
Abstract
Despite major advancements in lung cancer treatment, long-term survival is still rare and a deeper understanding of molecular phenotypes would allow the identification of specific cancer dependencies and immune-evasion mechanisms. Here we performed in-depth mass-spectrometry-based proteogenomic analysis of 141 tumors representing all major histologies of non-small cell lung cancer (NSCLC). We identified six distinct proteome subtypes with striking differences in immune cell composition and subtype-specific expression of immune checkpoints. Unexpectedly, high neoantigen burden was linked to global hypomethylation and complex neoantigens mapped to genomic regions, such as endogenous retroviral elements and introns, in immune-cold subtypes. Further, we linked immune evasion with LAG-3 via STK11 mutation-dependent HNF1A activation and FGL1 expression. Finally, we develop a data-independent acquisition mass-spectrometry-based NSCLC subtype classification method, validate it in an independent cohort of 208 NSCLC cases and demonstrate its clinical utility by analyzing an additional cohort of 84 late-stage NSCLC biopsy samples.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 digital issues and online access to articles
$119.00 per year
only $9.92 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout








Similar content being viewed by others

Data availability
MS proteomics data for DDA and DIA analyses have been deposited on the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifiers PXD020191 (DDA discovery cohort), PXD020548 (DIA discovery and late-stage cohorts) and PXD025560 (DIA validation cohort). For panel sequencing, sequence data have been deposited at the European Genome–phenome Archive, which is hosted by the European Bioinformatics Institute and the Centre for Genomic Regulation, under accession number EGAS00001005482. Previously published proteomics data that were re-analyzed in this study are available in PRIDE with the identifier PXD010429, in iProx Consortium with the subproject ID IPX0001804000 and CPTAC Data Portal (https://cptac-data-portal.georgetown.edu/study-summary/S056). Previously published gene expression data that were re-analyzed here are available under accession codes GSE60645 and GSE149521 and in ArrayExpress with the identifier E-MTAB-6043. The human (Pan-Cancer Atlas and LUAD gene expression) data were derived from the TCGA Research Network at http://cancergenome.nih.gov/. The dataset derived from this resource that supports the findings of this study is available at https://gdc.cancer.gov/access-data. Previously published resources of drug sensitivity in cancer cell lines data are available at https://www.cancerrxgene.org/. Source data are provided with this paper. All other data supporting the findings of this study are available from the corresponding author upon reasonable request.
Code availability
Custom code for the classifiers (SVM-RFE and k-TSP) can be found at https://github.com/lehtiolab/Code-Availability/tree/main/Lehtio_et_al_Nature_Cancer_2021.
References
Cancer Genome Atlas Research Network. Comprehensive genomic characterization of squamous cell lung cancers. Nature 489, 519–525 (2012).
Cancer Genome Atlas Research Network. Comprehensive molecular profiling of lung adenocarcinoma. Nature 511, 543–550 (2014).
Egeblad, M., Nakasone, E. S. & Werb, Z. Tumors as organs: complex tissues that interface with the entire organism. Dev. Cell 18, 884–901 (2010).
Stewart, P. A. et al. Proteogenomic landscape of squamous cell lung cancer. Nat. Commun. 10, 3578 (2019).
Gillette, M. A. et al. Proteogenomic characterization reveals therapeutic vulnerabilities in lung adenocarcinoma. Cell 182, 200–225 (2020).
Xu, J. Y. et al. Integrative proteomic characterization of human lung adenocarcinoma. Cell 182, 245–261 (2020).
Chen, Y. J. et al. Proteogenomics of non-smoking lung cancer in east asia delineates molecular signatures of pathogenesis and progression. Cell 182, 226–244 (2020).
Branca, R. M. et al. HiRIEF LC-MS enables deep proteome coverage and unbiased proteogenomics. Nat. Methods 11, 59–62 (2014).
Zhu, Y. et al. Discovery of coding regions in the human genome by integrated proteogenomics analysis workflow. Nat. Commun. 9, 903 (2018).
Karlsson, A. et al. Gene expression profiling of large cell lung cancer links transcriptional phenotypes to the new histological WHO 2015 classification. J. Thorac. Oncol. 12, 1257–1267 (2017).
Karlsson, A. et al. Genome-wide DNA methylation analysis of lung carcinoma reveals one neuroendocrine and four adenocarcinoma epitypes associated with patient outcome. Clin. Cancer Res. 20, 6127–6140 (2014).
Arbajian, E. et al. Methylation patterns and chromatin accessibility in neuroendocrine lung cancer. Cancers https://doi.org/10.3390/cancers12082003 (2020).
Gaujoux, R. & Seoighe, C. A flexible R package for nonnegative matrix factorization. BMC Bioinf. 11, 367 (2010).
Zhu, Y. et al. DEqMS: a method for accurate variance estimation in differential protein expression analysis. Mol. Cell Proteomics 19, 1047–1057 (2020).
Yoshihara, K. et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat. Commun. 4, 2612 (2013).
Helwak, A., Kudla, G., Dudnakova, T. & Tollervey, D. Mapping the human miRNA interactome by CLASH reveals frequent noncanonical binding. Cell 153, 654–665 (2013).
Giurgiu, M. et al. CORUM: the comprehensive resource of mammalian protein complexes-2019. Nucleic Acids Res. 47, D559–D563 (2019).
Schwanhausser, B. et al. Global quantification of mammalian gene expression control. Nature 473, 337–342 (2011).
Mayr, C., Hemann, M. T. & Bartel, D. P. Disrupting the pairing between let-7 and Hmga2 enhances oncogenic transformation. Science 315, 1576–1579 (2007).
Joshi, S., Kumar, S., Ponnusamy, M. P. & Batra, S. K. Hypoxia-induced oxidative stress promotes MUC4 degradation via autophagy to enhance pancreatic cancer cells survival. Oncogene 35, 5882–5892 (2016).
Ikink, G. J., Boer, M., Bakker, E. R. & Hilkens, J. IRS4 induces mammary tumorigenesis and confers resistance to HER2-targeted therapy through constitutive PI3K/AKT-pathway hyperactivation. Nat. Commun. 7, 13567 (2016).
Campanero, M. R. & Flemington, E. K. Regulation of E2F through ubiquitin-proteasome-dependent degradation: stabilization by the pRB tumor suppressor protein. Proc. Natl Acad. Sci. USA 94, 2221–2226 (1997).
Liu, J. et al. An integrative cross-omics analysis of DNA methylation sites of glucose and insulin homeostasis. Nat. Commun. 10, 2581 (2019).
Valkovicova, T., Skopkova, M., Stanik, J. & Gasperikova, D. Novel insights into genetics and clinics of the HNF1A-MODY. Endocr. Regul. 53, 110–134 (2019).
Charoentong, P. et al. Pan-cancer Immunogenomic analyses reveal genotype-immunophenotype relationships and predictors of response to checkpoint blockade. Cell Rep. 18, 248–262 (2017).
Dou, Y. et al. Proteogenomic characterization of endometrial carcinoma. Cell 180, 729–748 (2020).
Litchfield, K. et al. Meta-analysis of tumor- and T cell-intrinsic mechanisms of sensitization to checkpoint inhibition. Cell 184, 596–614 (2021).
Sautes-Fridman, C., Petitprez, F., Calderaro, J. & Fridman, W. H. Tertiary lymphoid structures in the era of cancer immunotherapy. Nat. Rev. Cancer 19, 307–325 (2019).
Cabrita, R. et al. Tertiary lymphoid structures improve immunotherapy and survival in melanoma. Nature 577, 561–565 (2020).
Attermann, A. S., Bjerregaard, A. M., Saini, S. K., Gronbaek, K. & Hadrup, S. R. Human endogenous retroviruses and their implication for immunotherapeutics of cancer. Ann. Oncol. 29, 2183–2191 (2018).
Chong, C. et al. Integrated proteogenomic deep sequencing and analytics accurately identify non-canonical peptides in tumor immunopeptidomes. Nat. Commun. 11, 1293 (2020).
Johansson, H. J. et al. Breast cancer quantitative proteome and proteogenomic landscape. Nat. Commun. 10, 1600 (2019).
Laumont, C. M. et al. Noncoding regions are the main source of targetable tumor-specific antigens. Sci. Transl. Med. https://doi.org/10.1126/scitranslmed.aau5516 (2018).
Almeida, L. G. et al. CTdatabase: a knowledge-base of high-throughput and curated data on cancer-testis antigens. Nucleic Acids Res. 37, D816–D819 (2009).
Simpson, A. J., Caballero, O. L., Jungbluth, A., Chen, Y. T. & Old, L. J. Cancer/testis antigens, gametogenesis and cancer. Nat. Rev. Cancer 5, 615–625 (2005).
Andrews, L. P., Yano, H. & Vignali, D. A. A. Inhibitory receptors and ligands beyond PD-1, PD-L1 and CTLA-4: breakthroughs or backups. Nat. Immunol. 20, 1425–1434 (2019).
Qin, S. et al. Novel immune checkpoint targets: moving beyond PD-1 and CTLA-4. Mol. Cancer 18, 155 (2019).
Wang, J. et al. Fibrinogen-like protein 1 is a major immune inhibitory ligand of LAG-3. Cell 176, 334–347 (2019).
Wei, J., Loke, P., Zang, X. & Allison, J. P. Tissue-specific expression of B7x protects from CD4 T cell-mediated autoimmunity. J. Exp. Med. 208, 1683–1694 (2011).
Jeon, H. et al. Structure and cancer immunotherapy of the B7 family member B7x. Cell Rep. 9, 1089–1098 (2014).
Zeqiraj, E., Filippi, B. M., Deak, M., Alessi, D. R. & van Aalten, D. M. Structure of the LKB1-STRAD-MO25 complex reveals an allosteric mechanism of kinase activation. Science 326, 1707–1711 (2009).
Kim, J. et al. CPS1 maintains pyrimidine pools and DNA synthesis in KRAS/LKB1-mutant lung cancer cells. Nature 546, 168–172 (2017).
Zhang, H. M. et al. AnimalTFDB 2.0: a resource for expression, prediction and functional study of animal transcription factors. Nucleic Acids Res. 43, D76–D81 (2015).
Cancer Genome Atlas Research Network et al. The Cancer Genome Atlas Pan-Cancer analysis project. Nat. Genet. 45, 1113–1120 (2013).
Yang, W. et al. Genomics of Drug Sensitivity in Cancer (GDSC): a resource for therapeutic biomarker discovery in cancer cells. Nucleic Acids Res. 41, D955–D961 (2013).
Shackelford, D. B. & Shaw, R. J. The LKB1-AMPK pathway: metabolism and growth control in tumour suppression. Nat. Rev. Cancer 9, 563–575 (2009).
Lim, S. B., Tan, S. J., Lim, W. T. & Lim, C. T. A merged lung cancer transcriptome dataset for clinical predictive modeling. Sci. Data 5, 180136 (2018).
Ott, P. A. et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature 547, 217–221 (2017).
Smith, C. C. et al. Alternative tumour-specific antigens. Nat. Rev. Cancer 19, 465–478 (2019).
Camidge, D. R., Doebele, R. C. & Kerr, K. M. Comparing and contrasting predictive biomarkers for immunotherapy and targeted therapy of NSCLC. Nat. Rev. Clin. Oncol. 16, 341–355 (2019).
Woo, S. R. et al. Immune inhibitory molecules LAG-3 and PD-1 synergistically regulate T-cell function to promote tumoral immune escape. Cancer Res. 72, 917–927 (2012).
Parra, E. R. et al. Immunohistochemical and image analysis-based study shows that several immune checkpoints are co-expressed in non-small cell lung carcinoma tumors. J. Thorac. Oncol. 13, 779–791 (2018).
Sica, G. L. et al. B7-H4, a molecule of the B7 family, negatively regulates T cell immunity. Immunity 18, 849–861 (2003).
Azuma, T. et al. Potential role of decoy B7-H4 in the pathogenesis of rheumatoid arthritis: a mouse model informed by clinical data. PLoS Med. 6, e1000166 (2009).
Simon, I. et al. B7-h4 is a novel membrane-bound protein and a candidate serum and tissue biomarker for ovarian cancer. Cancer Res. 66, 1570–1575 (2006).
Wei, B. et al. A protein activity assay to measure global transcription factor activity reveals determinants of chromatin accessibility. Nat. Biotechnol. 36, 521–529 (2018).
Courtois, G., Morgan, J. G., Campbell, L. A., Fourel, G. & Crabtree, G. R. Interaction of a liver-specific nuclear factor with the fibrinogen and α1-antitrypsin promoters. Science 238, 688–692 (1987).
Huang, P. et al. Direct reprogramming of human fibroblasts to functional and expandable hepatocytes. Cell Stem Cell 14, 370–384 (2014).
Simeonov, K. P. & Uppal, H. Direct reprogramming of human fibroblasts to hepatocyte-like cells by synthetic modified mRNAs. PLoS ONE 9, e100134 (2014).
Xu, L. et al. The Kinase mTORC1 promotes the generation and suppressive function of follicular regulatory T cells. Immunity 47, 538–551 e535 (2017).
Halvorsen, A. R. et al. TP53 mutation spectrum in smokers and never smoking lung cancer patients. Front. Genet. 7, 85 (2016).
Lehtiö, J. et al. Mass spectrometry-based proteomic analysis of NSCLC tumor and biopsy samples. Nat. Protoc. https://doi.org/10.21203/rs.3.pex-1560/v1 (2021).
Lehtiö, J. et al. Bioinformatics analysis of NSCLC multi-omics data. Nat. Protoc. https://doi.org/10.21203/rs.3.pex-1562/v1 (2021).
Lehtiö, J. et al. Immune landscape evaluation of NSCLC clinical samples. Nat. Protoc. https://doi.org/10.21203/rs.3.pex-1565/v1 (2021).
Benjamini, Y. & Hochberg, Y. Controlling the false discovery rate - a practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B Stat. Methodol. 57, 289–300 (1995).
Wilkerson, M. D. & Hayes, D. N. ConsensusClusterPlus: a class discovery tool with confidence assessments and item tracking. Bioinformatics 26, 1572–1573 (2010).
Butler, A., Hoffman, P., Smibert, P., Papalexi, E. & Satija, R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 36, 411–420 (2018).
Blondel, V. D., Guillaume, J.-L., Lambiotte, R. & Lefebvre, E. Fast unfolding of communities in large networks. J. Stat. Mech: Theory Exp. 2008, P10008 (2008).
Yu, G., Wang, L. G., Han, Y. & He, Q. Y. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 16, 284–287 (2012).
Travaglini, K. J. et al. A molecular cell atlas of the human lung from single-cell RNA sequencing. Nature 587, 619–625 (2020).
Liberzon, A. et al. The molecular signatures database (MSigDB) hallmark gene set collection. Cell Syst. 1, 417–425 (2015).
Hanzelmann, S., Castelo, R. & Guinney, J. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinf. 14, 7 (2013).
Ogata, H. et al. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 27, 29–34 (1999).
Lehtiö, J. et al. STK11 pathway in vitro validation. Nat. Protoc. https://doi.org/10.21203/rs.3.pex-1561/v1 (2021).
Acknowledgements
DNA sequencing was performed at SciLifeLab Clinical Genomics Facility, at Stockholm; and the MS analysis was supported by SciLifelab proteogenomics facility and Karolinska University Hospital Clinical proteomics facility. We thank M. Buggert for critical reading of the immune system regulation related parts. We thank J. Lindberg and V. Wirta for expert support on DNA sequencing analysis. pBABE-FLAG-LKB1 was a gift from L. Cantley (Addgene plasmid 8592). The study was funded by The Swedish Research Council, Swedish Cancer Society, The Cancer Research Funds of Radiumhemmet, European Council H2020 financing (projects Rescuer, OncoBiome, AipBAND, DART), The Swedish Foundation for Strategic Research, The Erling-Persson Family Foundation, the Sjöberg Foundation, the Fru Berta Kamprad Foundation, Karolinska Institutet’s funding for doctoral education, BioCARE a Strategic Research Program at Lund University, Stiftelsen Jubileumsklinikens Forskningsfond mot Cancer (Gustav V:s Jubilee Foundation) and The National Health Services (Region Skåne/ALF). C.G.H. laboratory is supported by a University of Edinburgh Chancellor’s Fellowship and the Worldwide Cancer Research. K.P.P. is funded by MRC Precision Medicine DTP Studentship.
Author information
Authors and Affiliations
Contributions
The project was conceived and supervised by J.L., M. Planck, J.S. and L.M.O. Clinical data review and inclusion of patients was conducted by S.I., M.K. and M. Planck. Clinical sampling, sample prep and transcriptomics data generation was performed by M.J., A.K. and J.S. Pathological evaluation and immunohistochemistry was performed by F.S., M.J. and H.B. Clinical sampling, inclusion of patients and clinical data review for the validation cohort was performed by O.T.B., V.D.H. and Å.H. In vitro cell line experiments were coordinated and performed by O.B. and L.M.O. STK11 rescue experiments were performed by K.P.P., R.C. and C.G.H. Proteomics sample preparation, MS data generation and searching was performed by Y.P., O.B., G.M., M. Pirmoradian, H.J.J. and R.M.B. Analysis of the sequencing data was performed by T.A., I.S., H.F.A. and D.T. DNA methylation data generation and analysis was conducted by E.A. and M.A. Proteogenomics analysis was performed by I.S., H.M.U., R.M.B and L.M.O. Classification was performed by T.A. and L.M.O. Integrative downstream analyses were performed by T.A., I.S., O.B. and L.M.O. The paper was written by J.L. and L.M.O.
Corresponding author
Ethics declarations
Competing interests
J.L. has received grant funding from AstraZeneca, Roche and Novartis (not financing this manuscript). J.L. and L.M.O. are shareholders of FenoMark Diagnostics. J.L., T.A., I.S. and L.M.O. are co-inventors on a patent application related to this work. J.L. and D.T. are associated with a Roche-financed Cancer Core Europe clinical trial (not associated with this manuscript). Since completing his contribution to the current work, M. Pirmoradian has become an employee of AstraZeneca. All other authors declare no competing interests.
Additional information
Peer review information Nature Cancer thanks the anonymous reviewers for their contribution to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Consensus clustering vs NMF clustering based on proteome data in NSCLC cohort.
Consensus clustering vs NMF clustering based on proteome data in NSCLC cohort. Clustering of NSCLC based on 9,793 proteins identified and quantified across all 141 samples in the cohort. a. ConsensusClusterPlus graphic output of Cumulative Distribution Function (CDF) plot, number of clusters k = 2:11. b. ConsensusClusterPlus graphic output for relative change in area (delta area) under the CDF curve, number of clusters k = 2:11. c. Cophonetic correlation coefficient for the different choice of rank (clusters) in the non-negative matrix factorization (NMF) clustering. d. Consensus clustering index and NMF membership index across the six subtypes in the NSCLC cohort. e. Overlap of samples in subtype assignment between Consensus clustering and NMF. f. Annotated heatmap showing the results of the consensus clustering including the six identified clusters. Annotations include: Histology, mRNA subtypes1–3, Stage, Age, Sex, Smoking, Tumor cell content (‘Purity’), Immune and Stromal Signatures as described in (Yoshihara et al. 2013), TMB calculated from panel sequencing data, selected putative functional mutations from panel sequencing analysis, PD-L1 from IHC, PD-L1 from MS, KI-67 from MS, and Histological subtype markers from MS (NCAM1, KRT5, NAPSA).
Extended Data Fig. 2 Enrichments for the NSCLC Proteome Subtypes.
Enrichments for the NSCLC Proteome Subtypes. Volcano plots showing the output from enrichment tests of NSCLC mRNA subtypes (a) and AC mRNA subtypes (Proximal Inflammatory (PI), Proximal Proliferative (PP) and Terminal Respiratory Unit (TRU)) (b). P-values were calculated using one-sided hypergeometric test with Benjamini-Hochberg adjustment. c. Scatter plot indicating the expression of SqCC markers KRT5 and KRT6A across the SqCC samples in the cohort (n = 25) colored by SqCC mRNA subtype (center) and proteome subtype (border). The associated Pearson’s correlation coefficient (Rho) and two-sided p-value from t-distribution with n − 2 degrees of freedom are provided. d. Network analysis of NSCLC proteome subtypes. UMAP plots for each proteome subtype separately. Colors indicate subtype median protein level (log2) for the 5,257 proteins. e. Module enrichment analysis performed against MSigDB Hallmarks gene sets. Indicated in the figure for each module are significantly enriched gene sets (One-sided hypergeometric test, Benjamini-Hochberg adjusted p-values < 0.05). f. Module enrichment analysis performed against cell subtypes gene sets gene sets. Indicated in the figure for each module are significantly enriched gene sets (One-sided hypergeometric test, Benjamini-Hochberg adjusted p-values < 0.05). g. Boxplot indicating the tumor cell content (‘purity’, calculated based on panel sequencing data) across the NSCLC Proteome Subtypes (n = 140). Green dotted line indicates cohort median. Middle line, median; box edges, 25th and 75th percentiles; whiskers, most extreme points that do not exceed ±1.5 × the interquartile range (IQR). P-value was calculated by Kruskal-Wallis test. Dunn’s multiple comparison tests with Benjamini–Hochberg adjustment are available in Supplementary Table 3. h. Volcano plots showing mutation enrichment analysis for the six NSCLC proteome subtypes. Horizontal red and green dotted lines in all volcano plots indicate p-value=0.01. P-values were calculated using Two-sided Fisher’s exact test with Benjamini-Hochberg adjustment.
Extended Data Fig. 3 CDRP outlier regulation level analysis.
CDRP outlier regulation level analysis. a. mRNA-protein correlation for genes (n = 8,865) divided based on annotation as either miRNA targets or not according to previously published data (Helwak et al. 2013). Statistical testing was performed using two-sided Welch’s t-test (exact p-value = 1.56 × 10−19). b. mRNA-protein correlation for genes (n = 1,674 gene symbols) divided based on mRNA and protein stability as previously determined (Schwanhausser et al. 2011). Statistical testing was performed using one-way analysis of variance (ANOVA) and pairwise two-sided Welch’s t-test uncorrected for multiple testing. c. mRNA-protein correlation for genes (n = 8865 gene symbols) divided based on corresponding proteins annotation as member of a protein complex according to CORUM (Giurgiu et al. 2019). Statistical testing was performed using two-sided Welch’s t-test (exact p-value = 1.13 × 10−56). d. Scatter plot showing promoter methylation to mRNA correlation vs mRNA to protein correlation for full gene-wise overlap (n = 9,018 gene symbols). Indicated on top and to the right are the corresponding density plots. e. Same as in a. but showing only CDRPs with quantification in at least 60 samples. f. Scatter plots indicating the mRNA and protein levels of IRS2 (n = 118 samples) and HNF1A (n = 66 samples). g. Scatter plot indicating the protein levels of IRS2 and HNF1A (n = 79 samples). For boxplots (a-c): middle line, median; box edges, 25th and 75th percentiles; whiskers, most extreme points that do not exceed ±1.5 × the interquartile range (IQR). Indicated in scatter plots is the number of samples with quantitative information at both mRNA and protein level (f), or for both proteins (g), a linear regression trendline (green) and outlier expression threshold (red). The associated Pearson’s correlation coefficients (Rho) and two-sided p-values from t-distribution with n − 2 degrees of freedom are provided.
Extended Data Fig. 4 Immunohistochemistry (IHC) evaluation of selected proteins.
Immunohistochemistry (IHC) evaluation of selected proteins. a. Examples of positive (high) and negative (low) CD3, CD8 and PD-L1 determined by IHC. Images showing example stainings for the immune cell markers CD3 (left) and CD8 (center), and PD-L1 (right). Top three rows show high stromal staining of CD3 and CD8 as well as cancer cell staining of PD-L1 as exemplified from three Subtype 2 samples. Bottom three rows show examples of low/negative staining for all three proteins from proteome Subtype 1 and Subtype 5. b. Immune cell marker expression in NSCLC proteome subtypes. Scatter plots showing MS-based quantification vs stromal staining determined by IHC for CD3E (left, n = 90 samples), and CD8A (right, n = 90 samples). IHC scores were based on at least 100 cells per sample and staining. Indicated in the plots are the linear regression trendlines in green. The associated Pearson’s correlation coefficients (Rho) and two-sided p-values from t-distribution with n − 2 degrees of freedom are provided.
Extended Data Fig. 5 Tertiary lymphoid structures (TLSs) and B-cell infiltration in NSCLC proteome subtypes.
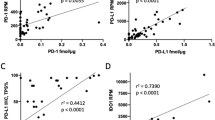
Tertiary lymphoid structures (TLSs) and B-cell infiltration in NSCLC proteome subtypes. a. Scatter plot indicating protein levels of PD-L1 vs the B-cell marker CD20 (MS4A1) in the entire NSCLC cohort (n = 141). b. Heatmap indicating mRNA expression levels of known TLS marker genes. Cohort samples are ordered as in main Fig. 1. c. Scatterplot indicating protein levels of PD-L1 vs the B-cell marker CD20 in cohort subset selected for whole section IHC evaluation (n = 19). d. TLS count (10 high power fields per sample) by subtype (n = 19 samples). e-f. IHC images showing examples of tertiary lymphoid structures from two different Subtype 3 samples (out of 11 stained samples). g. Boxplot indicating percent solid growth pattern in AC samples analyzed by whole section IHC (n = 16 samples). h. Boxplot indicating stromal signature in Subtype 2 and 3 samples analyzed by whole section IHC (n = 19 samples). i-n. IHC images showing examples of different growth patterns in six AC samples analyzed by whole section IHC (out of 16 stained samples). For boxplots: middle line, median; box edges, 25th and 75th percentiles; whiskers, most extreme points that do not exceed ±1.5 × the interquartile range (IQR). P-values in boxplots were calculated using two-sided Wilcoxon rank-sum test.
Extended Data Fig. 6 Proteogenomic analysis for detection of non-canonical peptides (NCPs) in the NSCLC cohort.
Proteogenomic analysis for detection of non-canonical peptides (NCPs) in the NSCLC cohort. a. Overview of the proteogenomic analysis. Six reading frame translation (6FT) database search was performed as previously described (Branca et al. 2014, Zhu et al. 2018) and search hits were filtered based on FDR < 1%; SpectrumAI for automatic MS2 spectrum inspection/validation of single-substitution peptide identifications; and outlier expression pattern. Resulting 651 NCPs showed low identification overlap across cohort samples indicating sample specific expression. Thirteen percent of corresponding genetic loci were supported by more than one unique peptide. b. Examples of mirror plots from NCP synthetic peptide validation for a peptide that passed the manual inspection (left) and a peptide that failed the manual inspection (right). For each example the upper part shows the annotated MS2 spectrum of the NCP identified in the original proteogenomic analysis, and the lower part shows the MS2 spectrum of the corresponding synthetic peptide. In the right figure, missing fragment ions in the spectrum of the synthetic peptide are indicated. Mirror plots of all 104 NCPs that were evaluated by synthetic peptides can be found in Supplementary Data 1. c. Pie chart indicating the results of the NCP synthetic peptide validation. d. Bar plot showing the results of the NCP synthetic peptide validation for each of the six NSCLC Subtypes. In total, the 104 NCPs evaluated were identified in 156 samples (the same NCP can be identified in several samples). e. Distribution of NCP synthetic peptide validation results per subtype indicating no statistically significant difference between subtypes. P value was calculated using two-sided Fisher’s exact test.
Extended Data Fig. 7 FGL1 and STK11 in NSCLC proteome landscape and TCGA dataset.
FGL1 and STK11 in NSCLC proteome landscape and TCGA dataset. a. Scatter plot showing protein vs mRNA level Pearson’s correlations in the NSCLC cohort for 9,244 genes where mRNA data and quantitative protein data was available for at least 70 samples. Red dotted lines indicate 5th and 95th percentiles of mRNA and protein level correlations. b. Scatterplot showing STK11 vs STRADA protein levels in NSCLC cohort colored by proteome subtype (n = 141 samples). c. Scatter plot showing STK11 vs FGL1 protein levels in NSCLC cohort colored by proteome subtype (n = 141 samples). Indicated by red circles are samples with STK11 mutations. d. Scatter plot showing protein level Pearson’s correlations in the NSCLC cohort vs mRNA level correlation in the TCGA PanCancer dataset for 10,447 genes where mRNA data and quantitative protein data were available for at least 70 samples. Red lines indicate 5th and 95th percentiles of mRNA and protein level correlations. e. Boxplots showing FGL1 (left) and CPS1 (right) mRNA levels by STK11 mutation status in the TCGA lung adenocarcinoma (LUAD) dataset (n = 504 samples). Middle line, median; box edges, 25th and 75th percentiles; whiskers, most extreme points that do not exceed ±1.5 × the interquartile range (IQR). P-values were calculated using two-sided Wilcoxon rank-sum test. f. Scatter plot showing STK11 vs FGL1 mRNA levels in the TCGA LUAD dataset colored by STK11 mutation status (n = 504 samples). g. Scatterplot showing FGL1 vs HNF1A mRNA levels in the TCGA LUAD dataset colored by STK11 mutation status (n = 504 samples). For scatter plots b, c, f, and g, linear regression trendlines are indicated in green. The associated Pearson’s correlation coefficients (Rho) and two-sided p-values from t-distribution with n − 2 degrees of freedom are provided.
Extended Data Fig. 8 Support-vector machine (SVM) and k-Top Scoring Pairs (k-TSP) based classification of NSCLC subtype.
Support-vector machine (SVM) and k-Top Scoring Pairs (k-TSP) based classification of NSCLC subtype. a. Sankey plot showing the SVM classification output from the SVM testing (100 Monte Carlo cross-validation (MCCV) iterations) with 94% accuracy. b. Stacked bar plots showing the subtype outlierness indicated by consensus index from the original clustering (top) and the classification output form the 100 MCCV iterations (bottom). Indicated by red arrows are seven samples that were frequently mis-classified by the SVM. c. DIA-MS analysis of the 141 samples resulted in the identification of 6,717 proteins (FDR < 1%) with a minimum of 2220 proteins per sample and a full overlap of 1202 proteins across all samples. Right part shows protein-wise and sample-wise correlation between DIA-MS based, and DDA-MS based quantifications. d. Selection of (k) for the k-TSP classifier was performed based on accuracy in test data, resulting in k = 13 feature pairs. e. k-TSP classifier feature pair importance evaluated by the frequency each feature pair was used across the 100 MCCV iterations. After training, the accuracy of the classifier was estimated using the test set samples. The overall accuracy was reported as the average accuracy of the 100 iterations. The 13 most frequently used feature pairs for each binary model (15 models), resulting in 195 final feature pairs, were used to build the final model. f. Sankay plot showing the classification output from the k-TSP test data (100 iterations) resulting in 87% accuracy. g. Stacked bar plots showing the subtype outlierness indicated by consensus index from the original clustering (top) and the classification output form the 100 MCCV iterations (bottom). Indicated by red arrows are 19 samples that were frequently mis-classified by the k-TSP.
Extended Data Fig. 9 SVM and k-TSP based classification of public domain AC transcriptomics and proteomics data.
SVM and k-TSP based classification of public domain AC transcriptomics and proteomics data. a. Output from SVM-based classification of the TCGA lung adenocarcinoma (LUAD) cohort based on mRNA-level data. Indicated below is sample annotation by mRNA subtype, mutation patterns and marker/signature levels. b. Kaplan-Meier plot showing overall survival in the TCGA LUAD cohort by classified subtype (n = 501 samples). P-value was calculated using log-rank test. c. Venn diagrams showing overlap between current early-stage NSCLC cohort and the Gillette et al. lung AC cohort in all identified proteins (top) and proteins with full overlap in respective cohorts (bottom). Indicated by red circle is the overlap with 250 most frequently used features from the SVM classifier optimization. d. Output from SVM-based classification of the Gillette et al. AC cohort (n = 111 samples). Indicated below is sample annotation by mRNA and protein subtype, mutation patterns and marker/signature levels. To the right, results are displayed by classified subtype including p-values from Kruskal-Wallis test (markers and signatures) or one-sided hypergeometric test with Benjamini-Hochberg adjustment (mutations). e. Output from k-TSP-based classification of the Xu et al. lung AC cohort (n = 99 samples). Indicated below is sample annotation by mutation patterns and marker/signature levels. To the right, results are displayed by classified subtype including p-values from Kruskal-Wallis test (markers and signatures) or one-sided hypergeometric test with Benjamini-Hochberg adjustment (mutations).
Extended Data Fig. 10 DIA-MS analysis and k-TSP based classification of NSCLC Validation and late-stage cohorts.
DIA-MS analysis and k-TSP based classification of NSCLC Validation and late-stage cohorts. a. DIA-MS analysis of the 208 samples in the NSCLC validation cohort resulted in the identification of 7,379 proteins (FDR < 1%), with a median number of identified proteins per sample of 3,552. b. Scatter plot showing k-TSP feature pair coverage vs number of identified proteins per sample. Red line indicate threshold for classification inclusion. c. k-TSP classifier output for the 188 samples where at least 50% of k-TSP feature pairs were covered colored by histological subgroup. d. Scatter plot indicating the levels of SqCC markers Keratin 5 (KRT5) and Keratin 6 A (KRT6A) in the SqCC subset of the NSCLC validation cohort color-coded by classified subtype as quantified by DIA-MS. e. (Left) Kaplan-Meier plot showing relapse-free survival in the NSCLC validation cohort by classified subtype (n = 171 samples). P-value was calculated using log-rank test. (Right) Pairwise statistics for relapse free survival in classified subtypes of the NSCLC validation cohort with p-values calculated by log-rank test with Benjamini-Hochberg adjustment. f. Bar plot showing the histologies of the 84 samples included in the late-stage cohort. g. Scatter plot showing mRNA and peptide yields from the sample prep of biopsy samples using Allprep kit followed by digestion, colored by biopsy type (n = 84 samples). h. Experimental setup for DIA-MS analysis of late-stage cohort samples. i. DIA MS analysis of the 84 samples resulted in the identification of 5,124 proteins (FDR < 1%), with a median number of identified proteins per sample of 2,494. j. Scatter plot showing peptide yield vs number of identified proteins per sample, colored by biopsy type (n = 84 samples). k. Scatter plot showing k-TSP feature pair coverage vs number of identified proteins per sample (n = 84 samples). Red line indicate threshold for classification inclusion. For scatter plots (b, g, and k), linear regression trendlines are indicated in green. The associated Pearson’s correlation coefficients (Rho) and two-sided p-values from t-distribution with n − 2 degrees of freedom are provided.
Supplementary information
Supplementary Information
Supplementary Figs. 1–16.
Supplementary Tables
Supplementary Tables 1–10.
Supplementary Data 1
Mirror plots from the validation of NCPs using synthetic peptides.
Source data
Source Data Fig. 1
Statistical source data.
Source Data Fig. 2
Statistical source data.
Source Data Fig. 3
Statistical source data.
Source Data Fig. 4
Statistical source data.
Source Data Fig. 5
Statistical source data.
Source Data Fig. 6
Statistical source data.
Source Data Fig. 6
Raw blot data.
Source Data Fig. 7
Statistical source data.
Source Data Fig. 8
Statistical source data.
Source Data Extended Data Fig. 1
Statistical source data.
Source Data Extended Data Fig. 2
Statistical source data.
Source Data Extended Data Fig. 3
Statistical source data.
Source Data Extended Data Fig. 4
Statistical source data.
Source Data Extended Data Fig. 5
Statistical source data.
Source Data Extended Data Fig. 6
Statistical source data.
Source Data Extended Data Fig. 7
Statistical source data.
Source Data Extended Data Fig. 8
Statistical source data.
Source Data Extended Data Fig. 9
Statistical source data.
Source Data Extended Data Fig. 10
Statistical source data.
Rights and permissions
About this article

Cite this article
Lehtiö, J., Arslan, T., Siavelis, I. et al. Proteogenomics of non-small cell lung cancer reveals molecular subtypes associated with specific therapeutic targets and immune-evasion mechanisms. Nat Cancer 2, 1224–1242 (2021). https://doi.org/10.1038/s43018-021-00259-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s43018-021-00259-9
This article is cited by
-
METTL3 drives NSCLC metastasis by enhancing CYP19A1 translation and oestrogen synthesis
Cell & Bioscience (2024)
-
HERC5 downregulation in non-small cell lung cancer is associated with altered energy metabolism and metastasis
Journal of Experimental & Clinical Cancer Research (2024)
-
The liver microenvironment orchestrates FGL1-mediated immune escape and progression of metastatic colorectal cancer
Nature Communications (2023)
-
Proteomics to study cancer immunity and improve treatment
Seminars in Immunopathology (2023)
-
Extracellular matrix profiles determine risk and prognosis of the squamous cell carcinoma subtype of non-small cell lung carcinoma
Genome Medicine (2022)
