
Abstract
The aims of this phase I/II study of docetaxel and S-1 were to determine the dose-limiting toxicity (DLT), maximum-tolerated dose (MTD), and recommended dose (RD) in the phase I part and to explore the tumour response, survival and safety in the phase II part. Patients with histologically- or cytologically confirmed unresectable or recurrent gastric cancer were eligible. Treatment consisted of intravenous docetaxel on day 1 (starting dose 50 mg m−2) and oral S-1 at a fixed dose of 40 mg m−2 twice daily on days 1–14, every 4 weeks up to six cycles. Nine patients took part in the phase I portion of the study. The MTD of docetaxel was determined to be 50 mg m−2, with the DLTs of grade 3 infection associated with grade 3 neutropenia and grade 4 neutropenia during S-1 administration. The RD of docetaxel was 40 mg m−2 in combination with S-1 40 mg m−2 b.i.d. The efficacy and safety of this regimen was therefore assessed in 46 patients with at least one measurable lesion. The overall response rate and estimated median overall survival were 46% (95% CI, 31–61%) and 14.0 months (8.3–17.3 months), respectively. The most common grade 3/4 toxicity was neutropenia (67% of patients), which was predictable and manageable. This regimen showed promising activity with moderate toxicities in advanced gastric cancer.
Similar content being viewed by others


Main
Although the incidence of gastric cancer (GC) has been declining, it remains one of the most common causes of cancer-related deaths (Jemal et al, 2005). Surgical resection at the early stage of disease is considered the most important intervention that leads to long-term disease-free survival, but many patients have recurrences or are diagnosed with more advanced stages of disease. For such patients, 1-year survival rates are approximately 50% in stage III disease, and <25% in stage IV disease (Alberts et al, 2003). Thus, considerable attention has been paid to the development of effective treatment for patients with advanced GC.
Cytotoxic chemotherapy including 5-fluorouracil (5-FU) is the most effective means of providing survival benefits and improvements in quality of life compared with best supportive care (Wöhrer et al, 2004). However, these improvements have been modest, at best, and thus far no combination has clearly provided a survival advantage over single agent 5-FU (Ohtsu, 2005). Therefore, although no accepted global standard regimen has been established, most physicians rely on 5-FU as monotherapy or as part of a combination strategy.
Docetaxel (Taxotere®, sanofi-aventis, Paris, France) is a semisynthetic taxoid derived from the European yew tree, Taxus baccata (Denis et al, 1990). A preclinical study of docetaxel showed a synergistic antitumour activity in combination with 5-FU (Bissery et al, 1995), which led to several clinical studies in advanced GC. In this setting, docetaxel has been evaluated both as a single agent (Sulkes et al, 1994; Einzig et al, 1996; Taguchi et al, 1998; Mai et al, 1999) and in combination with fluoropyrimidines (Constenla et al, 2002; Park et al, 2004), cisplatin (Roth et al, 2000), and cisplatin plus 5-FU (Van Custem et al, 2003). In a randomised phase III study, this latter regimen (TCF) improved overall survival compared to cisplatin plus 5-FU (Moiseyenko et al, 2005). The docetaxel containing combination regimens are associated with severe leucopenia and febrile neutropenia, and although these adverse events can be managed, efforts must be made to minimise such toxicities (Sulkes et al, 1994; Einzig et al, 1996; Taguchi et al, 1998; Mai et al, 1999).
The anticancer drug S-1 (TS-1®, Taiho Pharmaceutical Co. Ltd., Tokyo, Japan) is an oral formulation containing within each capsule tegafur, a 5-FU prodrug; gimeracil, an inhibitor of dihydropyrimidine dehydrogenase; and oteracil, which inhibits pyrimidine phosphoribosyl transferase specifically in the gastrointestinal tract and thereby decreases the phosphorylation of 5-FU in the intestine (Shirasaka et al, 1996). Phase II studies of S-1 monotherapy in patients with advanced GC showed overall response rates (ORRs) of 26–49% with the most relevant side-effects being diarrhoea in a European study and neutropenia in two Japanese studies (Sakata et al, 1998; Koizumi et al, 2000; Chollet et al, 2003).
Based on the clinical activity of both docetaxel and S-1, as well as the promising efficacy of docetaxel when combined with other fluoropyrimidines, we conducted a phase I/II study of docetaxel and S-1 in order to develop an effective treatment for patients with advanced GC that would improve on the safety profile of earlier taxane-fluoropyrimidine combinations.
Materials and methods
Study design
This was a multicentre, open-label, single-arm, phase I/II study conducted at 16 institutions in Japan. The objective of the phase I part was to determine the dose-limiting toxicity (DLT), maximum-tolerated dose (MTD) and recommended dose (RD) of docetaxel combined with a fixed dose of S-1. In the phase II part, the primary objective was to estimate the ORR of this combination at the RD. Secondary objectives were to assess progression-free survival, overall survival, 1-year survival rate, and adverse events. This study was conducted in accordance with the principles of the Declaration of Helsinki and Good Clinical Practice guidelines. The study protocol was reviewed and approved by the institutional review board of each participating centre. The objective response was independently reviewed by the extramural review committee, and the MTD and RD were determined by the independent data monitoring committee.
Patients
Patients were eligible if they signed informed consent and met all the following criteria: histologically- or cytologically confirmed unresectable or recurrent GC; at least one measurable lesion (patients without a measurable lesion, but evaluable nonmeasurable lesions were permitted in the phase I part of this study); Eastern Cooperative Oncology Group performance status of 0–1; good recovery from surgery (at least 28 days after the operation); up to one prior chemotherapy regimen except for prior taxane (paclitaxel or docetaxel) or S-1 (other fluoropyrimidines were allowed if at least 28 days elapsed after the last treatment); ⩾20 years of age; estimated life expectancy of at least 3 months; haemoglobin ⩾8.0 g dl−1; white blood cell count between 4000 and 12 000 mm−3; neutrophil count ⩾2000 mm−3; platelet count ⩾100 000 mm−3; serum bilirubin ⩽1.5 mg dl−1; aspartate aminotransferase and alanine aminotransferase ⩽2.5 times the upper limit of normal (ULN); and serum creatinine less than or equal to ULN.
Exclusion criteria were as follows: pregnant female or sexually active males/females unwilling to use contraception during the study; infection or suspected infection with fever; congestive heart failure; uncontrolled angina pectoris or arrhythmia; a history of myocardial infarction within the previous 3 months; uncontrolled diabetes or hypertension; interstitial pneumonia or lung fibrosis; peripheral neuropathy grade 2 or higher; pleural, peritoneal, or pericardial effusion that required treatment; gastrointestinal haemorrhage; symptomatic brain metastasis; diarrhoea; and active concomitant malignancy.
Phase I part
Patients received variable doses of intravenous docetaxel administered as a 1–2-h infusion on day 1 and oral S-1 administered at a fixed dose of 40 mg m−2 twice daily on days 1–14 every 4 weeks (one cycle). Patients were treated for up to six cycles unless disease progression or unacceptable toxicity was observed.
The initial starting dose of docetaxel was 50 mg m−2 (dose level 1), and step-wise dose increases to 60 and 70 mg m−2 were planned for successive patient cohorts (dose levels 2 and 3, respectively). In the case that dose level 1 was not acceptable, docetaxel 40 mg m−2 (dose level 0) would be explored.
At least three patients were to be started at dose level 1. If all three patients experienced DLTs, this dose level was determined to be the MTD, and dose level 0 would be explored. If two or fewer patients had DLT, an additional three patients were to be treated at the same dose level. If three or more than six patients treated at dose level 1 had DLTs, this dose level was determined to be the MTD, and dose level 0 would be explored. Dose escalation was planned until the MTD was reached, in which case the next lower dose level would be considered for further evaluation in the phase II part of the study. If three or more patients treated at dose level 0 experienced DLT, the regimen would be deemed not feasible.
Dose-limiting toxicitys were defined as follows: (1) grade 4 neutropenia lasting for 5 days or longer; (2) grade 4 neutropenia with fever (⩾38.5°C); (3) grade 4 thrombocytopenia; (4) grade 3/4 nonhaematological toxicities other than nausea/vomiting, anorexia, and general fatigue; and (5) any grade 4 haematological toxicity during S-1 administration. Assessment of DLTs was conducted only in the first treatment cycle. An independent data monitoring committee evaluated the safety results for each dose level and determined the MTD and RD.
Phase II part
Study treatment
Patients received the combination treatment with the RD of docetaxel on day 1 and oral S-1 40 mg m−2 twice daily in accordance with the treatment regimen described above. Cycles were repeated every 4 weeks for up to six cycles unless disease progression or unacceptable toxicity was observed. The protocol did not specify rules for interruption and resumption of S1 within each cycle. Such decisions were based on the clinical judgment of the investigator.
Chemotherapy was withheld for the following toxicities: (1) neutrophil count <1500 mm−3 or platelet count <100 000 mm−3; (2) aspartate aminotransferase or alanine aminotransferase >2.5 times ULN, or total serum bilirubin >1.5 mg dl−1; (3) body temperature ⩾38°C; (4) performance status ⩾2; (5) diarrhoea grade 2 or more; (6) neuropathy grade 2 or more; (7) oedema grade 2 or more. Treatment was restarted after recovery to baseline. If patients did not recover from these toxicities within 28 days of the last administration of S-1, they were withdrawn from the study.
If any of the following toxicities was observed, the dose of docetaxel was reduced by one level (phase I part) or by 10 mg m−2 (phase II part): (1) grade 3/4 neutropenia with fever (>38.5°C); (2) haemorrhage with grade 3/4 thrombocytopenia or requirement for a platelet transfusion; and (3) grade 3/4 nonhaematological toxicity other than nausea/vomiting, anorexia, general fatigue, and hypersensitivity. The dose of S-1 could be reduced by 20 mg per day if any of these toxicities were observed after the dose reduction of docetaxel or the RD was determined to be level 0.
Supportive care
Throughout this study, the prophylactic administration of granulocyte colony-stimulating factor (G-CSF), antiemetic agents, corticosteroids including premedication to docetaxel, or antihistamines was not allowed during the first treatment cycle. Use of these agents was allowed as secondary prevention of symptoms in all subsequent cycles.
Outcome measures
Tumour response was assessed according to the Response Evaluation Criteria in Solid Tumors (Therasse et al, 2000). Assessments by imaging studies were repeated every 4 weeks during the study. Progression-free survival was defined as the time from registration until objective tumour progression or death (censored at second-line chemotherapy) and overall survival was defined as the time from registration until death from any cause (censored at the time of last visit in patients who were lost to –follow up). Progression-free and overall survival, and 1-year survival rates were estimated using the Kaplan–Meier method. Adverse events were graded according to the National Cancer Institute Common Toxicity Criteria (version 2). Haematological and biochemical tests, performance status and clinical assessment of symptoms were monitored at least every week in the phase I part and every 2 weeks in the phase II part. The relative dose intensity (DI) was calculated as follows: actual DI=total dose (mg m−2)/treatment period (weeks); planned DI (PDI)=originally planned cumulative dose in the first course (mg m−2)/4 weeks; and relative DI=DI/PDI.
Statistical methods
The design of this study was based on a binominal distribution with no planned interim analysis. For the phase II part of this study, the primary end point was to determine the ORR. Assuming a null hypothesis of a 35% ORR and an alternative hypothesis of a 55% ORR, with one-sided type I error=0.05 and type II error=0.2, it was necessary to enrol a minimum 45 patients at the RD, including those treated at the RD during the phase I part of the study. All analyses were performed using SAS® version 8.2 (SAS Institute Inc., Cary, NC, USA).
Results
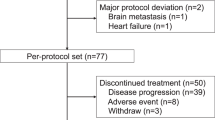
Between September 2002 and June 2004, a total of 50 patients were enrolled into the study. Nine patients (three in level 1 and six in level 0) were enrolled into the phase I part and 41 patients into the phase II part. Of the 41 patients enrolled into the phase II part of the study, one patient did not receive either docetaxel or S-1 because her disease had progressed rapidly and she could not take drugs orally before the initiation of treatment. This patient was excluded from all analyses according to the Full Analysis Set principle. Therefore, the population evaluable for efficacy and safety in the phase II analysis included six patients treated at the RD during the phase I part and 40 patients treated during the phase II part of the study.
Phase I part
The first cohort of three patients received docetaxel 50 mg m−2 combined with S-1 40 mg m−2 twice daily (dose level 1). Among these patients, one experienced grade 3 infection associated with grade 3 neutropenia on day 11, and two had grade 4 neutropenia on day 8 during S-1 administration. As all three patients treated at dose level 1 were deemed to have a DLT, the next cohort of patients was treated at dose level 0 (docetaxel 40 mg m−2). A total of six patients received 1–6 cycles of treatment at level 0. Among these, only one developed a DLT (grade 3 infection without neutropenia). From these results, the MTD and RD were determined to be level 1 and level 0, respectively.
Phase II part
Patients
Baseline characteristics of the 46 patients treated at the RD are shown in Table 1. Median age was 65 years (range, 42–79). All patients had at least one measurable lesion. Thirty-four patients (74%) had primary gastric lesions. Twenty-two patients (48%) had hepatic metastases. Sixteen patients (35%) had received a prior chemotherapy including 5-FU-containing regimens, irinotecan plus cisplatin or oral fluoropyrimidines.
Study treatment
A total of 165 cycles were administered, with a median of three cycles (range, 1–6). The median cumulative doses of docetaxel and S-1 were 120 mg m−2 (range, 40–240) and 3098 mg m−2 (range, 204–6368), respectively. Median relative dose intensities of docetaxel and S-1 were 99% (range, 75–101) and 82% (range, 18–97), respectively. Thirty-six patients (78%) who failed study treatment received the next-line chemotherapy. The main regimen was cisplatin plus irinotecan (17/36, 47%).
Efficacy
Tumour response results are shown in Table 2. The ORR was 46% (95% confidence interval ((CI), 31–61) with two patients (4%) showing a complete response and 19 (41%) a partial response. Figure 1 shows overall and progression-free survival. At a median follow-up of 12 months (range, 2–27), the median progression-free and estimated overall survival times were 4.2 months (95% CI, 2.2–5.2) and 14 months (95% CI, 8.3–17.3), respectively. The 1-year survival rate was 53% (95% CI, 38–67).

The cumulative probability of survivals. The solid and dotted lines present overall and progression-free survivals estimated by the Kaplan–Meier method in 46 patients, respectively.
Safety
Table 3 summarises adverse events observed during the phase II part of this study. The most common grade 3/4 haematological toxicities were neutropenia (31 patients, 67%) and leucopenia (19 patients, 41%). Among a total of 165 cycles, the median time from the treatment start to nadir was 14 days (range, 3–29) and the median time from nadir to recovery was 15 days (range, 2–27). The study treatments were delayed for the following reasons: neutropenia (6/165 cycles, 4%), thrombocytopenia (3/165, 2%), worsening of PS (2/165, 1%), and fever (1/165, 1%). The doses of S-1 were reduced in 16 patients (35%) mainly due to grade 4 neutropenia and grade 3 anorexia. Treatment with G-CSF as secondary prophylaxis for neutropenia was administered in 23 cycles (14%). The most common grade 3/4 nonhaematological toxicity was anorexia (10 patients, 22%). Fever and infection were observed in 10 patients (22%, grade 1 or 2) and two patients (4%, all grade 3), respectively. Oedema (grade 1) was observed in only three patients (7%), despite the absence of prophylactic corticosteroids or antihistamines. The majority of the nonhaematological toxicities were relatively mild. No treatment-related deaths were observed.
In five patients, combination treatment was discontinued due to the following adverse events: grade 4 neutropenia associated with grade 3 diarrhoea, grade 4 neutropenia associated with grade 2 thrombocytopenia, grade 3 neutropenia associated with unrecovered grade 2 anaemia, grade 4 cerebral infarction, and suspected grade 2 interstitial pneumonia.
Discussion
We evaluated the efficacy and safety of a chemotherapy combination regimen of docetaxel and S-1, two agents that separately have shown promise in the management of advanced GC. Response rates and safety data for S-1 have shown ORRs up to 49% with no grade 4 haematological toxicity (Sakata et al, 1998; Koizumi et al, 2000), while adding docetaxel to a standard chemotherapy regimen of cisplatin and 5-FU improves survival (TAX 325, Moiseyenko et al, 2005). Therefore, our hypothesis was that docetaxel combined with S-1 would confer a clinically meaningful improvement in ORR and median survival, with a manageable safety profile.
In the phase I part of our study, we identified docetaxel 40 mg m−2 on day 1 plus S-1 40 mg m−2 twice a day on days 1–14, every 28 days as the treatment schedule to recommend for further clinical evaluation. This dose of docetaxel is lower than that commonly used in the West to treat GC. However, we observed severe myelosuppression at dose level 1 (docetaxel 50 mg m−2), which was the MTD. In this study, the MTD was declared when all of the first three patients in a cohort experienced DLT or when at least three of six patients enrolled in a cohort experienced DLT. Although this definition differs from that employed in other studies, it is commonly accepted in Japan. Our use of this definition allowed us to find a RD that was tolerable for most patients enrolled in the phase II portion of this study.
We speculate the reason for the lower dose of docetaxel may be that the pharmacokinetic parameters (AUC and Cmax) of 5-FU increase according to the dose of docetaxel. For example, the respective mean AUC and Cmax of 5-FU were 522.5 ng h ml−1 and 100.3 ng ml−1 with docetaxel 40 mg m−2 and 857.2 ng h ml−1 and 155.8 ng ml−1, with docetaxel 50 mg m−2 (Yoshida et al, 2004). Synergy of this combination has been reported in vitro, suggesting that biochemical modulation of the two drugs occurs. This synergy may also result in increased toxicity (Wada et al, 2006). Perhaps higher doses of docetaxel would have conferred a greater response rate, but such doses were not feasible in our study population. Preliminary reports have described alternative dosing schedules for docetaxel in combination with S1 for the treatment of advanced GC. These have explored docetaxel dosing intervals ranging from every week to every 4 weeks with relative dose intensities from 10 to 20 mg m−2 per week. However, because final reports of these studies are not yet available, the feasibility and efficacy of these alternative regimens remains unknown (Yoshida et al, 2004; Kim et al, 2006; Park et al, 2006; Rino et al, 2006; Satoh et al, 2006; Takahashi et al, 2006).
In the phase II part of this study, we demonstrated that for patients with advanced GC, this regimen conferred an ORR of 46% and an estimated median survival of 14 months, with acceptable toxicity. The 46% ORR observed in this study was slightly lower than expected. We anticipated an ORR of 55% because results from phase II studies in Japan showed ORRs of 22% with docetaxel monotherapy (Taguchi et al, 1998; Mai et al, 1999) and 45% with S-1 (Sakata et al, 1998; Koizumi et al, 2000). A possible explanation is that we included a significant number of patients with liver metastasis (22/46, 47.8%) and intestinal-type histology (29/46, 63%), and many had previous exposure to chemotherapy (16/46, 34.8%). All of these features are associated with decreased response rates to chemotherapy (Taguchi et al, 1998, Mai et al, 1999, Koizumi et al, 2000, Ajani et al, 2005). Yet despite these unfavourable baseline characteristics, the combination of docetaxel and S-1 conferred an ORR that was similar to those obtained with other combination regimens (Ajani et al, 2005; Thuss-Patience et al, 2005).
At a median follow-up of 12 months, the median overall survival was estimated to be 14 months. Although cross-study comparisons should be taken with caution, the survival effect observed in our study compares favourably with other chemotherapy regimens, such as docetaxel (6–8 months (Taguchi et al, 1998; Mai et al, 1999)) or S-1 (7–8 months (Sakata et al, 1998; Koizumi et al, 2000)), or the combinations DC (11 months), DF (10 months), DCF (10 months), ECF (10 months) or FOLFOX-4 (11 months) (Ajani et al, 2005; De Vita et al, 2005; Thuss-Patience et al, 2005).
Patients enrolled in our study were, in some respects, less likely to respond to chemotherapy than patients in these other trials. All patients in our study had metastatic disease, 65% were chemo-naive, and 48% had liver metastases. In studies of DCF, ECF, and FOLFOX-4, the proportions of chemo-naive patients were 100, 100, and 84%, respectively; the rates of metastatic disease were 95, 98, and 92%, respectively; and the incidences of liver metastases were 80% (including peritoneal), 36, and 62%, respectively. Despite this, the ORR and survival observed in our study appeared higher than those reported for other combination regimens studied in more favourable populations. At a median follow-up of 12 months, the estimated median survival is 14 months, but this may improve over time.
Although survival data appear promising, there are limitations. Progression-free survival in this study was similar to those reported with conventional reference regimens containing 5-FU and cisplatin (Vanhoefer et al, 2000; Ohtsu et al, 2003). In addition, the median survival observed in our study may be partly influenced by poststudy chemotherapy, which was administered to 78% of patients.
The docetaxel/S1 regimen reported here was well tolerated and toxicities were manageable. The most common haematological toxicities were neutropenia and leucopenia. The incidence of grade 3/4 neutropenia was similar to those previously reported with docetaxel monotherapy (Sulkes et al, 1994; Einzig et al, 1996; Taguchi et al, 1998; Mai et al, 1999). Despite the lack of corticosteroid prophylaxis, only three patients developed fluid retention (all grade 1), and no patients had a hypersensitivity reaction. The majority of nonhaematological toxicities were mild.
The docetaxel/S1 regimen used in this study yielded a promising median survival time and manageable safety profile. Based partly on these results, a phase III study comparing S-1 alone to docetaxel plus S-1 in patients with advanced GC has begun enrolment in Japan to evaluate whether adding docetaxel to S1 improves clinical benefit.
In conclusion, the combination of docetaxel and S-1 is an active and well-tolerated regimen in patients with advanced GC. It is worthwhile to assess the survival benefit and quality of life in a phase III trial of this combination in advanced GC to establish the new regimen for the outpatient setting.
Change history
16 November 2011
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Ajani JA, Fodor MB, Tjulandin SA, Moiseyenko VM, Chao Y, Filho SC, Majlis A, Assadourian S, Van Cutsem E (2005) Phase II multi-institutional randomized trial of docetaxel plus cisplatin with or without fluorouracil in patients with untreated, advanced gastric, or gastroesophageal adenocarcinoma. J Clin Oncol 23: 5660–5667
Alberts SR, Cervantes A, van de Velde CJH (2003) Gastric cancer: epidemiology, pathology and treatment. Ann Oncol 14(Suppl 2): ii31–ii36
Bissery MC, Nohynek G, Sanderink GJ, Lavelle F (1995) Docetaxel (Taxotere): a review of preclinical and clinical experience. Part I: preclinical experience. Anticancer Drugs 6: 339–368
Chollet P, Schoffski P, Weigang-Kohler K, Schellens JH, Cure H, Pavlidis N, Grunwald V, De Boer R, Wanders J, Fumoleau P, EORTC Early Clinical Studies Group (2003) Phase II trial with S-1 in chemotherapy-naive patients with gastric cancer. A trial performed by the EORTC Early Clinical Studies Group (ECSG). Eur J Cancer 39: 1264–1270
Constenla M, Garcia-Arroyo R, Lorenzo I, Carrete N, Campos B, Palacios P (2002) Docetaxel, 5-fluorouracil, and leucovorin as treatment for advanced gastric cancer: results of a phase II study. Gastric Cancer 5: 142–147
Denis JN, Correa A, Greene AE (1990) An improved synthesis of the Taxol side chain and of RP56976. J Org Chem 55: 1957–1959
De Vita F, Orditura M, Matano E, Bianco R, Carlomagno C, Infusino S, Damiano V, Simeone E, Diadema MR, Lieto E, Castellano P, Pepe S, De Placido S, Galizia G, Di Martino N, Ciardiello F, Catalano G, Bianco AR (2005) A phase II study of biweekly oxaliplatin plus infusional 5-fluorouracil and folinic acid (FOLFOX-4) as first-line treatment of advanced gastric cancer patients. Br J Cancer 92: 1644–1649
Einzig AI, Neuberg D, Remick SC, Karp DD, O'Dwyer PJ, Stewart JA, Benson III AB (1996) Phase II trial of docetaxel (Taxotere) in patients with adenocarcinoma of the upper gastrointestinal tract previously untreated with cytotoxic chemotherapy: the Eastern Cooperative Oncology Group (ECOG) results of protocol E1293. Med Oncol 13: 87–93
Jemal A, Murray T, Ward E, Samuels A, Tiwari RC, Ghafoor A, Feuer EJ, Thun MJ (2005) Cancer statistics, 2005. CA Cancer J Clin 55: 10–30
Kim H, Kim YH, Shim BY, Shin SW, Moon H (2006) Phase I study for the escalation of docetaxel with fixed dose of TS1 in advanced gastric cancer. ASCO American Society of Clinical Oncology Gastrointestinal Cancers Symposium: Abstract No 87
Koizumi W, Kurihara M, Nakano S, Hasegawa K, for the S-1 Cooperative Gastric Cancer Study Group (2000) Phase II study of S-1, a novel oral derivative of 5-fluorouracil, in advanced gastric cancer. Oncology 58: 191–197
Mai M, Sakata Y, Kanamaru R, Kurihara M, Suminaga M, Ota J, Hirabayashi N, Taguchi T, Furue H (1999) A late phase II clinical study of RP56976 (docetaxel) in patients with advanced or recurrent gastric cancer: a cooperative study group trial (Group B). Jpn J Cancer Chemother 26: 487–496 (in Japanese with English abstract)
Moiseyenko VM, Ajani JA, Tjulandin SA, Majlis A, Constenla M, Boni C, Anelli A, Yver AJ, Van Cutsem E, on behalf of the TAX 325 Study Group (2005) Final results of a randomized controlled phase III trial (TAX 325) comparing docetaxel (T) combined with cisplatin (C) and 5-fluorouracil (F) to CF in patients (pts) with metastatic gastric adenocarcinoma (MGC). Proc Am Soc Clin Oncol 23: 308s (abstract 4002)
Ohtsu A (2005) Current status and future prospects of chemotherapy for metastatic gastric cancer: a review. Gastric Cancer 8: 95–102
Ohtsu A, Shimada Y, Shirao K, Boku N, Hyodo I, Saito H, Yamamichi N, Miyata Y, Ikeda N, Yamamoto S, Fukuda H, Yoshida S (2003) Randomized phase III trial of fluorouracil alone versus fluorouracil plus cisplatin versus uracil and tegafur plus mitomycin in patients with unresectable, advanced gastric cancer: the Japan Clinical Oncology Group Study (JCOG9205). J Clin Oncol 21: 54–59
Park SR, Chun JH, Yu MS, Kim YW, Lee JH, Choi IJ, Kim CG, Lee JS, Bae JM, Kim HKA (2006) Phase I study of S1 and weekly docetaxel in metastatic gastric cancer. ASCO American Society of Clinical Oncology Gastrointestinal Cancers Symposium: Abstract No 82
Park YH, Ryoo B-Y, Choi S-J, Kim H-T (2004) A phase II study of capecitabine and docetaxel combination chemotherapy in patients with advanced gastric cancer. Br J Cancer 90: 1329–1333
Rino Y, Takanashi Y, Yukawa N, Saeki H, Wada H, Kanari M, Yamada R, Satoh T, Yamamoto N, Imada TA (2006) Phase I study of bi-weekly combination therapy with S-1 and docetaxel for advanced or recurrent gastric cancer. Anticancer Res 26: 1455–1462
Roth AD, Maibach R, Martinelli G, Fazio N, Aapro MS, Pagani O, Morant R, Borner MM, Herrmann R, Honegger H, Cavalli F, Alberto P, Castiglione M, Goldhirsch A (2000) Docetaxel (Taxotere®)-cisplatin (TC): an effective drug combination in gastric carcinoma. Ann Oncol 11: 301–306
Sakata Y, Ohtsu A, Horikoshi N, Sugimachi K, Mitachi Y, Taguchi T (1998) Late phase II study of novel oral fluoropyrimidine anticancer drug S-1 (1M tegafur-0.4M gimestat-1M otastat potassium) in advanced gastric cancer patients. Eur J Cancer 34: 1715–1720
Satoh KT, Okamoto I, Miyazaki M, Shimizu T, Ozaki T, Nakagawa K, Fukuoka M (2006) S-1 plus docetaxel in patients with advanced gastric cancer: a phase I study. ASCO American Society of Clinical Oncology Gastrointestinal Cancers Symposium: Abstract No 84T
Shirasaka T, Shimamoto Y, Ohshimo H, Yamaguchi M, Kato T, Yonekura K, Fukushima M (1996) Development of a novel form of an oral 5-fluorouracil derivative (S-1) directed to the potentiation of the tumor selective cytotoxicity of 5-fluorouracil by two biochemical modulators. Anti-cancer Drugs 7: 548–557
Sulkes A, Smyth J, Sessa C, Dirix LY, Vermorken JB, Kaye S, Wanders J, Franklin H, LeBail N, Verweij J (1994) Docetaxel (Taxotere) in advanced gastric cancer: results of a phase II clinical trial. EORTC Early Clinical Trials Group. Br J Cancer 70: 380–383
Takahashi I, Emi Y, Kakeji Y, Tokunaga E, Ushiro S, Oki E, Watanabe M, Baba H, Maehara Y (2006) Phase I study of S-1 and biweekly docetaxel combination chemotherapy for advanced and recurrent gastric cancer. Oncol Rep 15: 849–854
Taguchi T, Sakata Y, Kanamaru R, Kurihara M, Suminaga M, Ota J, Hirabayashi N (1998) Late phase II clinical study of RP56976 (docetaxel) in patients with advanced/recurrent gastric cancer: a Japanese Cooperative Study Group trial (Group A). Jpn J Cancer Chemothr 25: 1915–1924 (in Japanese with English abstract)
Therasse P, Arbuck SG, Eisenhauer EA, Wanders J, Kaplan RS, Rubinstein L, Verweij J, Van Glabbeke M, van Oosterom AT, Christian MC, Gwyther SG (2000) New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J Natl Cancer Inst 92: 205–216
Thuss-Patience P, Kretzschmar A, Repp M, Kingreen D, Hemnesser D, Micheel S, Pink D, Dörken B, Reichardt P (2005) Docetaxel and continuous-infusion fluorouracil versus epirubicin, cisplatin, and fluorouracil for advanced gastric adenocarcinoma: a randomized phase II study. J Clin Oncol 23: 494–501
Van Custem E, Moiseyenko V, Tjulandin S, Fodor M, Boni C, Zuber E, Assadourian S (2003) Docetaxel (D), cisplatin (C), 5-fluorouracil (F) compared to cisplatin (C) and 5-fluorouracil (F) for chemotherapy-naïve patients with metastatic or locally recurrent, unresectable gastric carcinoma (MGC): interim results of a randomised phase III trial (V325). Eur J Cancer, (Suppl 1): s20 (abstract 51A)
Vanhoefer U, Rougier P, Wilke H, Ducreux MP, Lacave AJ, Van Cutsem E, Planker M, Dos Santos JG, Piedbois P, Paillot B, Bodenstein H, Schmoll HJ, Bleiberg H, Nordlinger B, Couvreur ML, Baron B, Wils JA (2000) Final results of a randomized phase III trial of sequential high-dose methotrexate, fluorouracil, and doxorubicin versus etoposide, leucovorin, and fluorouracil versus infusional fluorouracil and cisplatin in advanced gastric cancer: a trial of the European Organization for Research and Treatment of Cancer Gastrointestinal Tract Cancer Cooperative Group. J Clin Oncol 18: 2648–2657
Wada Y, Yoshida K, Suzuki T, Mizuiri H, Konishi K, Ukon K, Tanabe K, Sakata Y, Fukushima M (2006) Synergistic effects of docetaxel and S-1 by modulating the expression of metabolic enzymes of 5-fluorouracil in human gastric cancer cell lines. Int J Cancer Mar 23 [E-pub ahead of print]
Wöhrer SS, Raderer M, Hejna M (2004) Palliative chemotherapy for advanced gastric cancer. Ann Oncol 15: 1585–1595
Yoshida K, Hirabayashi N, Takiyama W, Ninomiya M, Takakura N, Sakamoto J, Nishiyama M, Toge T (2004) Phase I study of combination therapy with S-1 and docetaxel (TXT) for advanced or recurrent gastric cancer. Anticancer Res 24: 1843–1851
Acknowledgements
We thank Drs S Saitoh, A Sato, H Takiuchi, and N Hirabayashi for their extramural review to assess objective responses, and, T Sasaki, M Kurihara, and Y Ariyoshi for their peer review as members of the independent data monitoring committee, and T Taguchi and Y Ohashi for their special advice. We also acknowledge Y Kubota, N Fujimaki, A Hamajima, and S Katsuki for their contribution to this study, and K MacKenzie and Dr S Olsen for their constructive comments and editorial assistance. This study was sponsored by sanofi-aventis KK, Japan.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Yamaguchi, K., Shimamura, T., Hyodo, I. et al. Phase I/II study of docetaxel and S-1 in patients with advanced gastric cancer. Br J Cancer 94, 1803–1808 (2006). https://doi.org/10.1038/sj.bjc.6603196
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.bjc.6603196
Keywords
This article is cited by
-
Neoadjuvant camrelizumab and apatinib combined with chemotherapy versus chemotherapy alone for locally advanced gastric cancer: a multicenter randomized phase 2 trial
Nature Communications (2024)
-
Phase II study of S-1 plus docetaxel as first-line treatment for older patients with advanced gastric cancer (OGSG 0902)
International Journal of Clinical Oncology (2024)
-
Efficacy and safety of combination chemotherapy regimens containing taxanes for first-line treatment in advanced gastric cancer
Clinical and Experimental Medicine (2022)
-
A case of advanced gastric cancer achieved a pathological complete response by chemotherapy
Surgical Case Reports (2017)
-
Low-dosed docetaxel showed equivalent efficacy but improved tolerability compared with oxaliplatin in the S-1-based first-line chemotherapy regimen for metastatic or recurrent gastric adenocarcinoma
Medical Oncology (2015)
