
Abstract
Although prostate cancer (PrCa) is one of the most common cancers in men in Western countries, little is known about the inherited factors that influence PrCa risk. On the basis of the fact that BRIP1/FANCJ interacts with BRCA1 and functions as a regulator of DNA double-strand break repair pathways, and that germline mutations within the BRIP1/FANCJ gene predispose to breast cancer, we chose this gene as a candidate for mutation screening in familial and young-onset PrCa cases. We identified a truncating mutation, R798X, in the BRIP1/FANCJ gene in 4 out of 2714 UK PrCa cases enriched for familial (2 out of 641; 0.3%) and young-onset cases (2 out of 2073; 0.1%). On screening 2045 controls from the UK population, we found one R798X sequence alteration (0.05%; odds ratio 2.4 (95% CI 0.25–23.4)). In addition, using our data from a genome-wide association study, we analysed 25 SNPs in the genomic region of the BRIP1/FANCJ gene. Two SNPs showed evidence of association with familial and young-onset PrCa (rs6504074; Ptrend=0.04 and rs8076727; Ptrend=0.01). These results suggest that truncating mutations in BRIP1/FANCJ might confer an increased risk of PrCa and common SNPs might also contribute to the alteration of risk, but larger case–control series will be required to confirm or refute this association.
Similar content being viewed by others


Main
Prostate cancer (PrCa) aggregates in families, in a consistent manner with an important inherited component (reviewed in Edwards and Eeles, 2004). The genetic components underlying this familial risk have, however, proved difficult to identify. Genetic studies using linkage analysis in high-risk families have identified several possible susceptibility loci (Tavtigian et al, 2001; Carpten et al, 2002), but none of them have been definitively established (Easton et al, 2003). It seems likely now that susceptibility to PrCa is mediated, at least partially, through a combination of multiple low-penetrance loci (Haiman et al, 2007; Eeles et al, 2008; Thomas et al, 2008).
Genome-wide association studies (GWAS) have identified common variants in several regions that are associated with PrCa risk (Eeles et al, 2008; Gudmundsson et al, 2008; Thomas et al, 2008). In addition, resequencing of candidate genes, notably those involved in DNA double-strand break repair, has identified rarer variants associated with a more substantial risk. The most important of these is BRCA2 mutations, which confer a risk of approximately five-fold, but there is evidence that truncating mutations in NBS1 and CHEK2 also confer susceptibility to PrCa (Dong et al, 2003; Edwards et al, 2003; Cybulski et al, 2004; Agalliu et al, 2007).
The BRIP1/FANCJ gene encodes a helicase in which the C-terminal domain is reported to interact with BRCA1 (Cantor et al, 2001). The BRIP1 gene spans 180 kb, comprises 20 exons and encodes a protein of 1249 amino acids. It is located on chromosome 17q22, distal to BRCA1, which is at 17q21. BRIP1/FANCJ has an important role in BRCA-associated DNA damage repair functions, works as a DNA-dependent ATPase and a DNA helicase, and is essential for DNA repair and genomic stability. It forms a complex with the BRCT domain of BRCA1, and this is important for the role of BRCA1 in its DNA double-strand break repair function (Levitus et al, 2006).
Germline mutations in BRIP1/FANCJ are associated with Fanconi anaemia, a chromosomal instability syndrome characterised by developmental abnormalities and predisposition to cancer (Levitus et al, 2005). Truncating mutations in the BRIP1/FANCJ gene have recently been shown to be associated with a moderate risk of breast cancer (Seal et al, 2006). Given the observed associations of PrCa with other DNA repair genes, we considered BRIP1/FANCJ as an attractive candidate for PrCa-predisposition gene.
Materials and methods
Whole blood samples from PrCa cases were collected as part of the UK Genetic Prostate Cancer Study (UKGPCS), a national study of familial and young cases of PrCa. Familial samples were from 192 cases with two affected relatives (‘strong family history’) and 449 other samples with one affected relative (‘some family history’). One person per family was used for sequence analysis. Wherever possible, the youngest affected family member was considered. Young-onset cases comprised 2073 men with a diagnosis of PrCa at age ⩽60 years. All self-reported non-Caucasians were excluded. The study was approved by the multiregional ethics committee and all patients gave informed consent. All PrCa diagnosis was based on the verification of the histological report or death certificate.
Caucasian controls comprised 2045 blood DNA samples, which were selected through the ProtecT study. ProtecT is a national study of community-based PSA testing and a randomised trial of subsequent PrCa treatment. Men between the ages of 50 and 69 years are recruited through general practices in nine centres in the UK. Ethnically matched controls were sampled from these centres to match approximately the geographical distribution of the cases. DNA was extracted using standard methods as described in Eeles et al (2008).
The full coding sequence and exon–intron boundaries of the 192 familial samples were analysed by sequencing. The sequencing was carried out using the BigDyeTerminator v3.2 Cycle Sequencing kit and the 3700 automated sequencer (Applied Biosystems, Foster City, CA, USA).
All the additional samples, 2073 young-onset cases and 2045 UK population controls were tested for the R798X sequence variant by the 5′nuclease assay (Taqman™) using the ABIPrism 7900HT sequence detection system according to the manufacturer's instructions. Primers and probes were supplied directly by Applied Biosystems (http://www.appliedbiosystems.com/) as Assays-By-Design™.
We had earlier carried out a GWAS, involving 1854 of the PrCa cases and 1894 controls using the Illumina Infinium HumanHap550 array (Eeles et al, 2008). From these data, we identified 25 SNPs from the 270-kb region covering the genomic sequences of BRIP1/FANCJ, which were used to obtain evidence for associations between common variants and PrCa risk.
Significance tests, odds ratios (ORs) and confidence limits were computed using standard methods. For the SNPs, both Cochran–Armitage trend tests and 2-degrees-of-freedom tests were calculated. For Hardy–Weinberg equilibrium and Armitage trend testing, we used the public software developed by Tim M Strom and Thomas F Wienker (http://ihg.gsf.de/cgi-bin/hw/hwa1.pl).
Results
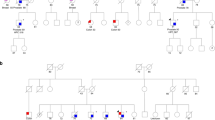
Initially, a set of genomic DNA samples representing the youngest affected individual from 192 multiple case PrCa families was screened. The full coding sequence and exon–intron boundaries of the BRIP1/FANCJ were analysed by sequencing. A truncating mutation, R798X (1 out of 192, 0.52%) was identified in the proband from a family with three affected brothers (Table 1). The pedigree and the result of the segregation analysis are shown in Figure 1. The available blood samples were tested for this germline mutation and one affected brother carried the mutation, whereas the unaffected brother did not. Only a paraffin-embedded tissue sample (biopsy) was available from the third affected brother, and sequencing the DNA sample from this did not show the mutation. Blood samples from his three sons were also tested and were all found to be non-carriers for the R798X mutation. In addition to this deleterious mutation, several missense mutations were identified in the 192 familial samples, as presented in Table 2. No other mutations that were predicted to truncate the protein were identified.

Prostate cancer (PC) family with the R798X nonsense mutation in BRIP1/FANCJ. Wild-type (WT) labels on individuals tested but non-carriers of R798X, Leu=leukaemia, SC=stomach cancer. (The pedigree has been modified to maintain confidentiality.)
To establish the contribution of the BRIP1/FANCJ R798X mutation to PrCa predisposition, we screened a further 449 samples with a single affected relative and 2073 young-onset PrCa cases, (individuals diagnosed with PrCa before 60 years of age with no family history) and 2045 controls. The R798X mutation was present in two of the young-onset cases (0.10%), in one of the familial cases (0.22%), and in one of the controls (0.05%) (Table 1). The ages of onset of the mutation carriers were 49 and 60 years. Neither case had a family history of PrCa. However, one of these mutation carriers had a family history of other cancers and malformations, which appear characteristic of Gorlin syndrome. The familial case had one affected second-degree relative with an unknown age of onset, and also had relatives affected with other cancers (colon and stomach cancer).
On the basis of our data, the estimated OR for PrCa was 2.4 (95% CI 0.25–23.4). If the control data of Seal et al (2006) are also considered in our analysis, the estimate is somewhat more precise (OR 2.5, 95% CI 0.41–14.7) but still not significantly different from 1.
We found five other amino-acid substitutions in the 192 familial samples sequenced (Table 2). All have been reported earlier (Seal et al, 2006; Guénard et al, 2008). All of these sequence variants were also found in the control series. The common variant p.Pro919Ser showed some evidence of an association with risk (Ptrend=0.034) in this set of cases with strong family history. However, this SNP (rs4986764) was also included on the Illumina 550k array used in our GWAS (Eeles et al, 2008). In this larger study of familial and young-onset cases there was no evidence of association with PrCa (Ptrend=0.48). None of the four rare variants showed a difference in frequency between cases and controls.
We also analysed the genotype frequencies of 25 SNPs from the Illumina 550k array in the genomic region of FANCJ/BRIP1 for 1853 PrCa cases and 1880 control individuals (Eeles et al, 2008). We found significant differences in genotype frequencies between cases and controls for two SNPs: rs6504074 and rs8076727 (Table 3). The estimated per allele OR for rs6504074 was 1.10 (Ptrend=0.029); this rose to 1.20 (Ptrend=0.01) when the analysis was restricted to familial cases. The estimated OR for rs8076727 was 1.29 (P=0.01); this rose to 1.39 (P=0.01) for familial cases.
Discussion
We have identified a recurrent deleterious mutation in the BRIP1/FANCJ gene in a set of familial and young-onset PrCa cases.
This truncating mutation has been reported earlier in four Fanconi anaemia subtype J families of diverse geographic origin (Levitus et al, 2005) and in five breast cancer families (Seal et al, 2006), suggesting that it is a relatively ancient founder mutation. The frequency of this mutation in our control population is almost identical to that found in the study by Seal et al (2006) (1 in 2081).
This recurrent R798X mutation in exon 17 predicts a truncated protein in which the helicase motif VI and the BRCA1 interacting regions are deleted. BRIP1/FANCJ binds directly to the C-terminal BRCA repeats of BRCA1 and this interaction is regulated in a cell-cycle-dependent manner. However, in the Fanconi anaemia pathway for DNA crosslink repair, BRIP1/FANCJ functions independently of this interaction (Bridge et al, 2005).
BRIP1/FANCJ encodes a DEAH-box helicase and can directly interact with DNA (Bridge et al, 2005). It can unwind DNA structures such as Holliday junctions formed during homologous recombination, suggesting an association with some DNA repair processes (Levitus et al, 2006). It is interesting that most of the Fanconi anaemia genes encode proteins that form a nuclear core complex with a function of monoubiquitination of FANCD2, but FANCD1 (BRCA2) and BRIP1/FANCJ function downstream of this and they both have a more defined role in DNA damage repair.
We have reported earlier that 2% of men diagnosed with PrCa at <55 years had a germline mutation in the BRCA2 gene (Edwards et al, 2003). BRCA2 is a high-risk breast cancer predisposition gene and is also known as FANCD1, responsible for causing FA-D1. It is therefore plausible that truncating mutations in BRID1/FANCJ also predispose to PrCa. Our results for the R798X mutation are consistent with a moderate risk of PrCa, comparable to that for breast cancer, but larger numbers will be required to confirm this.
We found five other amino-acid substitutions in the 192 familial samples sequenced (Table 2), all of which have been reported earlier (Seal et al, 2006; Guénard et al, 2008). Three of these variants are situated within the Rad3-related DNA helicase domain (res.1–888) and one, the common p.Pro919Ser variant, is within the BRCA1-binding domain (Bridge et al, 2005). The p.Arg173 is a strongly conserved residue in mammals, whereas the other variants are poorly conserved in other species (Guénard et al, 2008). We found no evidence for significant association between these sequence variants and PrCa risk.
We have also evaluated the association between 25 common SNPs and the risk of PrCa in a case–control design and found two SNPs significantly associated with PrCa risk. Both of the associated SNPs are in intron 6. These associations, although nominally significant, do not reach the stringent levels of significance appropriate for candidate gene or genome-wide scans, and could have occurred by chance. However, if verified in larger series, they could be markers for common variants in BRID1/FANCJ influencing the risk.
In conclusion, we report that deleterious mutations in BRIP1/FANCJ occur in a modest number of familial and young-onset PrCa cases. In addition, we found evidence of association with two common SNPs. These observations warrant further evaluation in independent case–control studies.
Change history
16 November 2011
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Agalliu I, Karlins E, Kwon EM, Iwasaki LM, Diamond A, Ostrander EA, Stanford JL (2007) Rare germline mutations in the BRCA2 gene are associated with early-onset prostate cancer. Br J Cancer 97: 826–831
Bridge WL, Vandenberg CJ, Franklin RJ, Hiom K (2005) The BRIP1 helicase functions independently of BRCA1 in the Fanconi anemia pathway for DNA crosslink repair. Nat Genet 37: 953–957
Cantor SB, Bell DW, Ganesan S, Kass EM, Drapkin R, Grossman S, Wahrer DC, Sgroi DC, Lane WS, Haber DA, Livingston DM (2001) BACH1, a novel helicase-like protein, interacts directly with BRCA1 and contributes to its DNA repair function. Cell 105: 149–160
Carpten J, Nupponen N, Isaacs S, Sood R, Robbins C, Xu J, Faruque M, Moses T, Ewing C, Gillanders E, Hu P, Bujnovszky P, Makalowska I, Baffoe-Bonnie A, Faith D, Smith J, Stephan D, Wiley K, Brownstein M, Gildea D, Kelly B, Jenkins R, Hostetter G, Matikainen M, Schleutker J, Klinger K, Connors T, Xiang Y, Wang Z, De Marzo A, Papadopoulos N, Kallioniemi OP, Burk R, Meyers D, Grönberg H, Meltzer P, Silverman R, Bailey-Wilson J, Walsh P, Isaacs W, Trent J (2002) Germline mutations in the ribonuclease L gene in families showing linkage with HPC1. Nat Genet 30: 181–184
Cybulski C, Górski B, Debniak T, Gliniewicz B, Mierzejewski M, Masojæ B, Jakubowska A, Matyjasik J, Złowocka E, Sikorski A, Narod SA, Lubiñski J (2004) NBS1 is a prostate cancer susceptibility gene. Cancer Res 64: 1215–1219
Dong X, Wang L, Taniguchi K, Wang X, Cunningham JM, McDonnell SK, Qian C, Marks AF, Slager SL, Peterson BJ, Smith DI, Cheville JC, Blute ML, Jacobsen SJ, Schaid DJ, Tindall DJ, Thibodeau SN, Liu W (2003) Mutations in CHEK2 associated with prostate cancer risk. Am J Hum Genet 72: 270–280
Easton DF, Schaid DJ, Whittemore AS, Isaacs WJ, International Consortium for Prostate Cancer Genetics (2003) Where are the prostate cancer genes? A summary of eight genome wide searches. Prostate 57: 261–269
Edwards SM, Eeles RA (2004) Unravelling the genetics of prostate cancer. Am J Med Genet C Semin Med Genet 129: 65–73
Edwards SM, Kote-Jarai Z, Meitz J, Hamoudi R, Hope Q, Osin P, Jackson R, Southgate C, Singh R, Falconer A, Dearnaley DP, Ardern-Jones A, Murkin A, Dowe A, Kelly J, Williams S, Oram R, Stevens M, Teare DM, Ponder BA, Gayther SA, Easton DF, Eeles RA, Cancer Research UK/British Prostate Group UK Familial Prostate Cancer Study Collaborators, British Association of Urological Surgeons Section of Oncology (2003) Two percent of men with early-onset prostate cancer harbor germline mutations in the BRCA2 gene. Am J Hum Genet 72: 1–12
Eeles RA, Kote-Jarai Z, Giles GG, Olama AA, Guy M, Jugurnauth SK, Mulholland S, Leongamornlert DA, Edwards SM, Morrison J, Field HI, Southey MC, Severi G, Donovan JL, Hamdy FC, Dearnaley DP, Muir KR, Smith C, Bagnato M, Ardern-Jones AT, Hall AL, O’Brien LT, Gehr-Swain BN, Wilkinson RA, Cox A, Lewis S, Brown PM, Jhavar SG, Tymrakiewicz M, Lophatananon A, Bryant SL, Horwich A, Huddart RA, Khoo VS, Parker CC, Woodhouse CJ, Thompson A, Christmas T, Ogden C, Fisher C, Jamieson C, Cooper CS, English DR, Hopper JL, Neal DE, Easton DF, UK Genetic Prostate Cancer Study Collaborators, British Association of Urological Surgeons’ Section of Oncology, UK ProtecT Study Collaborators (2008) Multiple newly identified loci associated with prostate cancer susceptibility. Nat Genet 40: 316–321
Gudmundsson J, Sulem P, Rafnar T, Bergthorsson JT, Manolescu A, Gudbjartsson D, Agnarsson BA, Sigurdsson A, Benediktsdottir KR, Blondal T, Jakobsdottir M, Stacey SN, Kostic J, Kristinsson KT, Birgisdottir B, Ghosh S, Magnusdottir DN, Thorlacius S, Thorleifsson G, Zheng SL, Sun J, Chang BL, Elmore JB, Breyer JP, McReynolds KM, Bradley KM, Yaspan BL, Wiklund F, Stattin P, Lindström S, Adami HO, McDonnell SK, Schaid DJ, Cunningham JM, Wang L, Cerhan JR, St Sauver JL, Isaacs SD, Wiley KE, Partin AW, Walsh PC, Polo S, Ruiz-Echarri M, Navarrete S, Fuertes F, Saez B, Godino J, Weijerman PC, Swinkels DW, Aben KK, Witjes JA, Suarez BK, Helfand BT, Frigge ML, Kristjansson K, Ober C, Jonsson E, Einarsson GV, Xu J, Gronberg H, Smith JR, Thibodeau SN, Isaacs WB, Catalona WJ, Mayordomo JI, Kiemeney LA, Barkardottir RB, Gulcher JR, Thorsteinsdottir U, Kong A, Stefansson K (2008) Common sequence variants on 2p15 and Xp11@22 confer susceptibility to prostate cancer. Nat Genet 40: 281–283
Guénard F, Labrie Y, Ouellette G, Joly Beauparlant C, Simard J, Durocher F, INHERIT BRCAs (2008) Mutational analysis of the breast cancer susceptibility gene BRIP1 /BACH1/FANCJ in high-risk non-BRCA1/BRCA2 breast cancer families. J Hum Genet 53: 579–591
Haiman CA, Patterson N, Freedman ML, Myers SR, Pike MC, Waliszewska A, Neubauer J, Tandon A, Schirmer C, McDonald GJ, Greenway SC, Stram DO, Le Marchand L, Kolonel LN, Frasco M, Wong D, Pooler LC, Ardlie K, Oakley-Girvan I, Whittemore AS, Cooney KA, John EM, Ingles SA, Altshuler D, Henderson BE, Reich D (2007) Multiple regions within 8q24 independently affect risk for prostate cancer. Nat Genet 39: 638–644
Levitus M, Joenje H, de Winter JP (2006) The Fanconi anemia pathway of genomic maintenance. Cell Oncol 28: 3–29
Levitus M, Waisfisz Q, Godthelp BC, de Vries Y, Hussain S, Wiegant WW, Elghalbzouri-Maghrani E, Steltenpool J, Rooimans MA, Pals G, Arwert F, Mathew CG, Zdzienicka MZ, Hiom K, De Winter JP, Joenje H (2005) The DNA helicase BRIP1 is defective in Fanconi anemia complementation group J. Nat Genet 37: 934–935
Seal S, Thompson D, Renwick A, Elliott A, Kelly P, Barfoot R, Chagtai T, Jayatilake H, Ahmed M, Spanova K, North B, McGuffog L, Evans DG, Eccles D, Easton DF, Stratton MR, Rahman N, Breast Cancer Susceptibility Collaboration (UK) (2006) Truncating mutations in the Fanconi anemia J gene BRIP1 are low-penetrance breast cancer susceptibility alleles. Nat Genet 38: 1239–1241
Tavtigian SV, Simard J, Teng DH, Abtin V, Baumgard M, Beck A, Camp NJ, Carillo AR, Chen Y, Dayananth P, Desrochers M, Dumont M, Farnham JM, Frank D, Frye C, Ghaffari S, Gupte JS, Hu R, Iliev D, Janecki T, Kort EN, Laity KE, Leavitt A, Leblanc G, McArthur-Morrison J, Pederson A, Penn B, Peterson KT, Reid JE, Richards S, Schroeder M, Smith R, Snyder SC, Swedlund B, Swensen J, Thomas A, Tranchant M, Woodland AM, Labrie F, Skolnick MH, Neuhausen S, Rommens J, Cannon-Albright LA (2001) A candidate prostate cancer susceptibility gene at chromosome 17p. Nat Genet 27: 172–180
Thomas G, Jacobs KB, Yeager M, Kraft P, Wacholder S, Orr N, Yu K, Chatterjee N, Welch R, Hutchinson A, Crenshaw A, Cancel-Tassin G, Staats BJ, Wang Z, Gonzalez-Bosquet J, Fang J, Deng X, Berndt SI, Calle EE, Feigelson HS, Thun MJ, Rodriguez C, Albanes D, Virtamo J, Weinstein S, Schumacher FR, Giovannucci E, Willett WC, Cussenot O, Valeri A, Andriole GL, Crawford ED, Tucker M, Gerhard DS, Fraumeni Jr JF, Hoover R, Hayes RB, Hunter DJ, Chanock SJ (2008) Multiple loci identified in a genome-wide association study of prostate cancer. Nat Genet 40: 310–315
Acknowledgements
We thank all the patients and control men who took part in this study. This work was supported by The Prostate Cancer Research Foundation and Cancer Research UK Grant C5047/A3354. DFE is a Principal Research Fellow of Cancer Research UK. We would also like to thank the following for funding: The Institute of Cancer Research and The Everyman Campaign, Prostate Research Campaign UK, The National Cancer Research Network UK, The National Cancer Research Institute (NCRI) UK and NHS funding to the NIHR Biomedical Research Centre. The ProtecT study is ongoing and is funded by the Health Technology Assessment Programme (projects 96/20/06, 96/20/99) and Cancer Research UK Grant Number C522/A8649. We would like to acknowledge the tremendous contribution of all members of the ProtecT study research group.
Author information
Authors and Affiliations
Consortia
Corresponding authors
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Kote-Jarai, Z., Jugurnauth, S., Mulholland, S. et al. A recurrent truncating germline mutation in the BRIP1/FANCJ gene and susceptibility to prostate cancer. Br J Cancer 100, 426–430 (2009). https://doi.org/10.1038/sj.bjc.6604847
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.bjc.6604847
Keywords
This article is cited by
-
Multimolecular characteristics and role of BRCA1 interacting protein C-terminal helicase 1 (BRIP1) in human tumors: a pan-cancer analysis
World Journal of Surgical Oncology (2023)
-
Spectrum of BRCA1 interacting helicase 1 aberrations and potential prognostic and therapeutic implication: a pan cancer analysis
Scientific Reports (2023)
-
Familiäres Prostatakarzinom und genetische Prädisposition
Der Urologe (2021)
-
BRIP1, RAD51C, and RAD51D mutations are associated with high susceptibility to ovarian cancer: mutation prevalence and precise risk estimates based on a pooled analysis of ~30,000 cases
Journal of Ovarian Research (2020)
-
Role of the DNA damage response in prostate cancer formation, progression and treatment
Prostate Cancer and Prostatic Diseases (2020)
