
Abstract
This study examined androgen receptor (AR) gene amplification and protein expression in 102 matched paired hormone sensitive and resistant tumours from 51 patients. AR gene amplification and X chromosome copy number were assessed by fluorescent in situ hybridisation, and protein expression was assessed by immunohistochemistry. All tumours were stained for PSA protein expression. Significantly more tumours exhibited AR amplification following the development of hormone resistance (20%, 10 out of 49) compared to matched hormone-sensitive tumours from the same patient (2%, one out of 48) (P=0.0085). The level of AR expression was significantly higher in hormone-resistant tumours compared to matched hormone-sensitive tumours from the same patient (130, interquartile range, 55–167 vs 94.5 interquartile range, 55–120, P=0.019). AR expression levels in hormone-resistant tumours with and without AR amplification were not significantly different. However, an increase in AR expression was seen with the development of AR amplification in paired tumours. The rate of AR gene amplification and/or an increase in AR protein expression during androgen resistant is too low to wholly explain the development of androgen resistance. Alternative mechanisms for modulating the function of the AR, or other signalling pathways, must be considered as key factors in the development of hormone-resistant prostate.
Similar content being viewed by others

Main
Prostate cancer is the second most frequent cause of male cancer-related deaths (Goktas et al, 1999). Androgens regulate prostate gland growth and differentiation by binding to the androgen receptor (AR), which regulates a network of androgen responsive genes for example, PSA. Prostate cancer growth is also stimulated by androgens (Gregory et al, 1998), and can be inhibited by AR antagonists (antiandrogens) or surgical castration (Avila et al, 2001). Approximately 70–80% of prostate cancer patients treated with antiandrogens respond favourably in the first instance (Goktas et al, 1999). This effect is however, transient (Newling et al, 1997) with the majority of patients developing androgen resistance (Newling et al, 1997). The mechanisms involved with the development of resistance are poorly understood, but AR mutations (Avila et al, 2001), AR amplification (Visakorpi et al, 1995), increased AR expression (Gregory et al, 1998) and activation of the AR by interaction with other signalling pathways have been implicated (Culig et al, 1998).
Androgen receptors are present in all epithelial cells of the prostate. Downregulation of AR expression during prostate cancer progression (Segawa et al, 2001) and increased expression with the development of hormone refractory tumours (Trapman and Cleutjens, 1997; Latil et al, 2001; Linja et al, 2001) have both been reported. AR expression has been reported to predict which patients will respond to hormone therapy and to correlate with tumour grade, stage and progression-free survival (Trapman and Cleutjens, 1997; Koivisto and Helin, 1999; Linja et al, 2001) . It has been postulated that AR protein expression is increased due to AR amplification by a gene dosage effect resulting in the development of androgen resistance. AR gene copy number has been demonstrated to correlate positively with PSA expression (Koivisto and Helin 1999). Polysomy of chromosome X ranges from 42 to 60% and amplification of the AR gene ranges from 20 to 30% in hormone-resistant prostate tumours (Koivisto et al, 1997; Bubendorf et al, 1999; Miyoshi et al, 2000; Edwards et al, 2001). However, there is still no clear investigation into AR gene amplification and AR protein expression in hormone-sensitive and hormone-resistant prostate cancer, using paired tumours from the same patient.
Materials and methods
Patients
A total of 51 patients (102 matched tumours) were retrospectively selected for analysis; all tumours had patient identification removed including block number and hospital number and were coded in order to make the database anonymous. In 20 cases, AR fluoresence in situ hybridisation results only have been previously reported (Edwards et al, 2001). Ethical approval was obtained from the multicentre research ethics committee (MREC) for Scotland and the appropriate local research and ethical committees (LREC) for use of matched hormone-sensitive and hormone-resistant tumours in our study. All patients received conventional androgen deprivation therapy (orchidectomy, antiandrogens or androgen ablation therapy). Patients were selected for analysis if they initially responded to treatment (response was defined by prostate-specific antigen (PSA) levels falling by at least 50%), but subsequently relapsed. Patients were classed as having hormone escaped cancer when sustained rising PSA levels were noted and were selected for study if a post-hormone relapse sample was available. The initial tumour sample was either a transurethral resection of the prostate (TURP) or a transrectal ultrasound-guided biopsy (TRUS); however, the relapsed tumour sample was always a TURP which was performed to treat clinical symptoms. PSA values and full clinical follow-up was available for each patient, tumours were assigned Gleason Scores by a single pathologist (KMG).
Fluorescence in situ hybridisation
Sections (5 μm) cut from archival formalin-fixed, paraffin-embedded tissue were placed on aminopropyltriethoxysilane (silane)-treated slides. The slides were pretreated on a VP2000 robotic slide processor (Vysis, UK, Ltd), as previously described14 using dual labelling (X chromosome, Spectrum Green™ labelled CEP X, Vysis, UK, Ltd and AR, Spectrum Orange™ labeled probe locus Xq11-13, Vysis, UK, Ltd).
The normal range for X chromosome and AR copy number were identified using the mean chromosomal copy number from 14 benign prostatic hyperplasia (BPH). Amplification was defined as an AR : X ratio greater than 1.5 (Edwards et al, 2001).
Immunohistochemistry (IHC)
Prostate-specific antigen and AR protein expression was determined in paraffin-embedded tissue sections by IHC with a standard immunoperoxidase procedure. Antigen retrial was performed by microwaving under pressure for 5 min in TE buffer. A purified immunoglobulin fraction of either a rabbit antiserum to human PSA (A 0562, DAKO, Denmark 2 μg ml−1) or AR (NCL-AR-2F12, Vector, UK, 1 μg ml−1) was used as the primary antibodies. In each case, an isotype-matched antibody was used as a negative control in which no staining was observed. Positive control sections were included in each IHC assay. Bound antibody was visualised by the streptavidin–biotin (ABC kit, Vector Labs, UK) method including diaminobenzidine as a chromogen (DAKO).
Staining was scored blind by two independent observers using a weighted histoscore method (Fraser et al, 2003), interclass correlation coefficients (ICCC) were calculated and confirmed acceptable correlation between observer scores (Armitage and Berry, 1995). ICCC values for histoscores were 0.67 (good) and 0.91 (excellent) for PSA and AR staining, respectively (Armitage and Berry, 1995). Staining was defined as increased or decreased only in those samples with a difference between paired samples of greater than 2 × the mean observer variation for the parameter under investigation.
Statistics
The Fisher's exact test was used to compare FISH results pre- and post-hormone resistance. Prostate-specific antigen protein expression and AR protein expression are shown as median and interquartile ranges. Paired Student's t-tests were used to compare PSA and AR expression between pre- and post-hormone-resistant tumours. Wilcoxon signed-rank test was used to compare Gleason sum between paired hormone-sensitive and hormone-resistant tumours. Comparisons between the resistant tumours with and without AR gene amplification were made with the Mann–Whitney test and χ2 test for linear trends. Correlations between AR gene copy number and PSA MHS (mean histoscore) and AR MHS or PSA MHS and AR MHS and Gleason sum were calculated using the Spearman rank test.
Results
Patient information
Of the 51 patients analysed, seven had bone metastases at the time of initial hormone therapy: 102 tumours were analysed. Gleason sum in hormone-sensitive tumours was significantly lower than in hormone-resistant tumours (P<0.001) (Table 1), although some commentators question the value of Gleason scores in hormone-relapsed disease. Mean patient age at diagnosis was 69 (41–83) years, and median follow-up was 1669 days (interquartile range, 1062–2428). Median time to hormone relapse was 899 (interquartile range, 537–1689).
FISH results
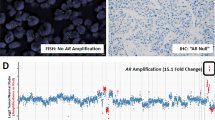
Figure 1 illustrates an example of a prostate cancer tumour with AR gene amplification. A total of 97 tumours were analysed by FISH (48 hormone-sensitive and 49 hormone-resistant tumours, five cases were not analysed as we were unable to successfully FISH them as they were fixed in Bouins). Significantly more hormone-resistant tumours had AR amplification (20%, 10 out of 49) than hormone-sensitive tumours (2%, one out of 48) (P=0.0085) (Table 2). In the amplified tumours, the median AR : X chromosome ratio was 3.11 (interquartile range, 2.4–5.8). In 22 patients, no abnormalities of either the X chromosome or AR copy number were detected in either the biopsy taken before or after hormone relapse. In all, 18 (38%) hormone-sensitive tumours and 23 (47%) hormone-relapsed tumours (P=0.46) had increased copies of the X chromosome (Table 2). A total of 15 (31%) hormone-sensitive tumours and 25 (51%) hormone-relapsed tumours (P=0.075) showed evidence of increased AR gene copies (Table 2). There was no significant difference between the level of gene or chromosome copy numbers with the development of hormone refractory disease. Gleason sum was not significantly different in hormone-resistant tumours with and without AR amplifications (Table 3). There was no significant difference in time to relapse in patients with AR amplifications (median 1143, interquartile range, 230–1731) compared to those without AR amplifications (median 857, interquartile range 541–1709).

A prostate cancer tumour with AR gene amplification is shown. AR amplification in prostate cancer nuclei (stained with DAPI, blue) showing increased copies of both AR (red) and chromosome X (green). Magnification × 1000.
IHC results
Figure 2 illustrates an example of AR protein expression in hormone-resistant prostate tumours. The median AR histoscore was 108 (interquartile range, 55–153), the level of AR expression was significantly higher in hormone-resistant tumours compared to matched hormone-sensitive tumours (median 130, interquartile range, 55–167 vs median 94.5 interquartile range, 55–120, P=0.019) (Table 2) with overlap between groups. There was no significant difference in time to relapse in patients where AR expression increased (median 907, interquartile range 485–1653) compared to those where AR expression was unchanged or decreased (median 836, interquartile range, 496–1914).

A prostate cancer tumour that expresses AR protein is shown. AR protein expression is coloured brown and is present in the tumour cell nuclei. Magnification × 400.
Four samples did not express AR protein. In one of the hormone-resistant cases, the previous hormone-sensitive biopsy showed both AR amplification (2.9) and a mean AR histoscore of 140. Following the development of androgen resistance, the AR amplification ratio did not change (3.09); however, no AR expression was detected in either of the two post-treatment biopsies from this patient. PSA expression remained unchanged in the transition from hormone-sensitive to hormone-resistant disease (Table 2).
AR expression levels in hormone-resistant tumours with and without AR amplification are not significantly different (Table 4). However, an increase in AR expression was seen in 80% (eight out of 10) of the cases (Figure 3A) with AR amplification compared to only 35% of cases without AR amplification (Figure 3B). Using Spearman's rank correlation, AR expression in hormone-sensitive and hormone-resistant prostate cancer did not correlate with either Gleason sum or PSA expression.

(A) The AR protein mean histoscore for matched hormone-sensitive and hormone-resistant tumours with AR amplification is shown. The cases shown with unbroken lines are those cases that developed AR amplification in the transition from hormone-sensitive to hormone-resistant disease. All cases had an increase in AR protein expression with the development of AR amplification, However, only eight out of the nine cases significantly increased. The case shown with the broken line had AR gene amplification in the hormone-sensitive and hormone-resistant tumour. This case had a significant decrease in AR protein expression with the development of hormone resistance. (B) The AR protein mean histoscore for 41 matched hormone-sensitive and hormone-resistant tumours with out AR amplification are shown. In 14 out of 41 (35%) cases, AR expression increased markedly with the development of hormone relapse.
All tumours included in this study expressed PSA. The median PSA histoscore was 146 (range=108–167), the level of PSA expression was significantly lower in hormone-resistant tumours compared to matched hormone-sensitive tumours (P=0.018; Table 2).
Discussion
Androgen receptor gene amplifications are uncommon in hormone-sensitive tumours and are present in 20–30% of hormone-resistant tumours (Visakorpi et al, 1995; Koivisto et al, 1997; Bubendorf et al, 1999; Miyoshi et al, 2000; Edwards et al, 2001). In the present study, we investigated AR amplification, AR protein expression and PSA expression in paired hormone-sensitive and hormone-resistant tumours from the same patient with documented initial responses to androgen deprivation therapy. We confirm here that a significant increase in AR gene amplification rates is seen in the transition from hormone-sensitive to hormone-resistant disease (P=0.0085); however, this is only observed in a proportion of cases. Based on our results and those from previous studies between 70 and 80% of cases with hormone relapse must therefore involve mechanisms other than amplification of the AR. While patients with AR gene amplification may represent a readily identifiable, and possibly treatable, group more research should be focused on alternative mechanisms of hormone escape. This is especially relevant as we have also identified a patient with AR amplification in the hormone-sensitive tumour, which responds fully to therapy, suggesting that AR amplification does not preclude a response to androgen deprivation therapy. In the light of this observation it is possible, though at present unproven and to our view unlikely, that AR amplification does not mediate hormone resistance.
It is hypothesised that AR gene amplification is involved in the development of hormone-resistant prostate cancer due to amplification, resulting in an increase in AR protein expression (Visakorpi et al, 1995). Real-time RT–PCR demonstrated that even one additional copy of the AR gene may increase AR expression (Linja et al, 2001), suggesting that even a small increase in relative gene dosage could have biological significance. However, in the study by Linja no correlation between AR mRNA and protein levels was performed. Similarly in a separate study, Latil et al, (2001) also describe increased AR mRNA levels in hormone escaped tumours. Both studies are important in that they demonstrate increases in AR mRNA in prostate cancers. However, neither study documents protein expression or makes use of paired samples from individual patients. An increase in AR protein expression is postulated to enable low circulating levels of androgens that are present following orchidectomy or treatment with LHRH agonists, to activate the AR even in the presence of antiandrogens (Koivisto et al, 1997). We therefore measured AR protein expression as well as AR amplification status in our patient cohort. As we have a unique data set of matched hormone-sensitive and hormone-resistant tumours from each patient and therefore enabling us to follow AR protein expression with the development of resistance in the same patient and relate this to AR amplification status, stringent quality controls (see above) were included to validate IHC results. Our study confirms that AR expression increases as the disease progresses to its hormone refractory state (P=0.019) and that 80% of cases in which AR amplification was observed also exhibited an increase in AR expression. However, an increase in AR expression was also seen in 35% of cases that did not develop AR amplification. This suggests that although an increase in AR expression is associated with AR amplification, an increase in expression may also be due to alternative mechanisms, for example, decrease in protein degradation or an increase in protein stabilisation. Almost half of all patients do not show any increase in AR expression during the development of hormone-resistant disease. We also identified a patient with AR amplification in both hormone-sensitive and hormone-resistant tumours and three cases with loss of AR protein expression at the development of hormone resistance, as observed by Gnanapragasam et al, (2002). Therefore, increased androgen receptor expression, by whatever mechanism, can at best only explain hormone resistance in a subgroup of prostate cancer cases. Alternative mechanisms of hormone resistance are activation of AR via phosphorylation (Zhu and Liu 1997; Lin et al, 2001), therefore increasing AR activity without effecting its expression levels or may bypass the AR, for example, AP-1 phosphorylation (Henttu and Vihko, 1998; Krishna et al, 2002).
While PSA expression was significantly lower in hormone-sensitive tumours compared to hormone-resistant tumours (P=0.018), this decrease was marginal with considerable overlap between values for pre- and post-hormone-resistant tumours (Table 1). The significance of this finding is therefore unclear and requires further investigation.
This is the first study to our knowledge that has correlated AR gene amplification, AR protein expression and PSA protein expression in matched hormone-sensitive and hormone-resistant prostate cancer. We have confirmed that AR gene amplification is associated with the development of hormone resistance in 20% of patients and this is related to an increase in AR expression in the majority of cases. We have also demonstrated that this is not the only mechanism by which AR expression is increased and also that in almost 60% of cases, AR expression remains unchanged or decreases. Therefore, in these cases, hormone resistance must be due to alternative mechanisms of AR activation or may be due to mechanisms that can stimulate AR responsive genes independent of the AR.
Change history
16 November 2011
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Armitage P, Berry G (1995) Statistical Methods in Medical Research, 3 rd ed. Oxford: Blackwell Science
Avila DM, Zoppi S, McPhaul (2001) The androgen receptor in syndromes of insensitivity and prostate cancer. J Steroid Biochem Mol Biol 76: 135–142
Bubendorf L, Kononen J, Koivisto P, Schraml P, Moch H, Gasser TC, Willi N, Mihatsch, MJ, Sauter G, Kallioniemi O-P (1999) Survey of gene amplifications during prostate cancer progression by high throughput fluorescence in situ hybridization on tissue microarrays. Cancer Res 59: 803–806
Culig Z, Hobisch A, Hittmair A, Peterziel H, Cato AC, Bartsch G, Klocker H (1998) Expression, structure, and function of androgen receptor in advanced prostatic carcinoma. Prostate 35: 63–70
Edwards J, Krishna NS, Mukherjee R, Watters AD, Underwood MA, Bartlett JMS (2001) Amplification of the androgen receptor may not explain the development of androgen-independent prostate cancer. Br J Urol Int 88: 633–637
Fraser JA, Reeves JR, Stanton PD, Black DM, Going JJ, Cooke TG, Bartlett JMS (2003) A role for BRCA1 in sporadic breast cancer. Br J Cancer 88: 1263–1270
Gnanapragasam VJ, Robson CN, Neal DE, Leung HY (2002) Regulation of FGF8 expression by the androgen receptor in human prostate cancer. Oncogene 21: 5069–5080
Goktas S, Ziada A, Crawford ED (1999) Combined androgen blockade for advanced prostatic carcinoma. Prostate Cancer Prostatic Dis 2: 172–179
Gregory CW, Hamil KG, Kim D, Hall SH, Pretlow TG, Mohler JL, French FS (1998) Androgen receptor expression in androgen independent prostate cancer is associated with increased expression of androgen regulated genes. Cancer Res 58: 5718–5724
Henttu P, Vihko P (1998) The protein kinase C activator, phorbol ester, elicits disparate functional responses in androgen-sensitive and androgen-independent human prostatic cancer cells. Biochem Biophyls Res Commun 244: 167–171
Koivisto P, Kononen J, Palmberg C, Tammela T, Hyytinen E, Isola J, Trapman J, Cleutjens K, Noordzij A, Visakorpi T, Kallioniemi O-P (1997) Androgen receptor gene amplification: a possible molecular mechanism for androgen deprivation therapy failure in prostate cancer. Cancer Res 57: 314–319
Koivisto PA, Helin HJ (1999) Androgen receptor gene amplification increases tissue PSA protein expression in hormone-refractory prostate carcinoma. J Pathol 189: 219–223
Krishna NS, Edwards J, Underwood MA, Grigor KM, Bartlett JMS (2002) AP-1 as a mediator of androgen refractory prostate cancer. Br J Cancer 86: s34
Lin HK, Yeh S, Kang HY, Chang C (2001) Akt suppresses androgen-induced apoptosis by phosphorylating and inhibiting androgen receptor. Proc Nat Acad Sci USA 98: 7200–7205
Latil A, Bieche I, Vidaud D, Lidereau R, Berthon P, Cussenot, Vidaud M (2001) Evaluation of androgen, estrogen (ERα and ERβ), and progesterone receptor expression in human prostate cancer by real-time quantitative reverse transcription–polymerase chain reaction assays. Cancer Res 61: 1919–1926
Linja MJ, Savinainen KJ, Saramaki OR, Tammela TL, Vessella RL, Visakorpi T (2001) Amplification and overexpression of androgen receptor gene in hormone-refractory prostate cancer. Cancer Res 61: 3550–3555
Miyoshi Y, Uemura H, Fujinami K, Mikata K, Harada M, Kitamura H, Koizumi Y, Kubota Y (2000) Fluorescence in situ hybridization evaluation of c-myc and androgen receptor gene amplification and chromosomal anomalies in prostate cancer in Japanese patients. Prostate 43: 225–232
Newling D, Fossa SD, Anderson L, Abrahamsson PA, Aso Y, Eisenberger MA, Khoury S, Kozlowski JS, Kelly K, Scher H, Hartley-Asp B (1997) Assessment of hormone refractory prostate cancer. Urology 49: 46–53
Segawa N, Mori I, Utsunomiya H, Nakamura M, Nakamura Y, Shan L, Kakudo K, Katsuoka Y (2001) Prognostic significance of neuroendocrine differentiation, proliferation activity and androgen receptor expression in prostate cancer. Pathol Int 51: 452–459
Trapman J, Cleutjens KBJM (1997) Androgen-regulated gene expression in prostate cancer. Semin Cancer Biol 8: 29–36
Visakorpi T, Hyytinen E, Koivisto P, Tanner M, Keinanen R, Palmberg C, Palotie A, Tammela T, Isola J, Kallioniemi OP (1995) In vivo amplification of the androgen receptor gene and progression of human prostate cancer. Nat Genet 9: 401–406
Zhu X, Liu JP (1997) Steroid-independent activation of androgen receptor in androgen-independent prostate cancer: a possible role for the MAP kinase signal transduction pathway? Mol Cell Endo 134: 9–14
Acknowledgements
We thank Dr W Angerson for the statistical advice he given. We also acknowledge Astra Zeneca for providing the funding for this study.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Edwards, J., Krishna, N., Grigor, K. et al. Androgen receptor gene amplification and protein expression in hormone refractory prostate cancer. Br J Cancer 89, 552–556 (2003). https://doi.org/10.1038/sj.bjc.6601127
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.bjc.6601127
Keywords
This article is cited by
-
Adjuvant enzalutamide for the treatment of early-stage androgen-receptor positive, triple-negative breast cancer: a feasibility study
Breast Cancer Research and Treatment (2022)
-
Branched worm-like nanoparticles featured with programmed drug release for synergistic castration-resistant prostate cancer therapy
Journal of Materials Science (2020)
-
Caffeic acid phenethyl ester suppresses androgen receptor signaling and stability via inhibition of phosphorylation on Ser81 and Ser213
Cell Communication and Signaling (2019)
-
IκBα mediates prostate cancer cell death induced by combinatorial targeting of the androgen receptor
BMC Cancer (2016)
-
Inhibiting androgen receptor nuclear entry in castration-resistant prostate cancer
Nature Chemical Biology (2016)
