian population has found an association between IL-8 −251 T/A polymorphism and risk of bladder cancer30. In a literature it was reported that the IL-8 −251 “AA” genotype and “A” allele is susceptible with higher risk for glioma31. Association between IL8 −251 T/A polymorphism and acne vulgaris infection was also found in a study population32. A cohort study suggested that SNP in promoter region of IL8 gene might be associated with an increased risk for recurrent Clostridium difficile infection. Linhartova et al. (2013) studied the relationship of four polymorphisms in IL8 gene with chronic (CP) and aggressive (AgP) periodontitis, an inflammatory disease that cause loss of connective tissue and destruction of alveolar bone10. These polymorphisms were mapped at chromosome coordinates rs2227532 (−845 T/C), rs4073 (−251 T/A), rs2227307 (+396T/G), and rs2227306 (+781C/T). The allele present in the IL8 polymorphic variants determines susceptibility to CP and AgP33. The −251TA heterozygote and +396TT homozygote genotype have been found associated with increase susceptibility to CP34, 35.
So far, most of the genetic analysis has been conducted on SNPs present in the IL8 gene promoter region and no study has been conducted on genetic analysis of SNPs in the coding region. Taking into account this consideration and the fact that IL-8 plays a central role in many infectious and inflammatory diseases, in the present study we aimed to predict the functional consequences of 50 SNPs associated with 41 rsIDs in the IL8 coding region as reported in dbSNP database. The study will provide in-depth understanding in relation to the role of nsSNP on IL-8 protein structure that may have potential role in IL-8 binding to its receptors on inflammatory cells and in disease susceptibility. In current study, we for the first time showed a strong correlation between IL-8 deregulation and the survival rate in cancer patients. Through our study, IL-8 deregulation has also appeared to be an important prognostic marker for the detection of patients with gastric and lung cancer but not for breast and ovarian cancer. This implicate that IL-8 deregulation does not impact the patient’s survival rate in case of sex or gender-specific cancer such as breast and ovarian cancer that are common among females.
Results
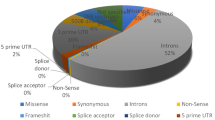
All the reported SNPs of IL8 gene were retrieved from NCBI dbSNP (http://www.ncbi.nlm.nih.gov/snp). A total of 734 SNPs were mapped in human IL8 gene sequence. The numbers of SNPs in different functional class are reported in Fig. 1. In NCBI dbSNP database, 41 rsIDs were mapped that were associated with SNPs in the coding region. We also mapped 100 SNPs in the 3′ UTR, 21 in the 5′ UTR region, 137 in the intronic regions and the rest 435 SNPs were other human active SNPs but the current study focuses only on SNPs mapped in the coding region. Since, some rsIDs showed multiple SNPs at a single nucleotide position; we recorded 50 SNPs associated with 41 rsIDs in the IL8 coding region, 3 synonymous and 47 non-synonymous (Table 1).

The pie-chart displays number of coding SNPs, 5′ UTR, 3′ UTR, intronic and other human active SNPs in human IL8 gene (based on the dbSNP database).
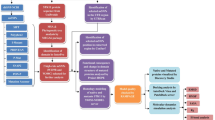
For ascertaining the structural and functional consequences of 50 SNPs on IL-8 protein structure, we used a multi-tier approach comprising of three steps. The objective of the step 1 is to collect a set of high confidence SNPs in IL8 gene. The step 1 has been performed in two sub-steps where the 50 SNPs mapped in IL8 gene are first subjected to sequence homology based SNP prediction using SIFT (Sorting Intolerant from Tolerant) and PROVEAN (Protein Variation Effect Analyzer) (step 1a); and then structural homology based SNP prediction using PolyPhen-2 (step 1b). The objective of the step 2 is to validate high confidence SNPs (identified in step 1) using several different in-silico approaches such as stability analysis of mutant proteins using ERIS (step 2a), identification of disease associated SNPs using MutPred and nsSNPAnalyzer (step 2b), association of SNPs with highly conserved buried (structural) and exposed (functional) amino acid residues in IL-8 protein using Clustal Omega and ConSurf (step 2c), and comparison of tertiary structure of the modeled mutant proteins with the wild-type so as to infer possible structural-functional consequences of nsSNPs in IL-8 protein (step 2d). Finally, in step 3 we attempt to see if there exist any correlation between IL-8 deregulation and the survival rate of patients with different cancer types (breast, gastric, lungs and ovarian cancer) using the gene expression data obtained from clinical databases such as the cancer genome atlas (TCGA) and gene expression omnibus (GEO). For this, we used Kaplan-Meier Plot analysis. A flow chart explaining different steps and approaches used in current study is depicted in Fig. 2.

An overview of the experimental design for in-silico identification/characterization (step 1), validation (step 2) of nsSNPs in IL8 gene and associating deregulation of IL8 gene with survival of cancer patients (step 3).
Prediction and collection of a set of damaging nsSNPs using sequence and structure homology based SNP prediction
The step 1 aimed to predict and collect a set of damaging nsSNPs using sequence and structure homology based prediction algorithms. The step 1 is indeed a complex step where we performed: 1) sequence homology-based damaging SNP prediction using SIFT and PROVEAN (step 1a); and 2) structural homology-based probably damaging SNP prediction using PolyPhen-2 (step 1b). Getting a set of damaging SNPs predicted using only a single approach may not always be sufficient and useful, therefore, we used two tools (SIFT and PROVEAN) in step 1a so as to collect as much as possible SNPs for subsequent analysis in step 1b. The rationale behind using two tools is that since each tool has its own threshold cut-off value for SNP classification (as damaging and non-damaging) and this may sometimes results in false prediction for a SNP that score prediction value close to the threshold cut-off value. The use of two tools (instead of one), to some extent, resolve such biases in SNP classification and prediction. Furthermore, the sequence homology based approach and the structural homology based approach of SNP prediction are used in a complimentary way, wherein sequence homology based approach predicts SNPs using genetic point of view; whereas, the structural homology based approach predicts SNPs using functional point of view. The same combination has already been used in same manner previously by many other studies conducted by other research groups36,37,38. In fact, clinical laboratories also employ in-silico prediction tools, either alone or in combination, to predict missense single nucleotide variants of uncertain pathogenicity39, 40. Using SIFT/PROVEAN (step 1a) and PolyPhen-2 (step 1b) in a complementary way is expected to provide a set of ‘high-confidence’ damaging SNPs that are common in both approaches (means ‘damaging/deleterious’ prediction in at least one of the two webservers (SIFT or PROVEAN) used in step 1a as well as ‘probably damaging’ prediction (PolyPhen-2) in step1b.
Prediction of damaging nsSNPs using SIFT and PROVEAN- sequence based approaches
Computational in-silico analysis using SIFT can predict 90% of damaging SNPs. Prediction of damaging effect of 50 SNPs mapped in genetic variants of IL8 gene was done using SIFT (http://sift.jcvi.org/). SIFT algorithm predicts damaging and tolerated (non-damaging) substitutions based on a sequence homology and physical properties of sequence submitted. The functional consequences of amino acid substitutions caused due to nsSNPs were predicted and ascertained using the respective SIFT score. In accordance with Ng and Henikoff (2003) criteria, amino acid substitutions at a given position with normalized probabilities of ≤0.05 in a tolerance index are predicted to be damaging; whereas those with normalized probabilities ≥0.05 are predicted to be tolerated41. The lower the tolerance index, the higher functional impact a particular SNP is likely to have on encoded amino acid residue, and vice versa.
Three synonymous SNPs (sSNPs) associated with rsIDs, namely rs147544998 (TGC-TGt, C61C), rs759032011 (AAG-AAa, K69K), and rs756294837 (AGG-AGa, R87R), were not included in further in-silico analysis since these do not entail any change in amino acid in protein sequence. These 3 synonymous SNPs were scored as “tolerated” in our SIFT analysis. Out of 47 nsSNPs, 8 were nonsense nsSNPs and have functional consequence either start lost or stop gain or stop lost and were also not included in further analysis. The SIFT doesn’t score tolerated or damaging status for 8 nonsense SNPs that represented either start lost (*1 V), stop gain (K30*, E31*, C61*, C77*, W84*, L93*) or stop lost (*100 L) nsSNPs. Remaining 39 nsSNPs were missense nsSNPs and were either scored as “damaging” or “tolerated”. Among the 39 missense (non-synonymous) SNPs analyzed, 20 were scored as damaging while 19 were scored as tolerated in SIFT analysis. The outcome of the SIFT server has been shown in Table 2.
In order to increase the confidence level of prediction, the estimation of deleterious effect of 50 SNPs mapped in genetic variants of IL8 gene was carried out using an additional web server namely PROVEAN (Protein Variation Effect Analyzer) (http://provean.jcvi.org). PROVEAN also works on a sequence based prediction algorithm42. The 3 synonymous SNPs that were scored as “tolerated” in SIFT analysis were scored as “neutral” in PROVEAN analysis also. PROVEAN also didn’t not scored the prediction for the 8 nonsense nsSNPs that represented start lost, stop gain, and stop lost. Out of 39 missense nsSNPs analyzed, 15 were scored as “deleterious” while 24 were scored as “neutral” in PROVEAN analysis (Table 2).
Rationale behind using two tools (SIFT and PROVEAN) in SNP prediction
The specific aim of using two web-servers for prediction of “damaging” or “deleterious” nsSNPs was to increase the confidence level of prediction analysis and to obtain a subset of nsSNPs that were predicted to be “damaging” or “deleterious” in both SIFT and PROVEAN analysis. Getting a set of damaging SNPs predicted using only a single approach may not always be sufficient and useful because some SNPs that score close to threshold cut-off value are prone to false prediction. The use of two tools, to some extent, resolve such biases in SNP classification and prediction. All such nsSNPs that are predicted to be damaging/deleterious by at least one of the two tools used were subjected to further analysis by structure homology based approach, PolyPhen-2. In this way, we increased the number of candidate nsSNPs that represent the causative mutations within the protein coding region and may have possible implication in disease phenotype. We found 12 missense nsSNPs that were scored as either “damaging” or “deleterious” in both analyses. The corresponding amino acid substitution for these missense nsSNPs in IL-8 protein sequence were L5Q, A8D, L14Q, E31K, S41Y, I55N, P59L, T64P, T64R, L70P, V88D, and V88A. However, some nsSNPs were predicted to be damaging in SIFT analysis; however, found neutral in PROVEAN and vice-versa (Supplemental Table 1). There were 11 such nsSNPs that entail amino acid substitutions such as K4T, E21D, S28R, E31A, Q35E, H45R, P59S, K69N (both AAG-AAc and AAG-AAt), E75D, and R87S. The subset of nsSNPs comprising 12 “damaging” or “deleterious” nsSNPs common to both SIFT and PROVEAN analysis and 11 nsSNPs that resulted in damaging/deleterious prediction in only one of the two analyses (SIFT and PROVEAN) were chosen for further in-silico analysis using PolyPhen-2.
Both servers (SIFT and PROVEAN) are species independent (can be used for humans as well as for other mammals such as cow, buffalo, and pig) and also gene-independent (can be used for any gene encoding a protein (UGT1A1), enzymes (cytochrome P450), transcription factors, cytokines TGF-β etc.)15, 38, 43. Similarly, other servers used by us also species and gene-independent. Therefore, using these two servers for prediction of functional consequences of non-synonymous mutation in IL8 gene seems to be scientifically logical.
Prediction of damaging missense nsSNPs using PolyPhen-2- sequence based approaches
The subset of 23 missense nsSNPs that were found “damaging” or “deleterious” and incongruent in SIFT and PROVEAN analysis were subjected to functional analysis using PolyPhen-2. The subset of 23 missense nsSNPs was subjected to PolyPhen-2 analysis to predict the damaging nsSNPs in three different categories: probably damaging, possibly damaging, and benign (means tolerant). Among the amino acid substitutions correspond to 23 missense nsSNPs analyzed, 9 amino acid substitutions (K4T, E31A, E31K, S41Y, I55N, P59L, P59S, L70P and V88D) were scored as probably damaging (score > 0.96), 10 amino acid substitutions (A8D, L14Q, E21D, S28R, H45R, T64P, T64R, K69N (both AAG-AAc and AAG-AAt), K69A and V88A) were scored as possibly damaging (score > 0.2 and <0.96), and 4 amino acid substitutions (L5Q, Q35K, E75D and R87S) was scored as benign (score < 0.2). The outcome of the PolyPhen-2 server has been shown in Table 3. The K4T substitution lies at the N-terminal region of the IL-8 protein that is lost during protein maturation. Rest 8 amino acid substitutions that were predicted to be ‘probably damaging’ in PolyPhen-2 analysis were considered as high confidence nsSNPs that may have potential structural-functional impact on IL-8 protein. These 8 high confidence nsSNPs were subjected to further validation analysis in step 2.
Validation of damaging effect of 8 high confidence nsSNPs using multiple approaches
The objective of the step 2 is to validate high confidence SNPs (identified in step 1) at using different approaches one by one (step 1- step 2a; step 1- step 2b; step 1- step 2c; step 1- step 2d) as depicted in Fig. 2. In step 2 of our work, we subjected the high confidence SNPs (identified in step 1) to four different in-silico approaches such as stability analysis of mutant proteins using ERIS (step 2a), identification of disease associated SNPs using MutPred and nsSNPAnalyzer (step 2b), association of SNPs with highly conserved buried (structural) and exposed (functional) amino acid residues in IL-8 protein using Clustal Omega and ConSurf (step 2c), and comparison of tertiary structure of the modeled mutant proteins with the wild-type so as to infer possible structural-functional consequences of nsSNPs in IL-8 protein (step 2d).
Effect of amino acid substitutions on mutant protein stability
It is widely known that the majority of disease associated nsSNPs affect the stability of the protein. We studied the effect of amino acid substitutions on the mutant protein stability using ERIS server. The server performs the structure based analysis of the mutant protein with substitution at a single amino acid residue and provides an estimation of free energy change in mutant protein with amino acid substitution at single site. The 8 amino acid substitutions, predicted to be ‘probably damaging’ in PolyPhen-2, were submitted to ERIS server to predict the ΔΔG stability (Table 4). We predicted ΔΔG with Eris using flexible backbone and with pre-relaxation setting because appropriate modeling of backbone of modeled proteins (as in flexible backbone setting) can significantly improve the free energy prediction by ERIS. We found that out of 8 mutants submitted to analysis, four mutant proteins (IL-8 E31A, IL-8 S41Y, IL-8 P59L, and IL-8 P59S) were predicted to have stabilizing effect while other four (IL-8 E31K, IL-8 I55N, IL-8 L70P, and IL-8 V88D) have destabilizing effect on the protein.
Identification of disease phenotype associated with nsSNPs using MutPred and nsSNPAnalyzer
The same subset of 8 amino acid substitutions scored as ‘probably damaging’ was subjected to in-silico phenotypic analysis using nsSNPAnalyzer. The nsSNPAnalyzer is web-tool to predict whether a nsSNPs has a disease phenotype. The nsSNPAnalyzer also provides additional useful information about the SNP to facilitate the interpretation of results, for instance, structural environment, area buried, fraction polar, and secondary structure. The analysis using nsSNPAnalyzer requires information contained in the multiple sequence alignment and information contained in the input PDB 3D protein structure to make predictions. We used the crystal structure of IL-8 protein (PDB ID: 5d14) retrieved from the Protein Data Bank. The analysis using nsSNPAnalyzer predicted L70P amino acid substitution to be associated with disease phenotype. The amino acid substitution was found associated with rsIDs, rs762899923. The outcome of the nsSNPAnalyzer server has been shown in Table 5. We compared the results obtained from the nsSNPAnalyzer and that obtained from SIFT, PROVEAN and PolyPhen-2.
For the MutPred, the same subset of 8 amino acid substitutions scored as ‘probably damaging’ was subjected to in-silico phenotypic analysis was used. The MutPred is web-tool to predict the diseased phenotype and to identify the molecular mechanisms that result amino acid substitution caused by nsSNPs44. The MutPred also provides additional useful information such as gain or loss of solvent accessibility, molecular recognition features (MoRFs), stability, catalytic sites, and post-translation modification sites etc. that further aid in the interpretation of obtained results. Scores with g-value > 0.5 and p-value < 0.05 are referred to as actionable hypotheses, whereas the scores with g-value > 0.75 and p-value < 0.05 are referred to as confident hypotheses. The g-value and p-value scores for the 8 amino acid substitutions has been shown in Table 6. In MutPred prediction, the L70P and V88D substitution showed highest g-values and lower p-values. We found that the L70P amino acid substitution was also predicted as “damaging” in SIFT, PROVEAN and PolyPhen-2 analysis and “diseased” in nsSNPAnalyzer with high score (Tables 2, 3 and 5).
Association of SNPs with highly conserved buried (structural) and exposed (functional) amino acid residues in IL-8 protein
From structural point of view, IL-8 expresses as a 99 amino acid long protein but its active and functional form contains only 72 residues that results from cleavage of first 27 amino acids at N-terminal. The monomeric unit in the three-dimensional structure reveals a highly flexible NH,-terminal region followed by three antiparallel β strands and a COOH-terminal α-helix.
Sequence based structural-functional analysis of IL-8 was performed using Clustal Omega based multiple sequence alignment analysis. For this analysis, the IL-8 protein sequence (Uniprot ID: P10145) was retrieved from Uniprot Knowledgebase. The IL-8 protein sequence was BLASTed against the UniprotKB/SwissProt entries and aligned using Clustal Omega with default settings. The results generated by the Clustal Omega tool consist of IL-8 protein sequence aligned with other phylogenetically close sequences from other organisms (Fig. 3). The results contain a colorimetric conservation score in range of 1–10. Multiple sequence alignment using Clustal Omega revealed that human IL-8 protein sequence contains a number of conserved residues and motifs. The highly conserved amino acid residues in human IL-8 protein were L5, A8, L10, A11, S16, L19, I37, P46, I49, N63, E65, I66, L70, L78, P80, and V89. There are five conserved cysteine residues, four are involved in intramolecular disulfide bridge formation. The C34 and C36 are part of the C-X-C motif and respectively form C61 and C77 disulfide bridge45. Besides this, we mapped three small conserved motifs sequences, ELR31–33, SGP57–59, and WVQ84–86 (Figs 3 and 4 panel A). The N-terminal of IL-8 protein is involved in receptor binding and includes a receptor binding site, the ELR31–33 motif45.

Amino acid alignment of human IL-8 protein (UniProt ID: P10145) along with its homologues in phylogenetically close species in mammals and fouls. Solid horizontal bars indicate conserved sequence motifs and residues with asterisk (*) mark indicate evolutionary conserved amino acids. The amino acid identities were colored according the Clustal color scheme, and the conservation index at each alignment position were provided by Jalview61.

Structural model of modeled human IL-8, wherein panel A shows two conserved sequence motifs, ELR31–33 and SGP57–59, that face each other and form a structural scaffold putatively involved in IL-8 binding to its receptor; and panel B shows the hydrophobic pocket formed of F44, F48, I49 and L70 on the surface of IL-8 protein having role in receptor binding.
We predicted 9 amino acid substitutions in IL-8 protein to be ‘probably damaging’ based on PolyPhen-2 analysis. In our PolyPhen-2 analysis, K4T, E31A, E31K, S41Y, I55N, P59L, P59S, L70P and V88D were found to be ‘probably damaging’. The impact of K4T substitution on IL-8 structure and function has not been interpreted because this residue lies in the N-terminal of IL-8 that gets cleaved off during process of mature IL-8 formation. The E31 is an important residue that form the part of highly conserved ELR31–33 motif, which act as an epitope for receptor binding. ConSurf analysis identified conserved residues in IL-8 protein and predicted residues to be exposed or buried in the IL-8 protein structure (Fig. 5). ConSurf analysis predicted E31 to be exposed and conserved residue, i.e., a functional residue. ConSurf analysis also predicted R33 to be exposed and conserved residue, i.e., a functional residue. We assume that these charged (E31 and R33) residues facilitate electrostatic interaction between IL-8 and IL-8 receptors. If so, we suggest that E31A and E31K substitutions would also entail some structural change in the ELR31–33 motif that are expected to putatively affect IL-8 binding with its receptor, i.e., its function.

Consurf analysis of human interleukin-8 protein (Uniprot ID: P10145).
Structure-based functional analysis
Next we compared the tertiary structure of the modeled mutant proteins with the wild-type so as to infer possible structural-functional consequences of nsSNPs in IL-8 protein (step 2d). We found that, the conserved ELR31–33 motif is immediately followed by conserved C-X-C motif and conserved I37 residue. These three structural features form a distinct region (a pocket) on the surface of IL-8 having role in receptor binding46. There is another distinct region, a hydrophobic pocket, in the IL-8 protein structure that comprises F44, F48, I49 and L70 and is also implicated in receptor binding (Fig. 4 panel B). Williams et al. (1996) through their mutagenesis study on IL-8 protein found that mutations in any residue belonging to these two regions (ELR31–33 epitope and hydrophobic pocket) on the surface cause no change in IL-8 protein structure but affects its receptor binding ability46. Williams et al. (1996) showed that Y40, F48, L70, S71 play important role in receptor binding46. Clark-Lewis et al. (1994) also demonstrated that G58 and P59 (residues belonging to SGP57–59 motif) are required for IL-8 activity in a functional study using a hybrid protein derived from IL-8 regions into IP10, a similar protein lacking IL-8 activity45. The C-terminal and the α-helical region spanning from N83 to E97 have no role in receptor binding45, 47.
Our structure-based approach for studying effect of amino acid substitution in IL-8 protein showed that both E31A and E31K substitution resulted in only local changes that mainly changed the orientation of N-terminal end of the mature IL-8 protein, which is putatively involved in electrostatic interaction between IL-8 and its receptors (Fig. 6). In congruent with this mutation studies such as alanine substitution of ELR31–33 motif showed that the motif is indispensable for IL-8 activity48. Chemically synthesized analogs containing single residue replacement in the ELR31–33 motif were also found to be sensitive to modification49. ELR31–33 motif also plays an important role in cancer angiogenesis50. The P59 is also an important residue that forms the part of highly conserved SGP57–59 motif implicated in receptor binding by Clark-Lewis et al. (1994)45. ConSurf analysis predicted P59 to be exposed and conserved residue, i.e., a functional residue. Our structure based functional analysis revealed that conserved ELR31–33 motif and SGP57–59 motif face each and form a structural scaffold that is presumably involved in receptor binding. Besides this, the 3D structure of IL-8 has identified the SGP57–59 motif as an essential conformation referred to as SGP57–59 turn that brings C34 and C61 in close proximity and proper geometry to allow disulfide bridge formation45 (Fig. 4 panel A). The interactive hydrophobic surface view of IL-8 visualized in Chimera 1.10.1 (Fig. 4 panel A) showed that the SGP57–59 motif is relatively exposed in the IL-8 structure (wild type) suggesting its direct involvement in IL-8 receptor binding. We found that, both P59L and P59S, substitutions entail considerable structural change in the SGP57–59 motif locally as well as the region on both side of the SGP57–59 motif suggesting that these substitutions are expected to putatively affect IL-8 binding with its receptor. L70 is an important residue and is part of hydrophobic pocket (formed by F44, F48, I49 and L70) on the IL-8 surface. ConSurf analysis predicted L70 to be buried and conserved residue, i.e., a structurally important residue. Our structure based approach revealed that L70P substitution result in high root mean square deviation in mutant protein as compared to the wild type protein. We, therefore, suggest that L70P substitution putatively entail structural change in the hydrophobic pocket formed by F44, F48, I49 and L70 (Fig. 6). Using nsSNPAnalyzer, L70P substitution has been predicted to be associated with disease phenotype. In fact, majority of disease associated nsSNPs had been found to be located in surface pockets of protein structures51.

Structural superimposition of modelled mutant proteins (in pink) on the wild type IL-8 protein (in peacock blue) using PyMOL.
We superimposed the 8 mutant IL-8 proteins onto the PDB structure of the wild type protein using SuperPose ver 1.0 server (Fig. 6). We found that the substitutions (such as E31A and E31K) in the N-terminal receptor binding ELR31–33 motif of the IL-8 protein entail local conformation changes as well as changes in the turn that harbors SGP57–59 motif, which has also been implicated in IL-8 binding with its receptor CXCR1. The substitutions such as P59L and P59S involves the proline residue which is most commonly found in turn and helps in the formation of beta-turns in protein. Substitution of proline with other amino acids is thus expected to effect the conformation of the SGP57–59 turn in IL-8 protein. This is in congruent with the high heavy chain RSMD for IL-8 P59L and IL-8 P59S obtained in structural superimposition analysis (Fig. 6). For remaining four amino acid substitutions in IL-8 proteins, i.e., S41Y, I55N, L70P and V88D, our structural analysis showed that the former two amino acid substitutions entail considerable structural change in the local structure and the latter two amino acid substitutions led to structural change in IL-8 protein, both locally and throughout the protein structure.
Molecular docking studies for studying the effect of E31A and E31K substitution on binding of IL8 to its receptor CXCR1
Based on our structure-based analysis, we hypothesized that E31A and E31K substitution may result in local changes that mainly changed the orientation of N-terminal end of the mature IL-8 protein, which is putatively involved in electrostatic interaction between IL-8 and its receptors. In order to support our hypothesis, we conducted docking studies on IL-8 E31A and IL-8 E31K mutant proteins with CXCR1 (PDB id: 2LNL). The docking was performed using ClusPro webserver using custom settings (https://cluspro.bu.edu/login.php)75. CXCR1 is a membrane receptor protein and the IL-8 is expected to bind it on the extracellular side. CXCR1 has seven transmembrane helices (TMHs); however, the transmembrane helices do not represent docking site for IL-8 protein, except the hydrophilic residues at the end of a TMH towards the extracellular side. On the contrary, the extracellular loops present between two adjacent TMHs could be the putative docking site for IL-8. Upon performing docking using default setting, we obtained mostly irrelevant docked poses in which the IL-8 wild type protein interacted with the transmembrane helices of the CXCR1. However, in a couple of poses, the C-terminal helix of IL-8 was found to interact with the extracellular side of the CXCR1. Previous studies showed that IL-8 protein contains the ELR31–33 and SGP57–59 motif that interact with CXCR145, 46. Therefore, we used custom setting in the ClusPro and performed docking in which the attraction of the ELR31–33 and SGP57–59 residues of the IL-8 for the CXCR1 was specified. In order to specify attraction, we entered the residues corresponding to ELR31–33 and SGP57–59 for the ligand IL-8 in the entry field provided in the ClusPro for all docking experiment performed.
For the IL-8 WT protein, entering the ELR31–33 and SGP57–59 residues in the attraction entry field resulted in ten most probable docked poses for each electrostatically-favored, hydrophobically-favored and Van der waals-electrostatically favored models. There was no single relevant docked pose in electrostatically favored and hydrophobically favored models suggesting that electrostatic and hydrophobic interaction alone are not sufficient for IL-8 interaction with its receptor CXCR1. However, in model that represented a combination of Van der waals- electrostatically favored models, there were six relevant poses that represented IL-8 docked onto CXCR1 on the extra-cellular side. All the six poses showed IL-8 interaction with its receptor CXCR1 through the C-terminal helix region (Fig. 7 panel A). For the IL-8 E31A mutant protein, entering the ELR31–33 and SGP57–59 residues in the attraction entry field resulted in nine out of ten docked poses to be relevant in Van der waals- electrostatically favored models (Fig. 7 panel B). This strongly suggests that E31A mutation in IL-8 has advantageous role in IL-8 interaction with its receptor CXCR1. This is in congruent with our ERIS based result wherein E31A mutation has been found to be stabilizing. On the contrary, one out of ten docked pose was also found to be relevant in electrostatically favored models (Fig. 7 panel C) and none found relevant in hydrophobically favored models. For the IL-8 E31K mutant protein, considering the ELR31–33 and SGP57–59 residues in the attraction field resulted in five out of ten docked poses to be relevant in Van der waals- electrostatically favored models (Fig. 7 panel D). This strongly suggests that E31K mutation in IL-8 does not have any beneficial role in IL-8 interaction with its receptor CXCR1. This is in congruent with our ERIS based result wherein E31K mutation has been found to be destabilizing. Additionally, there was not a single docked pose that represented relevant docking in both electrostatically favored and hydrophobically favored models. From our docking analysis it became imperative that besides electrostatic interaction, Van der waals forces may have important role in IL-8 binding with its receptor CXCR1.

The representative docking poses of IL-8 wild type (IL-8 WT) (Panel A) and mutant proteins (IL-8 E31A & IL-8 E31K) (Panel B–D) onto its receptor IL-8R1 (PDB id: 2LNL). The IL-8 WT and mutants proteins are red colored and the receptor CXCR1 is blue colored. The N- and C-terminal ends of both IL-8 WT/mutants and CXCR1 are also marked.
Clinical correlation between IL-8 deregulation and the survival rate of patients with different cancer types
In a separate analysis (step 3) in our work, we attempt to associate the deregulation in IL-8 with clinical databases so as to infer possible functional consequences of IL-8 deregulation in Cancer patients. For this, we performed the Kaplan-Meier Plot analysis based on the Affymetrix microarray gene expression data from Cancer Genome Atlas (TCGA) and Gene Expression Omnibus (GEO). In the analysis, we estimated the overall survival rate of the cancer patients (breast, gastric, lungs and ovarian cancer) with IL-8 deregulation. It is widely known that the polymorphisms in deregulation of IL8 gene render humans susceptible to inflammatory diseases (rheumatoid arthritis, inflammatory bowel diseases), cancer and visceral leishmaniasis11,12,13,14,15; however, in current study we, for the first time, associated the deregulation of the IL8 gene with the survival of the patients with gastric and lung cancer on the basis of the results from Kaplan-Meier Plot analysis. In current study, we for the first time showed a strong correlation between IL-8 deregulation and the survival rate in cancer patients. The analysis showed that IL-8 deregulation has different implications in different cancer types, for instance, in case of gastric cancer patients the high expression of IL8 gene is found to be associated with less number of patients at risk (more survival rate); whereas, in case of lung cancer patients the high expression of IL8 gene is found to be associated with more patients at risk (less survival rate) (Fig. 8). Through this study, the IL-8 deregulation has also appeared to be an important prognostic marker for the detection of patients with gastric and lung cancer but not for breast and ovarian cancer. This also implicate that IL-8 deregulation does not impact the patient’s survival rate in case of sex or gender-specific cancer such as breast and ovarian cancer that are common among females. Since, SNPs are also known to deregulate the encoded protein, we believe that the 8 nsSNPs identified in this study are expected to have functional consequences similar to as in IL-8 deregulation.

Microarray gene expression data based association of the deregulation of IL8 gene with survival of patients with different cancer types.
Discussion
There has been an explosion in the number of single nucleotide polymorphisms (SNPs) within public databases52. Single-nucleotide polymorphisms (SNPs) are considered to be the most common genetic changes that result from alterations in a single nucleotide. Among SNPs, non-synonymous SNPs (nsSNP) are associated with single amino acid substitution in the coding regions of a gene that may have the drastic effect on the structural and functional properties of the corresponding protein. These non-synonymous single nucleotide polymorphisms (nsSNPs) have been the subject of many recent studies and a large amount of data now exists in public repositories such as dbSNP53, HGVBase54 and HGMD55. The Swiss-Prot variant page and the ModSNP database provide a large resource for sequence and structure information on human protein variants56. Identification of single nucleotide polymorphisms in the coding region of a gene that have implications in inherited human diseases is the fundamental objective of research in medical genetics.
Several studies have attempted to predict the functional consequences of a nsSNP, namely whether it is disease related or neutral, based on attributes of the polymorphism. Some attributes depend only on the sequence information, for example the types of residue found at the SNP location. Single base changes in protein coding regions of DNA which lead to changes in amino acids have the potential to effect protein structure and function. In this study we focused on non-synonymous protein coding single nucleotide polymorphisms (nsSNPs), some associated with disease and others which are thought to be neutral. In our analysis, some nsSNPs are related to a disease condition but others are not associated with any change in phenotype and are regarded as neutral. A number of studies showed that the damaging/deleterious predictions scored for a particular SNP by the SIFT and PolyPhen-2 algorithms significantly correlate with the known phenotypes for that SNP based on experimental/laboratory data coming from site-directed mutagenesis studies and clinical association studies37, 40, 57. As found by other research groups, we also found that the combination of sequence homology based (step 1a) and structural homology based (step 1b) tools such as SIFT and PolyPhen-2 predict 70% of predicted damaging/deleterious nsSNPs correctly as damaging/deleterious43, 57. In current study, we have predicted the conserved amino acid residues in the IL-8 protein based on the sequence and position-specific evolutionary information using Clustal Omega and ConSurf. Structural phylogenetic analysis using ConSurf revealed that the functional residues are highly conserved in human IL-8; and most of the disease associated nsSNPs are within these conserved residues. We applied the general rule that mutations of conserved amino acid residues in a protein are deleterious while mutations of non-conserved amino acid residues are neutral. We identified the conserved amino acid residues in IL-8 protein sequence using multiple sequence alignment of IL-8 protein sequence with other phylogenetically close IL-8 sequence from other vertebrates (Fig. 3). Theoretical 3D structural models of human IL-8 were constructed using I-Tasser server and further refined and energy minimized using ModRefiner (Table 7). The structural consequences of nsSNPs can be easily ascertained by mapping nsSNPs onto the corresponding 3D structures of the wild type protein or onto the 3D structures of other proteins with high sequence similarity. If the modeled mutant proteins are different the wild type IL-8 there comes two possibilities. First is that that these mutations have destabilized the IL-8 protein or second that mutation has changed the conformation of the IL-8 protein that result in defective binding of IL-8 with its receptor CXCR1. Besides this, using stability analysis using ERIS server and docking of IL-8 wild type and mutant proteins we demonstrated that how 8 high-confidence nsSNPs affect mutant protein stability and its interaction with its receptor CXCR1. Finally, using a separate analysis (Kaplan-Meier plots) based on Affymetrix microarray gene expression data from Cancer Genome Atlas (TCGA) and Gene Expression Omnibus (GEO), we confirmed that the deregulation of IL8 gene may results drastic affect the survival rate of gastric and lung cancer patients. The results obtained from the analysis revealed that possible alteration in structure–function relationship of IL-8 protein. The mutations caused by these 8 SNPs may have similar functional consequences as seen in case of IL-8 deregulation that suggests that these 8 SNPs could possibly lead to prioritization of diseases, such as cancer.
In current study, we were able to predict a high confidence data regarding the impact of amino acid substitutions on IL-8 structure and function using solely bioinformatics and computational tools. In this study, nsSNPs with associated rsIDs such as rs138567132 (E31A), rs188378669 (E31K), rs749738011 (S41Y), rs202114642 (I55N), rs765951700 (P59L), rs373821605 (P59S), rs762899923 (L70P), and rs779068762 (V88D) were found to be high-confidence mutations. We have summarized possible structural and functional consequences of nsSNPs in IL-8 protein in Table 8. We believe that further in vitro functional studies are required to directly determine the effect of these mutant IL-8 proteins on IL-8 binding to its receptor (CXCR1 & 2) on surface of immune cells such as neutrophils. The crystal three-dimensional structures of the mutant proteins are necessary for further validation of structural changes that have occurred as a consequence of these amino acid substitutions. Besides this, in vitro functional studies are also required to directly measure the binding of mutant IL-8 protein to its receptor on immune cells such as neutrophils. Whether these amino acid modifications entail any differential change in IL-8 binding to its cognate receptors, CXCR1 and 2, needs to be studied out. Additional questions related to differences in chemotactic and exocytosis activity of IL-8 mutant proteins and wild type is also need to be addressed.
Materials and Methods
Sequence retrieval
The nucleotide sequence data on human IL8 gene was retrieved from Entrez Gene on National Centre for Biotechnology Information (NCBI) website. The amino acid sequence of human IL8 of was collected in FASTA format from UniProt knowledgebase (UniProt ID: P10145) (http://www.uniprot.org/).
Data mining and mapping of SNPs
All the reported SNPs of IL8 gene were retrieved from NCBI dbSNP (http://www.ncbi.nlm.nih.gov/snp). A total of 734 SNPs were mapped in human IL8 gene sequence. The numbers of SNPs in different functional class are reported in Fig. 1. In NCBI dbSNP database, 41 SNPs rsIDs were mapped in coding region, 100 occurred in the 3′ UTR, 21 occurred in 5′ UTR region, 137 occurred in intronic regions and the rest 435 SNPs were other human active SNPs.
Sequence homology-based prediction of damaging coding nsSNPs using SIFT
There were 50 SNPs found associated with the 41 rsIDs mapped from dbSNP database. Prediction of deleterious effect of 50 SNPs (synonymous, non-synonymous missense and non-synonymous nonsense) in genetic variants of human IL8 gene present on chromosome 4 was done using SIFT (http://sift.jcvi.org/). SIFT is an algorithm that predicts tolerable and intolerable (damaging) substitutions based on a sequence homology and physical properties of amino acids. The chromosome coordinates for each SNP were retrieved from dbSNP for SIFT analysis. The variants coordinates were first converted to Homo sapiens GRCh37 (also known as hg19) Ensembl 63 assembly/annotation version. The input format was comma separated residue based coordinate system (chromosome number, coordinate of the SNPs, orientation (1/-1), nucleotide substitution). Substitutions at a position with normalized probabilities of ≤0.05 in a tolerance index are predicted to be damaging; whereas those with normalized probabilities ≥0.05 are predicted to be tolerated41, 58.
Sequence homology-based prediction of damaging coding nsSNPs using PROVEAN
PROVEAN (Protein Variation Effect Analyzer) (http://provean.jcvi.org) is a sequence based prediction tool that estimates the effect of protein sequence variation on protein function42. The chromosome coordinates for each SNP were retrieved from dbSNP for PROVEAN analysis. The variants coordinates were first converted to Homo sapiens GRCh37 (also known as hg19) Ensembl 66 assembly/annotation version. The input format was comma separated residue based coordinate system (chromosome number, coordinate of the SNPs, nucleotide substitution). A nsSNP present in the coding region of a gene is predicted to be “deleterious” if the prediction score is below threshold value (cutoff is -2.5), and “neutral” if the predicted score is above the cutoff value.
Structural homology-based prediction of damaging coding missense nsSNPs using PolyPhen-2
PolyPhen-2 (Polymorphism Phenotyping-2) (http://genetics.bwh.harvard.edu/pph2/) is a web-based tool for annotation of coding non-synonymous SNPs. The tool employs a specific empirical rule, which comprises both physical and comparative considerations to predict the possible functional consequences of an amino acid substitution on the structure and function of a human protein. PolyPhen-2 uses query protein sequence in FASTA format as input and estimates the influence of a particular SNP or amino acid variant at a given position in query sequence59. The tool calculates the position-specific independent count (PSIC) score for every variant and calculates the score difference between variants.
Identification of conserved residues and sequence motifs
The UniProt protein sequence of human IL-8 protein was BLASTed against the UniprotKB/Swiss-Prot database in NCBI (http://blast.ncbi.nlm.nih.gov/Blast.cgi) and upto maximum 100 sequences displaying significant alignment were exported as Hit Table (CSV) files. Sequences showing more than 50% identity and E-value below 1.00E-20 were chosen for further computational analysis of conserved sequences and motifs using multiple sequence alignment with Clustal Omega60. The amino acid identities were colored according the Clustal color scheme, and the conservation index at each alignment position were provided by Jalview61.
Evolutionary phylogenetic analysis
Evolutionary conservation of amino acid in IL-8 protein was predicted by ConSurf web server62 by using a Bayesian algorithm (conservation scores: 1–4 variable, 5–6 intermediate, and 7–9 conserved)63, 64. The Clustal Omega MSA file was submitted and the conserved regions were predicted by means of coloring scheme and conservation score. Exposed and buried residues with high conservation levels were respectively scored as functional and structural residues in the human IL-8 protein sequence.
Analysis of effect of amino acid substitution on protein stability using ERIS
ERIS server (http://troll.med.unc.edu/eris/login.php) is an in-silico tool for calculating mutational effect of non-synonymous amino acids substitutions on protein stability. The PDB file of the human IL-8 protein serves as an input. ERIS provides the scores for free energy alterations ΔΔG, which is equal to difference between Gibb’s free energy of mutant IL-8 protein and wild type protein (ΔGF MT − ΔGF WT) in Kcal/mol, where ΔΔG < 0 is indicative of decrease in stability and vice-versa 65, 66.
Prediction of disease related amino acid substitution and phenotypes by MutPred and nsSNPAnalyzer
MutPred (http://mutpred.mutdb.org/) is an online server for prediction of molecular basis of the disease related amino acid substitution in a mutant protein44. It utilizes several attributes related to protein structure, function, and evolution. It uses three servers, SIFT58, PSI-BLAST67, and Pfam profiles68, 69, along with some structural disorder prediction algorithms, including TMHMM70, MARCOIL71, and DisProt72. Thus by combining the scores of all three servers, the accuracy of prediction rises to a greater extent.
The nsSNP Analyzer (http://snpanalyzer.uthsc.edu) is a web based tool to predict the phenotypic effect (disease-associated vs. neutral) of a nsSNP by using a machine learning method called RandomForest73. The nsSNPAnalyzer also extracts additional structural information from the SNP to facilitate the interpretation of results, for instance, structural environment, area buried, fraction polar, and secondary structure. The analysis using nsSNPAnalyzer requires information contained in the multiple sequence alignment and information contained in the input PDB 3D protein structure to make predictions.
Structural modeling and superimposition of wild type and mutant proteins
The three-dimensional structure models of the mutant proteins were constructed using I-Tasser (http://zhanglab.ccmb.med.umich.edu/I-TASSER/), which employs an integrated combinatorial approach comprising all three standard conventional methods for structure modeling that includes comparative sequence alignment, threading, and ab initio modeling74, and predicts protein 3D structure with almost no manual intervention. Finally, energy minimization of modeled mutant proteins was carried out using ModRefiner75, 76. This force field permits to evaluate the energy of the modeled structure as well as overhaul distorted geometries through energy minimization. The energy minimized 3D structures of mutant IL-8 were visualized in and generated by Chimera 1.10.1. The mutant proteins were superimposed onto IL-8 wild type protein and the corresponding RSMD values were generated using generated using SuperPose ver 1.0 (wishart.biology.ualberta.ca/Superpose/)77.
Molecular docking
The modeled IL-8 mutant proteins were docked onto IL-8 receptor protein, CXCR1 (PDB id: 2LNL) using ClusPro webserver (https://cluspro.bu.edu/login.php)78 using custom settings by entering residues in IL-8 and the receptor CXCR1 that we believe to participate in interaction. In order to specify attraction, we entered the residues corresponding to ELR31–33 and SGP57–59 for the ligand in the entry field provided in the ClusPro. The entries of the residues were in whitespace separated “chain-residue” format. For each docking experiment, the ClusPro generated 10 most-probable docking poses. The representative poses of each docking is represented in Fig. 7.
Kaplan-Meier plotter analysis
This tool offers an excellent meta-analysis based biomarker assessment for cancer patients (http://kmplot.com/analysis)79. This tool is capable of examining the potential effect of 54,675 genes on survival using 10,293 cancer patient’s (comprising 5,143 breast, 1,065 gastric, 2,437 lung and 1,648 ovarian cancer patients) microarray gene expression data. Kaplan-Meier Plotter uses three versions of the Affymetrix HG-U133 datasets (with 22,277 probe sets in common), and clinical data from Gene Expression Omnibus (GEO) and The Cancer Genome Atlas (TCGA) datasets. The probe used for the IL8 gene was ‘211506_s_at’. The overall survival analyses was run on 3951, 876, 1926, and 1306 breast, gastric, lungs, and ovarian cancer patients, respectively. Patient samples were split into two groups (high and low expression levels) based on the median value. These two groups of patients (high and low expression levels) for each breast, gastric, lung, and ovarian cancer were compared and the survival was assessed. For quality control, the biased arrays were excluded. The p-values below 0.05 were considered significant (Fig. 8).
References
Collins, F. S., Brooks, L. D. & Chakravarti, A. A DNA polymorphism discovery resource for research on human genetic variation. Genome Res 8, 1229–1231 (1998).
Dobson, R. J., Munroe, P. B., Caulfield, M. J. & Saqi, M. A. S. Predicting deleterious nsSNPs: an analysis of sequence and structural attributes. BMC Bioinformatics 7, 217, doi:10.1186/1471-2105-7-217 (2006).
Harada, A. et al. Essential involvement of interleukin-8 (IL-8) in acute inflammation. J Leukoc Biol 56, 559–564 (1994).
Luster, A. & Ravetch, J. Genomic characterization of a γ-interferon-inducible cytokine (IP-10) and identification of an interferon-inducible hypersensitive site. Molec Cell Biol 7, 3723–3731 (1987).
Mukaida, N., Shiroo, M. & Matsushima, K. Genomic structure of the human monocyte-derived neutrophil chemotactic factor IL-8. J Immunol 143, 1366–1371 (1989).
Remick, G. D. Interleukin-8. Crit Care Med 33, s646–s647 (2005).
Andia, D. C. et al. Genetic analysis of the IL8 gene polymorphism (rs4073) in generalized aggressive periodontitis. Arch Oral Biol 58, 211–217 (2013).
Marshall, R. M., Salerno, D., Garriga, J. & Graña, X. Cyclin T1 expression is regulated by multiple signaling pathways and mechanisms during activation of human peripheral blood lymphocytes. J Immunol 175, 6402–6411 (2005).
Hirao, Y., Kanda, T., Aso, Y., Mitsuhashi, M. & Kobayashi, I. Interleukin-8-An Early Marker for Bacterial Infection. Lab Med 31, 39–44 (2000).
Linhartova, P. B., Vokurka, J., Poskerova, H., Fassmann, A. & Holla, L. I. Haplotype analysis of interleukin-8 gene polymorphisms in chronic and aggressive periodontitis. Mediators Inflamm 2013, 342351, doi:10.1155/2013/342351 (2013).
Seitz, M., Dewald, B., Gerber, N. & Baggiolini, M. Enhanced production of neutrophil-activating peptide-1/interleukin-8 in rheumatoid arthritis. J Clin Invest 87, 463–469 (1991).
Grimm, M. C., Elsbury, S. K., Pavli, P. & Doe, W. F. 1996. Interleukin 8: cells of origin in inflammatory bowel disease. Gut 38, 90–98 (1996).
McCarron, S. L. et al. Influence of cytokine gene polymorphisms on the development of prostate cancer. Cancer Res 62, 3369–3372 (2002).
Ohyauchi, M. et al. The polymorphism interleukin 8 -251 A/T influences the susceptibility of Helicobacter pylori related gastric diseases in the Japanese population. Gut 54, 330–335 (2005).
Frade, A. F. et al. TGFB1 and IL8 gene polymorphisms and susceptibility to visceral leishmaniasis. Infect Genet Evol 11, 912–916 (2011).
Strieter, R. M. et al. The functional role of the ELR motif in CXC chemokine-mediated angiogenesis. J Biol Chem 270, 27348–27357 (1995).
Hébert, C. A. & Baker, J. B. Interleukin-8: a review. Cancer Invest 11, 743–750 (1993).
Elliott, C. L., Allport, V. C., Loudon, J. A., Wu, G. D. & Bennett, P. R. Nuclear factor-kappa B is essential for up-regulation of interleukin-8 expression in human amnion and cervical epithelial cells. Mol Hum Reprod 7, 787–790 (2001).
Manna, S. K. & Ramesh, G. T. Interleukin-8 induces nuclear transcription factor-κB through a TRAF6-dependent pathway. J Biol Chem 280, 7010–7021 (2005).
Hull, J., Thomson, A. & Kwiatkowski, D. Association of respiratory syncytial virus bronchiolitis with the interleukin 8 gene region in UK families. Thorax 55, 1023–1027 (2000).
Ren, Y. et al. Interleukin-8 serum levels in patients with hepatocellular carcinoma: correlations with clinicopathological features and prognosis. Clin Cancer Res 9, 5996–6001 (2003).
Garza-Gonzalez, E. et al. Assessment of the toll-like receptor 4 Asp299Gly, Thr399Ile and interleukin-8 −251 polymorphisms in the risk for the development of distal gastric cancer. BMC Cancer 7, 70, doi:10.1186/1471-2407-7-70 (2007).
Kamali-Sarvestani, E., Aliparasti, M. R. & Atefi, S. Association of interleukin-8 (IL-8 or CXCL8) −251 T/A and CXCR2+1208C/T gene polymorphisms with breast cancer. Neoplasma 54, 484–489 (2007).
Goverdhan, S. V. et al. Interleukin-8 promoter polymorphism -251A/T is a risk factor for age-related macular degeneration. Br J Ophthalmol 92, 537–540 (2008).
Hildebrandt, M. A. et al. Genetic variants in inflammation-related genes are associated with radiation-induced toxicity following treatment for non-small cell lung cancer. PLoS One 5, e12402, doi:10.1371/journal.pone.0012402 (2010).
Wang, N. et al. −251 T/A polymorphism of the interleukin-8 gene and cancer risk: a HuGE review and meta-analysis based on 42 case-control studies. Mol Biol Rep 39, 2831–2841, doi:10.1007/s11033-011-1042-5 (2012).
Cheng, D., Hao, Y., Zhou, W. & Ma, Y. Positive association between Interleukin-8 −251A> T polymorphism and susceptibility to gastric carcinogenesis: a meta-analysis. Cancer Cell Int 13, 100, doi:10.1186/1475-2867-13-100 (2013).
Liu, L. et al. Interleukin-8 −251 A/T gene polymorphism and gastric cancer susceptibility: a meta-analysis of epidemiological studies. Cytokine 50, 328–334 (2010).
Walczak, A. et al. The lL-8 and IL-13 gene polymorphisms in inflammatory bowel disease and colorectal cancer. DNA Cell Biol 31, 1431–1438 (2012).
Ahirwar, D. K., Mandhani, A. & Mittal, R. D. IL-8 -251 T>A polymorphism is associated with bladder cancer susceptibility and outcome after BCG immunotherapy in a northern Indian cohort. Arch Med Res 41, 97–103 (2010).
Liu, H. et al. Association between interleukin 8–251 T/A and +781 C/T polymorphisms and glioma risk. Diagn Pathol 10, 138, doi:10.1186/s13000-015-0378-x (2015).
Sabir, H. et al. Polymorphism in the IL-8 gene promoter and the risk of acne vulgaris in a Pakistani population. Iranian J Allergy Asthma Immunol 14, 443–449 (2015).
Li, G. et al. Association of matrix metalloproteinase (MMP)-1, 3, 9, interleukin (IL)-2, 8 and cyclooxygenase (COX)-2 gene polymorphisms with chronic periodontitis in a Chinese population. Cytokine 60, 552–560 (2012).
Andia, D. C. et al. Interleukin-8 gene promoter polymorphism (rs4073) may contribute to chronic periodontitis. J Periodontol 82, 893–899 (2011).
Scarel-Caminaga, R. M. et al. Haplotypes in the Interleukin 8 gene and their association with chronic periodontitis susceptibility. Biochem Genet 49, 292–302 (2011).
Flanagan, S. E., Patch, A. M. & Ellard, S. Using SIFT and PolyPhen to predict loss-of-function and gain-of-function mutations. Genet Test Mol Biomarkers 14, 533–537 (2010).
Dong, C., Wei, P., Jian, X., Gibbs, R. & Boerwinkle, E. et al. Comparison and integration of deleteriousness prediction methods for nonsynonymous SNVs in whole exome sequencing studies. Hum Mol Gen 24, 2125–2137 (2015).
Rodrigues., C., Santos-Silva, A., Costa, E. & Bronze-da-Rocha, E. Performance of in silico tools for the evaluation of UGT1A1 missense variants. Hum Mutat 36, 1215–1225 (2015).
Richards, S., Aziz, N., Bale, S., Bick, D. & Das, S. et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17, 405–424 (2015).
Karbassi, I., Maston, G. A., Love, A., Di Vincenzo, C. & Braastad, C. D. et al. A standardized DNA variant scoring system for pathogenicity assessments in Mendelian disorders. Hum Mutat 37, 127–134 (2016).
Ng, P. C. & Henikoff, S. SIFT: predicting amino acid changes that affect protein function. Nucleic Acids Res 31, 3812–3814 (2003).
Choi, Y., Sims, G. E., Murphy, S., Miller, J. R. & Chan, A. P. Predicting the functional effect of amino acid substitutions and indels. PLoS ONE 7, e46688, doi:10.1371/journal.pone.0046688 (2012).
Wang, L. L., Li, Y. & Zhou, S. F. A bioinformatics approach for the phenotype prediction of nonsynonymous single nucleotide polymorphisms in human cytochromes P450. Drug Metab Dispos 37, 977–991 (2009).
Li, B. et al. Automated inference of molecular mechanisms of disease from amino acid substitutions. Bioinformatics 25, 2744–2750 (2009).
Clark-Lewis, I., Dewald, B., Loetschef, M., Mosef, B. & Baggiolinin, B. Structural requirements for Interleukin-8 function identified by design of analogs and CXC chemokine hybrids. J Biol Chem 269, 16075–16081 (1994).
Williams, G. et al. Mutagenesis Studies of Interleukin-8. J Biol Chem 271, 9579–9586 (1996).
Clark-Lewis, I., Schumacher, C., Baggiolini, M. & Moser, B. J Biol Chem 266, 23128–23134 (1991).
Hébert, C. A., Vitangcol, R. V. & Baker, J. B. Scanning mutagenesis of interleukin-8 identifies a cluster of residues required for receptor binding. J Biol Chem 266, 18989–18994 (1991).
Clark-Lewis, I., Dewald, B., Geiser, T., Moser, B. & Baggiolini, M. Proc Natl Acad Sci USA 90, 3574–3577 (1993).
Strieter, R. M., Belperio, J. A., Roderick, J. P. & Keane, M. P. CXC chemokines in angiogenesis of cancer. Semin Cancer Biol 14, 195–200 (2004).
Stitziel, N., Binkowski, T., Tseng, Y., Kasif, S. & Liang, J. topoSNP: a topographic database of non-synonymous single nucleotide polymorphisms with and without known disease association. Nucleic Acids Res 32, D520–5222 (2004).
Sachidanandam, R., Weissman, D., Schmidt, S. C., Kakol, J. M. & Stein, L. D. A map of human genome sequence variation containing 1.42 million single nucleotide polymorphisms. Nature 409, 928–933 (2001).
Smigielski, E. M., Sirotkin, K., Ward, M. & Sherry, S. T. dbSNP: a database of single nucleotide polymorphisms. Nucleic Acids Res 28, 352–355, doi:10.1093/nar/28.1.352 (2000).
Fredman, D. et al. HGVbase: a human sequence variation database emphasizing data quality and a broad spectrum of data sources. Nucleic Acids Res 30, 387–391 (2002).
Stenson, P. D. et al. Human Gene Mutation Database (HGMD). Hum Mutat 21, 577–581 (2003).
Yip, Y. L. et al. The Swiss-Prot variant page and the ModSNP database: a resource for sequence and structure information on human protein variants. Hum Mutat 23, 464–470, doi:10.1002/humu.20021 (2004).
Wang, Z. & Moult, J. SNPs, protein structure, and disease. Hum Mutat 17, 263–270 (2001).
Kumar, P., Henikoff, S. & Ng, P. C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc 4, 1073–1081 (2009).
Ramensky, V., Bork, P. & Sunyaev, S. Human non-synonymous SNPs: server and survey. Nucleic Acids Res 30, 3894–3900 (2002).
Sievers, F. et al. Fast, scalable genera on of high-quality protein multiple sequence alignments using. Clustal Omega. Mol Syst Biol 7, 539, doi:10.1038/msb.2011.75 (2011).
Waterhouse, A. M. et al. Jalview Version 2-a multiple sequence alignment editor and analysis workbench. Bioinformatics 25, 1189–1191 (2009).
Ashkenazy, H., Erez, E., Martz, E. & Pupko, T. and Ben-Tal N. ConSurf 2010: calculating evolutionary conservation in sequence and structure of proteins and nucleic acids. Nucleic Acids Res 38, W529–533, doi:10.1093/nar/gkq399 (2010).
Mayrose, I., Graur, D., Ben-Tal, N. & Pupko, T. Comparison of site-specific rate-inference methods for protein sequences: empirical Bayesian methods are superior. Mol Biol Evol 21, 1781–1791 (2004).
Pupko, T., Bell, R. E., Mayrose, I., Glaser, F. & Ben-Tal, N. Rate4Site: an algorithmic tool for the identification of functional regions in proteins by surface mapping of evolutionary determinants within their homologues. Bioinformatics 18, S71–S77 (2002).
Yin, S., Ding, F. & Dokholyan, N. V. Eris: an automated estimator of protein stability. Nat Methods 4, 466–467 (2007).
Yin, S., Ding, F. & Dokholyan, N. V. Modeling backbone flexibility improves protein stability estimation. Structure 15, 1567–1576 (2007).
Altschul, S. F. et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25, 3389–3402 (1997).
Finn, R. D. et al. The Pfam protein families database: towards a more sustainable future. Nucleic Acids Res 44, D279–D285 (2016).
Punta, M. et al. The Pfam protein families database. Nucleic Acids Res 40, D290–D301 (2012).
Krogh, A., Larsson, B., von Heijne, G. & Sonnhammer, E. L. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol 305, 567–580 (2001).
Dolrenzi, M. & Speed, T. An HMM model for coiled-coil domains and a comparison with PSSM-based predictions. Bioinformatics 18, 617–625 (2002).
Sickmeier, M. DisProt: the database of disordered proteins. Nucleic Acids Res 35, D786–D793 (2007).
Bao, L., Zhou, M. & Cui, Y. nsSNPAnalyzer: identifying disease-associated non synonymous single nucleotide polymorphisms. Nucleic Acids Res 33, 480–482 (2005).
Roy, A., Kucukural, A. & Zhang, Y. I-TASSER: a unified platform for automated protein structure and function prediction. Nat Protoc 5, 725–738 (2010).
Xu, D. & Zhang, Y. Improving the physical realism and structural accuracy of protein models by a two-step atomic-level energy minimization. Biophys J 101, 2525–2534 (2011).
Zhang, J., Liang, Y. & Zhang, Y. Atomic-Level Protein Structure Refinement Using Fragment-Guided Molecular Dynamics Conformation Sampling. Structure 19, 1784–1795 (2011).
Maiti et al. SuperPose: a simple server for sophisticated structural superimposition. Nucleic Acid Res 32, W590–W594 (2004).
Kozakov, D. et al. The ClusPro web server for protein-protein docking. Nat Protoc 12, 255–278 (2017).
Szasz, A. M. et al. Cross-validation of survival associated biomarkers in gastric cancer using transcriptomic data of 1,065 patients. Oncotarget 7, 49322–49333 (2016).
Acknowledgements
T.C.D. acknowledges all faculty members and the research scholars of the Biosciences Department for their support and encouragement. N.V.D. acknowledges the NIH support grants R01GM11401, R01GM064803 and R01GM123247.
Author information
Authors and Affiliations
Contributions
T.C.D. conceived the idea. T.C.D. designed the experimental methodologies. T.C.D., D.K., G.D., V.Y., and A.K. performed the experiments. T.C.D. analyzed the results. T.C.D. wrote the manuscript. T.C.D., A.K. and N.V.D. revised the manuscript. All authors reviewed and approved the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Dakal, T.C., Kala, D., Dhiman, G. et al. Predicting the functional consequences of non-synonymous single nucleotide polymorphisms in IL8 gene. Sci Rep 7, 6525 (2017). https://doi.org/10.1038/s41598-017-06575-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-06575-4
This article is cited by
-
Analysis of nonsynonymous SNPs in candidate genes that influence bovine temperament and evaluation of their effect in Brahman cattle
Molecular Biology Reports (2024)
-
In silico screening of non-synonymous SNPs in human TUFT1 gene
Journal of Genetic Engineering and Biotechnology (2023)
-
A computational analysis reveals eight novel high-risk single nucleotide variants of human tumor suppressor LHPP gene
Egyptian Journal of Medical Human Genetics (2023)
-
Non-synonymous SNPs variants of PRKCG and its association with oncogenes predispose to hepatocellular carcinoma
Cancer Cell International (2023)
-
Assessment of genetic homogeneity of somatic embryo-derived plants and seed-derived plants of a robusta coffee cultivar using molecular markers and functional genes sequencing
Plant Cell, Tissue and Organ Culture (PCTOC) (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
