
Abstract
The apoptosis stimulating proteins of p53 (ASPP) family consists of three members, ASPP1, ASPP2 and iASPP. They bind to proteins that are key players in controlling apoptosis (p53, Bcl-2 and RelA/p65) and cell growth (APCL, PP1). So far, the best-known function of the ASPP family members is their ability to regulate the apoptotic function of p53 and its family members, p63 and p73. Biochemical and genetic evidence has shown that ASPP1 and ASPP2 activate, whereas iASPP inhibits, the apoptotic but not the cell-cycle arrest function of p53. The p53 tumour suppressor gene, one of the most frequently mutated genes in human cancer, is capable of suppressing tumour growth through its ability to induce apoptosis or cell-cycle arrest. Thus, the ASPP family of proteins helps to determine how cells choose to die and may therefore be a novel target for cancer therapy.
Similar content being viewed by others

Main
The recently discovered Apoptosis Stimulating Proteins of p53 (ASPP) family consists of three members, ASPP1, ASPP2 and iASPP. The most evolutionary conserved member, iASPP, is the inhibitory form and the only ASPP family member identified in Caenorhabditis elegans (Bergamaschi et al, 2003b). Mammals, including mice and humans, have evolved two additional members, ASPP1 and ASPP2 (Samuels-Lev et al, 2001). All three proteins share sequence similarity in their C-termini which contain their signature sequences of Ankyrin repeats, SH3 domain and Proline-rich region (Figure 1). Thus, ASPP also stands for Ankyrin repeats, SH3 domain, and Proline-rich region containing Protein. The C-terminus is the preferred binding site for ASPP partners including p53 (Gorina and Pavletich, 1996), RelA/p65 (subunit 3 of nuclear factor-κB) (Yang et al, 1999a), Bcl-2 (Naumovski and Cleary, 1996), adenomatous polyposis coli-like (APCL) (Nakagawa et al, 2000), Hepatitis-C core protein (Cao et al, 2004), amyloid-β-precursor protein-binding protein 1 (APPBP1) (Chen et al, 2003), YES-associated protein-1 (YAP1) (Espanel and Sudol, 2001), Sp1 (Ohno et al, 1997) and SAM68 (Thornton et al, 2006). The N-terminus is only conserved in the pro-apoptotic members, ASPP1 and ASPP2.

The human ASPP family structure. The amino-acid length of each ASPP member is indicated on the right, and individual structural elements are highlighted. Abbreviations: α, α-helical domain; Pro, proline-rich region; Ank, ankyrin repeats; SH3, SH3 domain.
To date, the most well-known function of the ASPP family of proteins is their ability to regulate apoptosis mediated by p53 and its family members, p63 and p73 (Bergamaschi et al, 2004). ASPP1 and ASPP2 enhance the ability of p53 to stimulate specifically the expression of pro-apoptotic target genes but not genes involved in cell-cycle arrest (Samuels-Lev et al, 2001; Bergamaschi et al, 2004), whereas iASPP inhibits p53-mediated apoptosis (Bergamaschi et al, 2003b). This implies that iASPP can compete with ASPP1 and ASPP2 for binding to p53, thereby inhibiting the ability of ASPP1 and ASPP2 to stimulate the apoptotic function of p53.
The p53 tumour suppressor plays an important role in regulating the proliferation of cells through either cell-cycle arrest or apoptosis (Rowan et al, 1996). p53 has been shown to be lost or mutated in over half of human cancers (http://www-p53.iarc.fr/). In the other 50% of tumours, where wild-type p53 is intact, the regulation of the p53 pathway is abnormal. Understanding why p53 is unable to perform its role as a tumour suppressor in these wild-type tumours has been the focus of extensive research. It is hoped that restoration of the tumour suppressor and apoptotic functions of p53 in tumours expressing wild-type p53 will enable the development of new therapeutic strategies in the treatment of cancer. In this review, we will focus on the recent understanding of the biological function of the ASPP family of proteins and their potential role in tumorigenesis.
iASPP is a proto-oncogene whereas ASPP1 and ASPP2 are tumour suppressors
Depletion of the only homologue of the ASPP family, iASPP, in C. elegans by RNAi specifically increased the number of apoptotic germ cell corpses (Bergamaschi et al, 2003b). Because of this resulting phenotype, the gene was termed ape-1 for apoptosis enhancer-1, whose gene product is Ce-iASPP. These studies provided the initial genetic evidence implicating iASPP as an antiapoptotic gene. This unique phenotype further revealed that Ce-iASPP is a key inhibitor of p53 in C. elegans as RNAi of iASPP failed to show any effect in p53 null worms. Furthermore, Ce-iASPP was identified as an inhibitor of p53 in a library screen for p53 inhibitors (Lettre et al, 2004). Therefore, Ce-iASPP is one of the most conserved inhibitors of p53 identified so far.
Unlike iASPP, ASPP2 is pro-apoptotic and is the most well-characterised member of the ASPP family. Existing studies have suggested that ASPP2 plays a central role in the regulation of apoptosis and cell growth. The most compelling evidence comes from the ASPP2 knockout mouse study (Vives et al, 2006). The group examined the role of ASPP2 in tumorigenesis and its relationship with p53 tumour suppression in homozygous or heterozygous mice lacking ASPP2 or ASPP2 and p53 alleles. ASPP2 knockout mice exhibit postnatal lethality. ASPP2 heterozygous mice have 45% tumour incidence over their lifespans, whereas wild-type mice have 15% tumour incidence. Compared to the wild-type mice, the heterozygous mice develop twice as many spontaneous lymphomas, as well as rhabdomyosarcomas and squamous cell carcinomas. The ASPP2 heterozygous mice also have an increased susceptibility to the onset of tumour development in response to ionising radiation. The failure to generate a p53 and ASPP2 double knockout mouse demonstrates that ASPP2 and p53 deficiency are synthetic lethal. This phenotype also suggests that, genetically, ASPP2 shares overlapping functions with p53 in mouse development. Consistent with this, a combination of p53 and ASPP2 heterozygosity accelerated the onset of tumour development. The mouse model demonstrates unequivocally that ASPP2 is a tumour suppressor that functions as an activator of the tumour suppressor function of p53.
Biochemically, ASPP2-mediated apoptosis has been shown to be inhibited by nuclear factor-κB and Bcl-2 family proteins, suggesting that the antiapoptotic functions of these proteins result from blocking the pro-apoptotic function of ASPP2 (Yang et al, 1999b; Takahashi et al, 2005). The observation that ASPP2 can localise to the mitochondria suggests that ASPP2 might play a role in regulating apoptosis through a mitochondrial-dependent pathway (Takahashi et al, 2005). Furthermore, treatment of wild-type and p53 null cell lines with ASPP1 and ASPP2 antisense RNA reduces apoptosis induced by DNA-damaging agents such as cisplatin, etoposide and UV (Samuels-Lev et al, 2001; Bergamaschi et al, 2003b). Cell lines stably expressing iASPP have been produced and are more resistant to apoptosis induced by cisplastin or UV compared to the parental cell lines. In addition, co-expression of ASPP1 and ASPP2 inhibits, whereas expression of iASPP enhances, the transforming activity of the oncogenes RAS and E1A in rat embryo fibroblasts, further implicating ASPP1 and ASPP2 as tumour suppressors, whereas iASPP is likely to be a proto-oncogene (Bergamaschi et al, 2003b).
Altered ASPP expression in human cancers is linked to tumour progression
Initial evidence suggesting the involvement of ASPP in human cancer came from the analysis of the crystal structure of the C-terminal ankyrin repeats and SH3 domains of ASPP2 and the DNA-binding domain of p53. The amino acids in p53 responsible for binding to ASPP2, namely 178His, 181Arg, 243Met and 247Arg, are all mutated to a relatively high frequency in human cancer (Gorina and Pavletich, 1996). Importantly, the two most frequently mutated residues, 248Arg and 273Arg, contact both DNA and ASPP2.
Recent studies have shown altered expression of the ASPP family members in a variety of human cancers. The expression of ASPP1 and ASPP2 is frequently downregulated in human breast tumours (Samuels-Lev et al, 2001) and tumour cell lines (Liu et al, 2005) expressing wild-type p53, suggesting that there is a selective advantage for loss of ASPP1 and ASPP2 expression in human breast tumours expressing wild-type p53. Reduced levels of ASPP2 have also been detected in human lung cancer cell lines (Mori et al, 2000), and ASPP1 downregulation was observed in one non-small-cell lung carcinoma cell line and two mesothelioma cell lines (Mori et al, 2004). Furthermore, in leukaemia cancer cell lines a concomitant downregulation of ASPP1 with an upregulation of iASPP has been observed, suggesting that abnormal ASPP expression could be an important step in the formation of human haematopoietic neoplasms and might be a useful molecular marker for the diagnosis of leukaemia (Liu et al, 2004). Significantly higher levels of iASPP were detected in human breast carcinomas (Bergamaschi et al, 2003a), leukaemia cell lines and acute leukaemias (AL) (Zhang et al, 2005). Interestingly, the level of iASPP gene expression was significantly higher in AL than in cells from normal donors or AL patients in complete remission, suggesting that overexpression of iASPP may promote leukaemogenesis and/or disease progression of AL (Zhang et al, 2005).
Other studies have linked reduced ASPP2 levels with poor prognosis and metastasis. A lowered ASPP2 expression was linked to a poor clinical outcome in diffuse large B-cell lymphoma (Lossos et al, 2002) and with poor distant recurrence-free survival in breast cancer patients (Cobleigh et al, 2005). Reduced expression of ASPP2 has also been observed in a microarray study of metastatic breast cancer samples compared to non-metastatic samples (Sgroi et al, 1999), suggesting that ASPP2 might be associated with breast cancer progression. Furthermore, an association between the ASPP2 (TP53BP2) locus and gastric cancer susceptibility has also been reported (Ju et al, 2005). Four single nucleotide polymorphisms (g.206692C>T, g.198267A>T, g.164895G>A and g.152389A>T) within TP53BP2 showed a significant link with gastric cancer susceptibility.
Regulation of ASPP expression
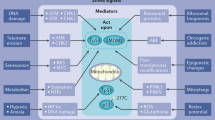
Our current understanding is that three main mechanisms are involved in the regulation of the ASPP family members: inactivation and gene silencing by methylation, activation by E2F transcription factors and regulation of ASPP2 at the protein level. The mechanism of action and the regulation of the ASPP family members in tumour cells is summarised in Figure 2.

Regulation of ASPP family of proteins. Stress signals including DNA damage, upregulation of the E2F transcription factors and inhibition of the proteasome activate ASPP1 and ASPP2 to interact with p53 family members and induce apoptosis. iASPP prevents this, instead allowing cell proliferation to occur. Gene silencing of ASPP1 and ASPP2 by methylation also prevents p53-dependent apoptosis.
ASPP1 and ASPP2 gene silencing
DNA methylation is the main epigenetic modification in humans, and changes in the methylation status of genes play an important role in tumorigenesis. Aberrant methylation of normally unmethylated CpG islands in the promoters of various tumour suppressor genes has been reported (Merlo et al, 1995). Initial evidence for methylation-dependent regulation of ASPP2 expression came from the observation that methyl-CpG-binding protein 1 (MBD1) is required for the formation of the CAF-1/MBD1/SETDB1 complex, which facilitates methylation during replication-coupled chromatin assembly (Sarraf and Stancheva, 2004). In the absence of MBD1, methylation at multiple genomic loci is lost. This results in the activation of the ASPP2 gene, which is normally repressed by MBD1, demonstrating that ASPP2 expression is regulated by methylation.
Furthermore, hypermethylation of the CpG islands in the 5′ untranslated regions of the ASPP1 and ASPP2 genes and reduced expression of ASPP1 and ASPP2 is reported in HEPG-2, MCF-7 and A549 cell lines which express wild-type p53 (Liu et al, 2005). In contrast, in normal fibroblasts, the ASPP1 and ASPP2 genes are unmethylated. This suggests that epigenetic changes of ASPP1 and ASPP2 occur in tumours that are wild type for p53 and that downregulation of ASPP1 and ASPP2 expression correlates with abnormal methylation of the ASPP1 and ASPP2 genes. Expression of ASPP1 in acute lymphoblastic leukaemia (ALL) is also significantly reduced as a consequence of the hypermethylation of the ASPP1 gene promoter (Agirre et al, 2006). In addition, methylation of ASPP1 is significantly higher in adult compared to childhood ALL, and T-cell compared to B-cell ALL. Relapse rate and mortality are significantly higher in patients expressing methylated ASPP1. Finally, methylation of the ASPP1 gene promoter is indicative of a poor prognosis in ALL patients. Consistent with these findings, another study shows aberrant methylation of ASPP1 in 34% of the 50 patients with de novo T-cell ALL (Roman-Gomez et al, 2005).
ASPP1 and ASPP2 are transcriptional targets of E2F1
The E2F1 transcription factor is a downstream target of the tumour suppressor retinoblastoma (Rb). When activated, E2F1 induces cell proliferation. E2F1 can also mediate apoptosis via p53-dependent and independent mechanisms. ASPP1 and ASPP2 have been identified as transcriptional targets of E2F, and increased expression of E2F-1 was shown to induce the expression of ASPP1 and ASPP2 at both mRNA and protein levels (Stanelle et al, 2002; Chen et al, 2005; Fogal et al, 2005; Hershko et al, 2005). Many tumour types exhibit defective Rb pathways and consequently an increased E2F activity. As a result, they also have an increased expression of ASPP1, ASPP2 or both, thereby enhancing the cellular sensitivity to p53-mediated apoptosis. This may be one possible explanation as to why tumour cells are more sensitive to p53-dependent apoptosis than primary cells.
Regulation of ASPP2 expression at the protein level
The proteasome, a major component of the ubiquitin-mediated protein degradation pathway, has recently become a therapeutic target in cancer therapy. Treatment of cells with bortezomib, a specific inhibitor of the proteasome which is clinically active in different tumour types, increased ASPP2 protein and protein half-life but not RNA levels (Zhu et al, 2005). Treatment with anthracycline, a chemotherapy agent that can induce DNA damage and inhibit the proteasome, also increased ASPP2 protein but not RNA levels. In addition, knockdown of ASPP2 by siRNA attenuated bortezomib-induced apoptosis in wild-type p53 cells. Importantly, the central region of ASPP2 is ubiquitinated. Taken together, these data demonstrate that ASPP2 is a direct proteasomal substrate, and that proteasomal degradation modulates ASPP2 protein levels and apoptotic function. Furthermore, post-translational control of ASPP2 following DNA damage and stress induction may be an important mechanism by which ASPP2 stimulates apoptosis under these conditions.
iASPP modulates the apoptotic function of p53 codon 72 polymorphic variants
The most common p53 polymorphism whereby either an arginine (p53Arg72) or a proline (p53Pro72) is expressed at codon 72 only exists in humans and p53Arg72 is human-specific. The frequency of the allele encoding p53Pro72 is closely linked to latitude and is much higher in populations living near the equator suggesting that p53Pro72 is selected in an environment with high levels of UV light (Sjalander et al, 1995). A novel mechanism by which the ASPP family members selectively regulate the apoptotic function of p53Pro72 and p53Arg72 has been described (Bergamaschi et al, 2006). iASPP has a higher binding affinity to p53Pro72 than to p53Arg72, and preferentially modulates the apoptotic potential of p53Pro72 over p53Arg72. Furthermore, p53Arg72 is more active than p53Pro72 in the induction of apoptosis, and the greater apoptosis inducing activity of p53Arg72 results from its ability to escape iASPP inhibition. Thus, the most efficient way to inactivate the apoptotic function of p53Arg72 in human tumorigenesis is by intragenic mutation. An alternative way to inactivate the p53Pro72 isoform is by reduced expression of ASPP1, ASPP2 or overexpression of iASPP in addition to mutation in p53 itself. Therefore, the ASPP family provides an additional level of regulation of p53. This work suggests that the expression levels of the ASPP family members taken together with the expression of polymorphic p53 variants can provide clues about cancer susceptibility, prognosis and therapy.
Conclusion
Inactivation of ASPP1 and ASPP2, proteins that cooperate with p53 to induce apoptosis, and activation of the inhibitor of p53, iASPP, appear to be common themes in human tumours and are linked to poor prognosis and metastasis. Reduced expression of ASPP1 and ASPP2, or increased expression of iASPP, appear to be mechanisms involved in preventing wild-type p53 from working effectively. Hence, the ASPP family of proteins may be novel prognostic markers for human cancers. Identification of molecules that target the altered expression of the ASPP family members, whereby ASPP1 and ASPP2 expression could be enhanced or iASPP expression reduced, will allow us to develop new therapeutic strategies to treat cancer. For instance, a nine amino-acid peptide derived from ASPP2 (CBP3) was found to bind to the p53 core domain and stabilise it in vitro (Friedler et al, 2002). CBP3 was capable of restoring the apoptotic function of mutant p53, thereby killing mutant p53-expressing cells. From a chemical library screen, a small molecule, RITA (reactivation of p53 and induction of tumour cell apoptosis), was identified, which bound to p53 and induced its accumulation in tumour cells (Issaeva et al, 2004). RITA was shown to disrupt the interaction between p53 and MDM2, and was able to reactivate p53 in tumours that have aberrant MDM2 expression. The compound was also able to inhibit the interaction between iASPP and p53 and hence induce apoptosis.
The role of ASPP1, ASPP2 and iASPP in exerting positive and negative effects on the activity of p53 needs to be further explored in a tissue- and cell-type context. The altered expression of p53 activators such as ASPP1 and ASPP2 and inhibitors of p53 such as iASPP during tumour development in different tissue and cell types may explain the variation in p53 mutation rates in different tumour types. Furthermore, by taking into account both ASPP expression pattern and p53 codon 72 polymorphism, specific chemotherapeutic regimes could be tailored to different tumour types and individual patients.
Change history
16 November 2011
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Agirre X, Roman-Gomez J, Jimenez-Velasco A, Garate L, Montiel-Duarte C, Navarro G, Vazquez I, Zalacain M, Calasanz MJ, Heiniger A, Torres A, Minna JD, Prosper F (2006) ASPP1, a common activator of TP53, is inactivated by aberrant methylation of its promoter in acute lymphoblastic leukemia. Oncogene 25: 1862–1870
Bergamaschi D, Gasco M, Hiller L, Sullivan A, Syed N, Trigiante G, Yulug I, Merlano M, Numico G, Comino A, Attard M, Reelfs O, Gusterson B, Bell A, Heath V, Tavassoli M, Farrell P, Smith P, Lu X, Crook T (2003a) p53 polymorphism influences response in cancer chemotherapy via modulation of p73-dependent apoptosis. Cancer Cell 3: 387–402
Bergamaschi D, Samuels Y, Jin B, Duraisingham S, Crook T, Lu X (2004) ASPP1 and ASPP2: common activators of p53 family members. Mol Cell Biol 24: 1341–1350
Bergamaschi D, Samuels Y, O'Neil NJ, Trigiante G, Crook T, Hsieh JK, O'Connor DJ, Zhong S, Campargue I, Tomlinson ML, Kuwabara PE, Lu X (2003b) iASPP oncoprotein is a key inhibitor of p53 conserved from worm to human. Nat Genet 33: 162–167
Bergamaschi D, Samuels Y, Sullivan A, Zvelebil M, Breyssens H, Bisso A, Del Sal G, Syed N, Smith P, Gasco M, Crook T, Lu X (2006) iASPP preferentially binds the proline rich region of p53 and modulates the apoptotic function of codon 72 polymorphic p53. Nat Genet 38: 1133–1141
Cao Y, Hamada T, Matsui T, Date T, Iwabuchi K (2004) Hepatitis C virus core protein interacts with p53-binding protein, 53BP2/Bbp/ASPP2, and inhibits p53-mediated apoptosis. Biochem Biophys Res Commun 315: 788–795
Chen D, Padiernos E, Ding F, Lossos IS, Lopez CD (2005) Apoptosis-stimulating protein of p53-2 (ASPP2/53BP2L) is an E2F target gene. Cell Death Differ 12: 358–368
Chen Y, Liu W, Naumovski L, Neve RL (2003) ASPP2 inhibits APP-BP1-mediated NEDD8 conjugation to cullin-1 and decreases APP-BP1-induced cell proliferation and neuronal apoptosis. J Neurochem 85: 801–809
Cobleigh MA, Tabesh B, Bitterman P, Baker J, Cronin M, Liu ML, Borchik R, Mosquera JM, Walker MG, Shak S (2005) Tumor gene expression and prognosis in breast cancer patients with 10 or more positive lymph nodes. Clin Cancer Res 11: 8623–8631
Espanel X, Sudol M (2001) Yes-associated protein and p53-binding protein-2 interact through their WW and SH3 domains. J Biol Chem 276: 14514–14523
Fogal V, Kartasheva NN, Trigiante G, Llanos S, Yap D, Vousden KH, Lu X (2005) ASPP1 and ASPP2 are new transcriptional targets of E2F. Cell Death Differ 12: 369–376
Friedler A, Hansson LO, Veprintsev DB, Freund SM, Rippin TM, Nikolova PV, Proctor MR, Rudiger S, Fersht AR (2002) A peptide that binds and stabilizes p53 core domain: chaperone strategy for rescue of oncogenic mutants. Proc Natl Acad Sci USA 99: 937–942
Gorina S, Pavletich NP (1996) Structure of the p53 tumor suppressor bound to the ankyrin and SH3 domains of 53BP2. Science 274: 1001–1005
Hershko T, Chaussepied M, Oren M, Ginsberg D (2005) Novel link between E2F and p53: proapoptotic cofactors of p53 are transcriptionally upregulated by E2F. Cell Death Differ 12: 377–383
Issaeva N, Bozko P, Enge M, Protopopova M, Verhoef LG, Masucci M, Pramanik A, Selivanova G (2004) Small molecule RITA binds to p53, blocks p53-HDM-2 interaction and activates p53 function in tumors. Nat Med 10: 1321–1328
Ju H, Lee KA, Yang M, Kim HJ, Kang CP, Sohn TS, Rhee JC, Kang C, Kim JW (2005) TP53BP2 locus is associated with gastric cancer susceptibility. Int J Cancer 117: 957–960
Lettre G, Kritikou EA, Jaeggi M, Calixto A, Fraser AG, Kamath RS, Ahringer J, Hengartner MO (2004) Genome-wide RNAi identifies p53-dependent and -independent regulators of germ cell apoptosis in C. elegans. Cell Death Differ 11: 1198–1203
Liu ZJ, Lu X, Zhang Y, Zhong S, Gu SZ, Zhang XB, Yang X, Xin HM (2005) Downregulated mRNA expression of ASPP and the hypermethylation of the 5′-untranslated region in cancer cell lines retaining wild-type p53. FEBS Lett 579: 1587–1590
Liu ZJ, Zhang Y, Zhang XB, Yang X (2004) Abnormal mRNA expression of ASPP members in leukemia cell lines. Leukemia 18: 880
Lossos IS, Natkunam Y, Levy R, Lopez CD (2002) Apoptosis stimulating protein of p53 (ASPP2) expression differs in diffuse large B-cell and follicular center lymphoma: correlation with clinical outcome. Leuk Lymphoma 43: 2309–2317
Merlo A, Herman JG, Mao L, Lee DJ, Gabrielson E, Burger PC, Baylin SB, Sidransky D (1995) 5′ CpG island methylation is associated with transcriptional silencing of the tumour suppressor p16/CDKN2/MTS1 in human cancers. Nat Med 1: 686–692
Mori S, Ito G, Usami N, Yoshioka H, Ueda Y, Kodama Y, Takahashi M, Fong KM, Shimokata K, Sekido Y (2004) p53 apoptotic pathway molecules are frequently and simultaneously altered in nonsmall cell lung carcinoma. Cancer 100: 1673–1682
Mori T, Okamoto H, Takahashi N, Ueda R, Okamoto T (2000) Aberrant overexpression of 53BP2 mRNA in lung cancer cell lines. FEBS Lett 465: 124–128
Nakagawa H, Koyama K, Murata Y, Akiyama T, Nakamura Y (2000) APCL, a central nervous system-specific homologue of adenomatous polyposis coli tumour suppressor binds to 53-binding protein 2 and translocates it to the perinucleus. Cancer Res 60: 101–105
Naumovski L, Cleary ML (1996) The p53-binding protein 53BP2 also interacts with bcl2 and impedes cell cycle progression at G2/M. Mol Cell Biol 16: 3884–3892
Ohno A, Hirashima T, Kubo A, Masuda N, Takada M, Fujiwara H, Yasumitsu T, Kiku M, Fukuoka M, Nakagawa K (1997) p53 status and prognosis in stage I-IIIa non-small cell lung cancer. Int J Oncol 10: 521–528
Roman-Gomez J, Jimenez-Velasco A, Agirre X, Prosper F, Heiniger A, Torres A (2005) Lack of CpG island methylator phenotype defines a clinical subtype of T-cell acute lymphoblastic leukemia associated with good prognosis. J Clin Oncol 23: 7043–7049
Rowan S, Ludwig RL, Haupt Y, Bates S, Lu X, Oren M, Vousden KH (1996) Specific loss of apoptotic but not cell-cycle arrest function in a human tumour derived p53 mutant. EMBO 15: 827–838
Samuels-Lev Y, O'Connor DJ, Bergamaschi D, Trigiante G, Hsieh JK, Zhong S, Campargue I, Naumovski L, Crook T, Lu X (2001) ASPP proteins specifically stimulate the apoptotic function of p53. Mol Cell 8: 781–794
Sarraf SA, Stancheva I (2004) Methyl-CpG binding protein MBD1 couples histone H3 methylation at lysine 9 by SETDB1 to DNA replication and chromatin assembly. Mol Cell 15: 595–605
Sgroi DC, Teng S, Robinson G, Le Vangie R, Hudson JR, Elkahloun AG (1999) In vivo gene expression profile analysis of human breast cancer progression. Cancer Res 59: 5656–5661
Sjalander A, Birgander R, Kivela A, Beckman G (1995) p53 polymorphisms and haplotypes in different ethnic groups. Hum Hered 45: 144–149
Stanelle J, Stiewe T, Theseling CC, Peter M, Putzer BM (2002) Gene expression changes in response to E2F1 activation. Nucleic Acids Res 30: 1859–1867
Takahashi N, Kobayashi S, Kajino S, Imai K, Tomoda K, Shimizu S, Okamoto T (2005) Inhibition of the 53BP2S-mediated apoptosis by nuclear factor kappaB and Bcl-2 family proteins. Genes Cells 10: 803–811
Thornton JK, Dalgleish C, Venables JP, Sergeant KA, Ehrmann IE, Lu X, Saunders PT, Elliott DJ (2006) The tumour-suppressor protein ASPP1 is nuclear in human germ cells and can modulate ratios of CD44 exon V5 spliced isoforms in vivo. Oncogene 25: 3104–3112
Vives V, Su J, Zhong S, Ratnayaka I, Slee E, Goldin R, Lu X (2006) ASPP2 is a haploinsufficient tumor suppressor that cooperates with p53 to suppress tumor growth. Genes Dev 20: 1262–1267
Yang J-P, Hori M, Sanda T, Okamoto T (1999a) Identification of a novel inhibitor of nuclear factor-kB, RelA-associated inhibitor. J Biol Chem 274: 15662–15670
Yang J-P, Hori M, Takahashi N, Kawabe T, Kato H, Okamoto T (1999b) NF-kB subunit p65 binds to 53BP2 and inhibits cell death induced by 53BP2. Oncogene 18: 5177–5186
Zhang X, Wang M, Zhou C, Chen S, Wang J (2005) The expression of iASPP in acute leukemias. Leuk Res 29: 179–183
Zhu Z, Ramos J, Kampa K, Adimoolam S, Sirisawad M, Yu Z, Chen D, Naumovski L, Lopez CD (2005) Control of ASPP2/(53BP2L) protein levels by proteasomal degradation modulates p53 apoptotic function. J Biol Chem 280: 34473–34480
Acknowledgements
We thank the Ludwig Institute for Cancer Research for supporting the work and Drs D O'Connor and E Slee for reading the manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
This article is cited by
-
Evaluation of apoptosis stimulating protein of TP53-1 (ASPP1/PPP1R13B) to predict therapy resistance and overall survival in acute myeloid leukemia (AML)
Cell Death & Disease (2024)
-
Regulation of immunological tolerance by the p53-inhibitor iASPP
Cell Death & Disease (2023)
-
Knockout of ASPP2 promotes DEN-induced hepatocarcinogenesis via the NF-κB pathway in mice
Cancer Gene Therapy (2022)
-
Both IRBIT and long-IRBIT bind to and coordinately regulate Cl−/HCO3− exchanger AE2 activity through modulating the lysosomal degradation of AE2
Scientific Reports (2021)
-
ASPP2 suppression promotes malignancy via LSR and YAP in human endometrial cancer
Histochemistry and Cell Biology (2020)
