
Abstract
Background:
Telomeres, located at chromosome ends, are progressively shortened during each cell cycle by replication-dependent loss of DNA termini. Although maintenance of telomere length is critical for cell-replicative potential and tumourigenesis, the erosion of telomeres can lead to genetic instability, a pivotal mechanism in the neoplastic process.
Patients and methods:
A total of 118 colorectal cancer (CRC) samples (53 right-colon, 30 left-colon, and 35 rectal tumours) and corresponding adjacent non-cancerous tissues were evaluated for telomere length, p53 mutation, and microsatellite instability (MSI). Telomere length was estimated by real-time PCR.
Results:
Telomeres were significantly shorter in CRCs than in adjacent tissues, regardless of tumour stage and grade, site, or genetic alterations (P<0.0001). Moreover, in normal tissues, but not in tumours, telomere length inversely correlated with age (r=−0.24, P=0.017). Telomere length in CRCs did not differ with tumour progression or p53 status; however, in CRCs carrying the wild-type p53, telomeres were significantly shorter in tumours with MSI than in those with stable microsatellites (P=0.027). Furthermore, telomere length differed according to tumour location, being longer in rectal cancers (P=0.03).
Conclusions:
These findings suggest that telomere shortening is a key initial event in colorectal carcinogenesis. The extent of telomere erosion is related to tumour origin site and may be influenced by the mismatch repair pathway.
Similar content being viewed by others


Main
Colorectal cancers (CRCs) encompass a heterogeneous complex of diseases differing in molecular pathways and biological characteristics, arising through a multistep carcinogenic process, of which several genetic and epigenetic events have been characterised. There are at least two major pathways by which molecular events can lead to CRC: loss of heterozygosity with chromosomal instability, and a deficient DNA mismatch repair pathway with microsatellite instability (MSI) (Grady and Carethers, 2008; Ogino and Goel, 2008; Koopman et al, 2009). Although p53 and K-ras genes are known to be involved in chromosomal instability-associated carcinogenesis, MSI is frequently associated with the CpG island methylator phenotype (Barault et al, 2008). These two pathways are known to have a different origin and clinical outcome; right-sided proximal colon tumours are often MSI-associated lesions and have a better prognosis than left-sided distal tumours (Iacopetta, 2002; Gervaz et al, 2004; Walther et al, 2008). Rectal tumours are thought to arise through mechanisms similar to left-colon cancers; recently, it has been proposed that they constitute a third entity displaying specific characteristics (Li and Lai, 2009).
Telomere dysfunction has emerged as having a causative role in carcinogenesis by promoting genetic instability. Indeed, telomeres are specialised DNA structures located at the end of chromosomes; they are composed of (TTAGGG)n tandem repeats and are essential for stabilising chromosomes by protecting them from end-to-end fusion and DNA degradation. Telomeres are progressively shortened during each cell-replication cycle because of end-replication problems of DNA polymerase, and telomere shortening induces somatic cells to undergo senescence and apoptosis (reviewed in Blackburn et al, 2006). Further erosion of telomeres may impair their function in protecting chromosome ends, resulting in genetic instability. Although maintenance of telomere length by reverse transcriptase telomerase (hTERT) is critical to preserving the replicative potential of cancer cells (Hiyama and Hiyama, 2007; Artandi and DePinho, 2010), telomere erosion has been proposed to have two conflicting roles: tumour suppression and genetic instability, a key event in the initiation of carcinogenesis (Hackett and Greider, 2002; Meeker et al, 2004; Perera et al, 2008).
The role of telomere dysfunction in colorectal carcinogenesis is still largely undefined. Several studies demonstrated that telomeres were shorter in CRCs than in adjacent normal mucosa (Hastie et al, 1990; Takagi et al, 1999; Gertler et al, 2004; Garcia-Aranda et al, 2006), but this finding was not confirmed by other studies (Katayama et al, 1999; O'Sullivan et al, 2006). Telomeres were also found to be significantly shorter in high-grade dysplastic lesions (Raynaud et al, 2008) and large adenomas (O'Sullivan et al, 2006) than in normal adjacent mucosa; subsequent activation of telomerase may explain the increase of telomere length from preneoplastic lesions to invasive carcinoma (Raynaud et al, 2008). However, the relationship between telomere length and tumour progression remains to be assessed. Moreover, few studies have addressed the relationship of telomere length with the site of tumour origin and/or molecular pathways of colorectal carcinogenesis. The suggestion that right-colon cancers have shorter telomeres than tumours arising in other sites (Takagi et al, 1999; Garcia-Aranda et al, 2006) has not been confirmed in other studies (Engelhardt et al, 1997; Gertler et al, 2004). One study suggested that telomeres were shorter in MSI tumours than in cancers with stable microsatellites (MSS) (Takagi et al, 2000), but a relationship between telomere length and markers of chromosome instability in MSS tumours has not been established yet.
Telomere length has traditionally been estimated by Southern blotting using enzymes with restriction sites in subtelomeric DNA. However, the large amounts of DNA required by this method limit its use in many biological samples. Using a real-time quantitative PCR method, we analysed telomere length in a large series of CRCs and we then determined the association of this parameter with tumour stage, location, and markers of genetic instability.
Patients and methods
Patients
For this study, 118 cases were selected from patients with a histopathological confirmation of CRC who underwent surgical resection at a single institution. The criteria of selection were the availability of adjacent non-cancerous mucosa and data on p53 gene mutation and MSI status (n=193). In all, 28 tumour samples (14.5%) were found to have a high degree of MSI (MSI-H); this percentage reflected that reported in literature (Kim et al, 2007; Ogino et al, 2009) and all 28 were included in this study. In agreement with previous reports (Kim et al, 2007; Grady and Carethers, 2008; Ogino et al, 2009), most of the MSI-H samples (24 of 28) had the wild-type p53 gene. An equal number of age-matched MSS tumours were selected with the wild-type (n=45) or mutated (P=45) p53 gene. The median age of selected patients was 65 years (range 41–82 years); 49 were women and 69 were men. According to the American Joint Committee on Cancer staging (Greene et al, 2002), 23 tumours were classified as stage I (T1–T2, N0, M0), 37 as stage II (T3–T4, N0, M0), 29 as stage III (any T, N1–N2, M0), and 29 as stage IV (any T, any N, M1). Moreover, 18 tumours were classified as grade 1, 78 as grade 2, 17 as grade 3, and 5 were mucinous. Grading was assessed independently by two pathologists. Tumours were located in the right colon in 53 cases (44.9%), in the left colon in 30 cases (25.4%), and in the rectum in 35 cases (29.7%). At the time of sampling, each patient signed the informed consent form approving the use of his or her biological samples for research purposes. The study was approved by the local ethical committee.
Samples
Tissues specimens were obtained at the time of surgery, immediately shock frozen in liquid nitrogen, and stored at −80°C until use. Cryostat sections of 6 μm thickness from each tissue sample were prepared using a 1720 Digital cryostat (Leitz, Germany). One section of each sample was stained with haematoxylin–eosin for histopathology. All tumour samples analysed for telomere length contained >80% tumour cells. DNA was extracted by the standard phenol/chloroform method from frozen tissues.
Telomere length measurement by quantitative real-time PCR
Telomere length was determined using real-time PCR (Cawthon, 2002; O'Callaghan et al, 2008) with minor modifications. Two PCRs were performed for each sample, one to determine the cycle threshold (Ct) value for telomere (T) amplification and the other to determine the Ct value for the amplification of a single-copy (S) control gene (acidic ribosomal protein P0, RPLP0). The primer sequences for telomere amplification were TEL1B 5′-CGGTTTGTTTGGGTTTGGGTTTGGGTTTGGGTTTGGGTT-3′ and TEL2B 5′-GGCTTGCCTTACCCTTACCCTTACCCTTACCCTTACCCT-3′ (O'Callaghan et al, 2008) and those for RPLP0 amplification were RPLP01 5′-CAGCAAGTGGGAAGGTGTAATCC-3′ and RPLP02 5′-CCCATTCTATCATCAACGGGTACAA-3′ (Boulay et al, 1999). Each PCR reaction was performed using a 10 μl sample (1 ng of DNA per μl) and a 40 μl mixture containing 1.25 U AmpliTaq Gold DNA polymerase (Applied Biosystems, Foster City, CA, USA), 150 nM 6-ROX, 0.2 × SYBRGreen I nucleic acid stain 10 000 × (Invitrogen, Milan, Italy), 50 mM KCl, 2 mM MgCl2, 0.2 mM of each deoxynucleoside triphosphate (Applied Biosystems), 5 mM dithiothreitol, 1% dimethyl sulphoxide, and 15 mM Tris–HCl pH 8.0, as well as primer pair TEL1B (300 nM) and TEL2B (900 nM) or primer pair RPL01 (300 nM) and RPL02 (500 nM). A reference curve was generated at each PCR run, consisting of reference DNA from the RAJI cell line (Nishikura et al, 1985) serially diluted from 10 to 0.41 ng μl−1. All real-time PCR reactions were carried out using the ABI Prism 7900 HT Sequence Detection System (Applied Biosystems). Telomere and RPLP0 sequences were amplified using the following conditions: 95°C for 10 min to activate the AmpliTaq Gold DNA polymerase, and then 25 cycles each at 95°C for 15 s and 54°C for 2 min for telomere; 40 cycles each at 95°C for 15 s and 58°C for 1 min for RPLP0. ABI Prism software version 2.3 was used for analysis. Intra- and inter-assay reproducibility of both telomere and RPL0 PCR results was evaluated initially in a series of experiments using dilutions of the reference curve. The s.d. of Ct values was ⩽0.189 (% coefficient of variation ⩽1.13) in six replicates of samples amplified in the same PCR run, and ⩽0.251 (% coefficient of variation ⩽1.58) among mean values of triplicates in different PCR runs. Both reference and sample DNAs were analysed in duplicate. Variation of Ct values in the sample was ⩽0.3 Ct (s.d. ⩽0.212; % coefficient of variation ⩽1.25) in both telomere and RPL0 PCR runs. Mean Ct values were used to calculate the relative telomere length using the telomere/single-copy-gene ratio (T/S) according to the formula: ΔCtsample=Cttelomere−Ctcontrol, ΔΔCt=ΔCtsample−ΔCtreference curve (where ΔCtreference curve=Cttelomere−Ctcontrol ) and then T/S=2−ΔΔCt (Cawthon 2002; O'Callaghan et al, 2008).
Telomere length measurement by Southern blotting
In an initial series of samples, telomere length was also determined by Southern blotting. Aliquots of 5 μg DNA were digested with Hinf1 and Rsa1 restriction enzymes (20 U each) for 3 h at 37°C. Digested telomere restriction fragments were separated by 0.8% agarose gel electrophoresis. Gels were transferred to a positively charged nylon membrane and were UV cross-linked for 5 min. Hybridisation was performed with the TeloTAGG telomere length assay kit (Roche Diagnostics, Mannheim, Germany) according to the manufacturer's recommendations. Briefly, filters were hybridised with a digoxigenin (DIG)-labelled telomere-specific probe for 3 h at 42°C, washed twice, and then incubated with a DIG-specific antibody coupled to alkaline phosphatase (AP) for 30 min. Hybridisation was finally visualised using AP-metabolising CDP-Star (Disodium 2-chloro-5-(4-methoxyspiro {1,2-dioxetane-3,2'-(5'-chloro)tricyclo-[3.3.1.13,7]decan}-4-yl)-1-phenyl phosphate; Sigma, St Louis, MO, USA), a highly sensitive chemiluminescent substrate. Membranes were scanned and telomere lengths were estimated by densitomer analysis (Gel Doc 100, Biorad, Milan, Italy).
Quantification of hTERT transcripts
Extraction of RNA from CRC samples and reverse transcription of RNA into cDNA were carried out as previously described (Terrin et al, 2008). The quantification of all hTERT transcripts (hTERT-AT) in CRC samples was carried out by real-time PCR, exactly as previously described (Terrin et al, 2007, 2008), and normalised for 103 copies of the housekeeping hypoxanthine guanine phosphoribosyl transferase 1 (HPRT1) gene (Terrin et al, 2008).
P53 and MSI analyses
P53 gene mutations were detected by PCR single-strand conformational polymorphism (SSCP) analysis of exons 4–8, and DNA sequencing of samples with abnormal SSCP results (Bertorelle et al, 1995). The MSI status was determined by analysing five microsatellites of the Bethesda recommended panel (BAT-25, BAT-26, D2S123, D5S346, and D17S250) (Pucciarelli et al, 2003), and defining high MSI (MSI-H) tumours as those with two or more altered markers (Boland et al, 1998).
Statistical analysis
Comparisons of telomere length in normal and neoplastic tissues according to gender, site of disease, tumour stage and grade, MSI and p53 status were performed using the Kruskal–Wallis test. The relationship between telomere length and age was explored with the Spearman's rank correlation coefficient. The diagnostic performance of T/S values was evaluated using a receiver-operating characteristic curve; the cutoff point corresponding to the maximum Younden index was selected to better discriminate between normal and cancerous tissue. Results were reported with their 95% confidence intervals (CI). Multiple linear regression analyses were used to determine the adjusted association of telomere length with tumour location, age, p53 and MSI status. All P-values were two-sided, and P-value of <0.05 was considered statistically significant. Statistical analyses were performed using SAS stastical software (SAS Institute, Cary, NC, USA).
Results
Telomere length in cancer tissues and adjacent non-cancerous mucosa
In 118 tumour samples and the corresponding adjacent non-cancerous tissues, telomere length was determined by real-time PCR. For 42 cases, telomere length was also assessed by Southern blotting. A significant linear correlation was found between the T/S values provided by real-time PCR and the telomere length estimated by Southern blot (rs=0.860, 95% CI 0.77–0.92, P<0.0001, Figure 1). Overall, the median level of T/S values in cancer samples was 0.64 (interquartile range (IQR), 0.47–0.99), lower than that estimated in adjacent mucosa (median 1.84(1.47–2.63); P<0.0001) (Figure 2A). Conversion of T/S values in Kb, using the relationship shown in Figure 1, indicated that telomere length varied from 3.38 to 7.44 Kb with a median (IQR) of 4.10 (3.83–4.64) Kb in tumours, and from 3.61 to 11.55 Kb with a median of 5.93(5.36–7.15) Kb in normal tissues.

Correlation between telomere lengths estimated by Southern blotting (Kb) and real-time PCR (T/S value). A significant correlation was found (r=0.860, P<0.0001), giving the mathematical relation y=1.53x+3.12.

Relative telomere lengths, expressed as T/S values, in tumours (K) and adjacent normal mucosa (N) in (A) all samples, and (B) according to tumour stage I, II, III, or IV. Boxes and whiskers represent the 25th–75th and 10th–90th percentiles, respectively; the median is the central line in each box. *P<0.0001. (C) Correlation between age and relative telomere lengths in tumours and normal mucosa. (D) Receiver-operating characteristics (ROC) curve for telomere lengths in tumours and normal mucosa using T/S values. Sensitivity (y axis) was plotted against the false-positive fraction (100-specificity). The area under the ROC curve (AUC) is 0.91 (95% confidence intervals (CI) 0.87–0.95).
Telomere lengths were shorter in cancers than in adjacent non-cancerous mucosa for all tumour stages (P<0.0001; Figure 2B) and grades (median (IQR) 0.63(0.47–0.97) vs 2.04(1.40–2.41), 0.67(0.46–1.02) vs 1.87(1.49–2.78), and 0.59(0.45–0.78) vs 1.68(1.45–2.21) for grades 1, 2, and 3, respectively; tumours vs normal tissues P<0.0001). Although no correlation was found between age and telomere length in cancers, telomere length in normal tissues inversely correlated with age (r=−0.24, 95% CI −0.42 −0.05, P=0.017; Figure 2C). A patient-by-patient comparison of matched tissue samples showed that all but four cases had longer telomeres in non-cancerous mucosa than in CRC samples with a median difference of 1.22 (0.61–1.81); these differences inversely correlated with age (r=−0.23, 95% CI −0.41–0.04, P=0.023). From the receiver-operating characteristic curve analysis, the T/S value that best discriminated between the telomere length of neoplastic and normal tissues was 1.14. By using this cutoff value, sensitivity and specificity of the assay for cancer-associated telomere were 83% (95% CI 75–90%) and 92% (95% CI 85–96%), respectively (Figure 2D).
Relationship between telomere length and tumour characteristics
Telomere lengths did not significantly differ with tumour stage. Median (IQR) T/S values were 0.66 (0.56–0.66), 0.69(0.49–0.93), 0.55(0.39–1.02), and 0.64 (0.51–0.98) in tumour stages I, II, III, and IV, respectively (Figure 2B, overall, P=0.472). A similar observation was found by comparing T/S values with tumour grade; T/S values did not differ between well-differentiated (median 0.63, IQR 0.47–0.97), moderately differentiated (0.67, 0.46–1.02), and poorly differentiated tumours (0.59, 0.45–0.78) (overall, P=0.561).
The CRCs were then stratified according to p53 and MSI status: an equal number of MSS tumours had the wild-type (n=45) or mutated (n=45) p53 gene; 24 MSI-H tumours had the wild-type p53 gene, whereas four tumours showed the rare pattern of both MSI-H and the mutated p53 gene. Median (IQR) T/S values were 0.65(0.50–0.98) and 0.62(0.43–0.97) in CRCs with the wild-type (n=69) and mutated p53 gene (n=49), respectively, and 0.66(0.46–1.03) and 0.56(0.43–0.73) in MSS (n=90) and MSI-H tumours (n=28), respectively (P=0.065) (Figures 3A and B). Furthermore, in tumours carrying the wild-type p53 gene, telomeres were significantly shorter in MSI-H (n=24) than in MSS cases (n=45) (0.56(0.43–0.73) vs 0.70(0.50–1.113), P=0.027) (Figure 3C). In MSS tumours, those carrying the mutated p53 gene had shorter telomeres than those carrying the wild-type p53 gene, but this difference was not statistically significant (0.62(0.43–0.97) vs 0.70(0.50–1.113), P=0.218) (Figure 3C).

Relative telomere lengths, expressed as T/S values, in tumours according to (A) mutated (n=49) or wild-type (n=69) p53 gene, and (B) high microsatellite instability (MSI-H) (n=28) or stable microsatellite (MSS) (90) status. (C) Relative telomere lengths, expressed as T/S values, in p53 mutated MSS (n=45), p53 wild-type MSI-H (n=24), and p53 wild-type MSS (n=45) tumours. Boxes and whiskers represent the 25th–75th and 10th–90th percentiles, respectively; the median is the central line in each box.
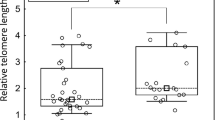
Telomere lengths also differed according to tumour location. Median (IQR) T/S values were 0.57 (0.41–0.82), 0.61(0.44–0.98), and 0.78(0.62–1.09) in the 53 right-colon, 30 left-colon, and 35 rectal carcinomas, respectively (overall, P=0.03; right colon vs rectum P=0.009) (Figure 4). Normal tissues surrounding colon and rectal tumours did not significantly differ (median (IQR) 1.82 (1.48–2.51), 2.13(1.49–2.86), and 1.87(1.25–2.61) in right-colon, left-colon, and rectal tissues, respectively; P=0.81). It can be noted that 49% (26 of 53) of right-colon tumours but only 3% (2 of 65) of left-colon and rectal cancers showed MSI. The four cases with both MSI and the p53-mutated gene were also right-colon cancers. In contrast, p53 mutation occurred in a higher percentage in left-colon and rectal tumours (60 and 43%, respectively) than in right-colon cancers (23%). With multivariate analyses, telomere length remained significantly associated with tumour location after adjustment for age and p53 status (P=0.04), as well as after adjustment for age and p53/MSI status (P=0.05). It is interesting that even among MSS tumours, right-colon cancers had shorter telomeres than left-colon and rectal cancers (median (IQR) 0.58(0.40–1.05) vs 0.62(0.44–0.98) and 0.80 (0.60–1.20), respectively; P=0.09; right-colon vs rectum P=0.047).

Relative telomere lengths, expressed as T/S values, in tumours according to the site of origin: right colon (n=53), left colon (n=30), rectum (n=35). Boxes and whiskers represent the 25th–75th and 10th–90th percentiles, respectively; the median is the central line in each box.
Relationship between hTERT expression and tumour characteristics
Quantification of TERT transcripts was available for 65 CRC samples. No relationship was found between hTERT levels and MSI status or tumour location. Indeed, median (IQR) levels of hTERT were 82 (34–294) and 144(65–464) copies in MSI (n=13) and MSS CRCs (n=52), respectively (P=0.195), and 114(39–360), 172(71–1192), and 85(37–282) copies in right-colon (n=22), left-colon (26), and rectal CRCs (n=17), respectively (P=0.301). In contrast, hTERT levels significantly varied with tumour progression; median (IQR) hTERT levels were 41(25–183), 82(60–161), 128(72–236), and 660(268–2543) copies in tumour stages I (n=20), II (n=13), III (n=11), and IV (n=21), respectively (P<0.0001), and 36(22–240), 102(70–322), and 1854(630–5739) copies in well-differentiated (n=14), moderately differentiated (n=41), and poorly differentiated tumours (n=7), respectively (P<0.0001). No correlation was found between telomere length and hTERT level.
Discussion
The role of telomere erosion in colorectal carcinogenesis is still largely undefined. Using a real-time PCR assay, which requires a considerably low quantity of biological samples, we estimated telomere length in a large number of CRCs and in corresponding adjacent non-cancerous mucosa. For the first time, we determined the relationship between telomere length and tumour progression and location, as well as its relationship to MSI and p53 mutation status, two markers of major pathways of genetic instability in CRCs. Overall, our results indicated that (i) telomeres in CRCs were consistently shorter than those of adjacent non-cancerous mucosa, regardless of tumour stage and grade, site, or genetic alterations; (ii) telomeres were shorter in MSI than in MSS carcinomas, and the difference was significant between MSI and MSS tumours carrying the wild-type p53 gene; (iii) regardless of genetic alterations, telomeres were shorter in tumours arising from the right colon and were longer in rectal cancers.
In agreement with previous studies (Hastie et al, 1990; Gertler et al, 2004), we found that telomere length decreased with ageing in normal mucosa, but not in tumours. It has been proposed that telomere shortening with age may increase the risk of cancer (Blasco, 2005; Risques et al, 2008); this concept may be supported by our results, as telomere shortening between normal and neoplastic tissues was found to be inversely correlated with age. Although telomere length in somatic cells primarily reflects cellular proliferation, telomere length in tumour cells reflects the balance between cellular proliferation with telomere loss and telomerase activity with de novo synthesis of telomeric sequences. Evidence that telomeres were shorter in CRCs than in adjacent mucosa, even in well-differentiated tumours, strongly supports the concept that telomere erosion is a critical initial event in colorectal carcinogenesis. It is likely that stabilisation and maintenance of telomeres, essential to preventing cellular senescence and conferring unlimited replicative potential, occur after initial extensive cellular proliferation. It is noteworthy that, although several studies agree that there is a shortening of telomeres in preneoplastic lesions (O'Sullivan et al, 2006; Raynaud et al, 2008), the relationship between telomere length and tumour progression is still controversial (Engelhardt et al, 1997; Gertler et al, 2004; O'Sullivan et al, 2006). We did not find any relationship between telomere length and tumour progression. In agreement with previous studies (Terrin et al, 2008), levels of hTERT increased with tumour progression; it is conceivable that telomere length may stabilise with tumour progression because of an increase in telomerase activity compensating for replicative telomere loss.
It should be pointed out that ∼15% of CRCs presented MSI-H, whereas the p53 gene is the known major genetic alteration in CRCs with MSS (Kim et al, 2007; Ogino et al, 2009). A link between telomere shortening and MSI-H has been suggested in a previous study of 55 CRCs; all MSI tumours (n=8) but only half of the remaining MSS tumours had shorter telomeres than those found in adjacent normal mucosa (Takagi et al, 2000). Our study conducted in a larger number of CRCs demonstrated that both MSI-H and MSS tumours had shorter telomeres than those found in adjacent mucosa and that MSI-H had shorter telomeres than MSS cancers. The MSI pathway involves a failure of the mismatch repair system (Aaltonen et al, 1993), which maintains genetic stability not only by repairing DNA replication errors but also by preventing chromosomal recombinations; a deficiency of mismatch repair helps cells overcome cellular crises caused by critical shortening of telomeres (Bechter et al, 2004). Thus, MSI cells may undergo more proliferative cycles and a more pronounced shortening of telomeres before stabilising than MSS cells. It is of interest that the difference was particularly high and significant when MSI tumours were compared with MSS tumours carrying the wild-type p53 gene. It can be noted that MSS tumours with the mutated p53 gene had slightly shorter telomeres than MSS tumours with the wild-type p53 gene. In cells with mutated p53, telomeres may protract their shortening along with cell proliferation, However, p53 is a well-known negative regulator of hTERT promoter, and mutated p53 may also result in hTERT activation (Liu et al, 2004); thus, stabilisation of telomeres may occur earlier than in MSI tumours.
We found that right-colon tumours displayed significantly shorter telomeres than tumours arising from other sites. In agreement with previous studies (Iacopetta, 2002; Gervaz et al, 2004; Walther et al, 2008), we found that MSI status was strongly associated with tumour origin in the right colon. However, this association could only partially explain the finding that telomeres were shorter in right-colon cancers, as half of these tumours were MSS. Interestingly, even in MSS tumours, those arising in the right colon had significantly shorter telomeres than those arising in the rectum. The prognosis of rectal cancers is worse than that of colon cancer and it is currently under debate whether rectal cancers are actually a distinct entity. Telomere length distinguishes colon from rectal cancers; as telomere lengths of normal tissues surrounding colon and rectal cancers did not differ, it is likely that the different telomere length in cancers is due to a different kinetics of telomere erosion/stabilisation in colon and rectal carcinogenesis. Although no association was found between hTERT level and the tumour origin site, earlier activation of hTERT may contribute to these differences between rectal and colon cancers. Future studies on these aspects may be of help in the design of anti-neoplastic strategies.
References
Aaltonen LA, Peltomäki P, Leach FS, Sistonen P, Pylkkänen L, Mecklin JP, Järvinen H, Powell SM, Jen J, Hamilton SR (1993) Clues to the pathogenesis of familial colorectal cancer. Science 260: 812–816
Artandi SE, DePinho RA (2010) Telomeres and telomerase in cancer. Carcinogenesis 31: 9–18
Barault L, Charon-Barra C, Jooste V, de la Vega MF, Martin L, Roignot P, Rat P, Bouvier AM, Laurent-Puig P, Faivre J, Chapusot C, Piard F (2008) Hypermethylator phenotype in sporadic colon cancer: Study on a population-based series of 582 cases. Cancer Res 68: 8541–8546
Bechter OE, Zou Y, Walker W, Wright W, Shay JW (2004) Telomeric recombination in mismatch repair deficient human colon cancer cells after telomerase inhibition. Cancer Res 64: 3444–34451
Bertorelle R, Esposito G, Del Mistro A, Belluco C, Nitti D, Lise M, Chieco-Bianchi L (1995) Association of p53 gene and protein alterations with metastases in colorectal cancer. Am J Surg Pathol 19: 463–471
Blackburn EH, Greider CW, Szostak JW (2006) Telomeres and telomerase: the path from maize, Tetrahymena and yeast to human cancer and aging. Nat Med 12: 1133–1138
Blasco MA (2005) Telomeres and human disease: ageing, cancer and beyond. Nat Rev Genet 6: 611–622
Boland CR, Thibodeau SN, Hamilton SR, Sidransky D, Eshleman JR, Burt RW, Meltzer SJ, Rodriguez-Bigas MA, Fodde R, Ranzani GN, Srivastava S (1998) A National Cancer Institute Workshop on microsatellite instability for cancer detection and familial predisposition: development of international criteria for the determination of microsatellite instability in colorectal cancer. Cancer Res 58: 5248–5257
Boulay JL, Reuter J, Ritschard R, Terracciano L, Hermann R, Rochlitz C (1999) Gene dosage by quantitative real-time PCR. Biotechniques 27: 228–230
Cawthon RM (2002) Telomere measurement by quantitative PCR. Nucleic Acids Res 30: e47
Engelhardt M, Drullinsky P, Guillem J, Moore AS (1997) Telomerase and telomere length in the development and progression of premalignant lesions to colorectal cancer. Clin Cancer Res 3: 1931–1941
Garcia-Aranda C, de Juan C, Diaz-Lopez A, Sanchez-Pernaute A, Torres AJ, Diaz-Rubio E, Balibrea JL, Benito M, Iniesta P (2006) Correlations of telomere length, telomerase activity, and telomeric-repeat binding factor 1 expression in colorectal carcinoma. Cancer 106: 541–551
Gertler R, Rosenberg R, Stricker D, Friederichs J, Hoos A, Werner M, Ulm K, Holzmann B, Nekarda H, Siewert JR (2004) Telomere length and human telomerase reverse transcriptase expression as markers for progression and prognosis of colorectal carcinoma. J Clin Oncol 22: 807–814
Gervaz P, Bucher P, Morel P (2004) Two colons-two cancers: paradigm shift and clinical implications. J Surg Oncol 88: 261–266
Grady WM, Carethers JM (2008) Genomic and epigenetic instability in colorectal cancer pathogenesis. Gastroenterology 135: 1079–1099
Greene FL, Page DL, Fleming ID, Fritz A, Balch CM, Haller DG (2002) American Joint Committee on Cancer Staging Manual. 6th edn, Springer: Philadelphia
Hackett JA, Greider CW (2002) Balancing instability: dual roles for telomerase and telomere dysfunction in tumorigenesis. Oncogene 21: 619–626
Hastie ND, Dempster M, Dunlop MG, Thompson AM, Green DK, Allshire RC (1990) Telomere reduction in human colorectal carcinoma and with aging. Nature 346: 866–868
Hiyama E, Hiyama K (2007) Telomere and telomerase in stem cells. Br J Cancer 96: 1020–1024
Iacopetta B (2002) Are there two sides to colorectal cancer? Int J Cancer 101: 403–408
Katayama S, Shiota G, Oshimura M, Kawasaki H (1999) Clinical usefulness of telomerase activity and telomere length in the preoperative diagnosis of gastric and colorectal cancer. J Cancer Res Clin Oncol 125: 405–410
Kim GP, Colangelo LH, Wieand HS, Paik S, Kirsch IR, Wolmark N, Allegra CJ, National Cancer Institute (2007) Prognostic and predictive roles of high-degree microsatellite instability in colon cancer: a National Cancer Institute-National Surgical Adjuvant Breast and Bowel Project Collaborative Study. J Clin Oncol 25: 767–772
Koopman M, Kortman GA, Mekenkamp L, Ligtenberg MJ, Hoogerbrugge N, Antonini NF, Punt CJ, van Krieken JH (2009) Deficient mismatch repair system in patients with sporadic advanced colorectal cancer. Br J Cancer 100: 266–273
Li F, Lai M (2009) Colorectal cancer, one entity or three. J Zhejiang Univ Sci B 10: 219–229
Liu L, Lai S, Andrews LG, Tollefsbol TO (2004) Genetic and epigenetic modulation of telomerase activity in development and disease. Gene 340: 1–10
Meeker AK, Hicks JL, Iacobuzio-Donahue CA, Montgomery EA, Westra WH, Chan TY, Ronnett BM, De Marzo AM (2004) Telomere length abnormalities occur early in the initiation of epithelial carcinogenesis. Clin Cancer Res 10: 3317–3326
Nishikura K, Erikson J, Ar-Rushdi A, Huebner K, Croce CM (1985) The translocated c-myc oncogene of Raji Burkitt lymphoma cells is not expressed in human lymphostoid cells. Proc Natl Acad Sci USA 82: 2900–2904
O'Callaghan NJ, Dhillon VS, Thomas P, Fenech M (2008) A quantitative real-time PCR method for absolute telomere length. Biotecniques 44: 807–809
O'Sullivan J, Risques RA, Mandelson MT, Chen L, Brentnall TA, Bronner MP, Macmillan MP, Feng Z, Siebert JR, Potter JD, Rabinovitch PS (2006) Telomere length in the colon declines with age: a relation to colorectal cancer? Cancer Epidemiol Biomarkers Prev 15: 573–577
Ogino S, Goel A (2008) Molecular classification and correlates in colorectal cancer. J Mol Diagn 10: 13–27
Ogino S, Nosho K, Irahara N, Shima K, Baba Y, Kirkner GJ, Meyerhardt JA, Fuchs CS (2009) Prognostic significance and molecular associations of 18q loss of heterozygosity: a cohort study of microsatellite stable colorectal cancers. J Clin Oncol 27: 4591–4598
Perera SA, Maser RS, Xia H, McNamara K, Protopopov A, Chen L, Hezel AF, Kim CF, Bronson RT, Castrillon DH, Chin L, Bardeesy N, Depinho RA, Wong KK (2008) Telomere dysfunction promotes genome instability and metastatic potential in a K-ras p53 mouse model of lung cancer. Carcinogenesis 29: 747–753
Pucciarelli S, Agostini M, Viel A, Bertorelle R, Russo V, Toppan P, Lise M (2003) Early-age at onset colorectal cancer and microsatellite instability as markers of hereditary nonpolyposis colorectal cancer. Dis Colon Rectum 46: 305–312
Raynaud CM, Jang SJ, Nuciforo P, Lantuejoul S, Brambilla E, Mounier N, Olaussen KA, André F, Morat L, Sabatier L, Soria JC (2008) Telomere shortening is correlated with the DNA damage response and telomeric protein down-regulation in colorectal preneoplastic lesions. Ann Oncol 19: 1875–1881
Risques RA, Lai LA, Brentnall TA, Li L, Feng Z, Gallaher J, Mandelson MT, Potter JD, Bronner MP, Rabinovitch PS (2008) Ulcerative colitis is a disease of accelerated colon aging: evidence from telomere attrition and DNA damage. Gastroenterology 135: 410–418
Takagi S, Kinouchi Y, Hiwatashi N, Chida M, Nagashima F, Takahashi S, Negoro K, Shimosegawa T, Toyota T (1999) Telomere shortening and the clinicopathologic characteristics of human colorectal carcinomas. Cancer 86: 1431–1436
Takagi S, Kinouchi Y, Hiwatashi N, Nagashima F, Chida M, Takahashi S, Negoro K, Shimosegawa T, Toyota T (2000) Relationship between microsatellite instability and telomere shortening in colorectal cancer. Dis Colon Rectum 43: 12–17
Terrin L, Rampazzo E, Pucciarelli S, Agostini M, Bertorelle R, Esposito G, DelBianco P, Nitti D, De Rossi A (2008) Relationship between tumor and plasma levels of hTERT mRNA in patients with colorectal cancer: implications for monitoring of neoplastic disease. Clin Cancer Res 14: 7444–7451
Terrin L, Trentin L, Degan M, Corradini I, Bertorelle R, Carli P, Maschio N, Bo MD, Noventa F, Gattei V, Semenzato G, De Rossi A (2007) Telomerase expression in B-cell chronic lymphocytic leukemia predicts survival and delineates subgroups of patients with the same IgVH mutation status and different outcome. Leukemia 21: 965–972
Walther A, Houlston R, Tomlinson I (2008) Association between chromosomal instability and prognosis in colorectal cancer: a meta-analysis. Gut 57: 941–950
Acknowledgements
This work was supported by the Associazione Italiana per la Ricerca sul Cancro, and by Ministero della Salute ‘Progetto Oncologico di Medicina Molecolare’. RE has a fellowship from the Associazione Italiana per la Ricerca sul Cancro. We thank Pierantonio Gallo for artwork, and Lisa Smith for editorial assistance.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
This work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Rampazzo, E., Bertorelle, R., Serra, L. et al. Relationship between telomere shortening, genetic instability, and site of tumour origin in colorectal cancers. Br J Cancer 102, 1300–1305 (2010). https://doi.org/10.1038/sj.bjc.6605644
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.bjc.6605644
Keywords
This article is cited by
-
The crosstalk between telomeres and DNA repair mechanisms: an overview to mammalian somatic cells, germ cells, and preimplantation embryos
Journal of Assisted Reproduction and Genetics (2024)
-
The dynamics of telomere length in primary and metastatic colorectal cancer lesions
Scientific Reports (2023)
-
Age-specific oncogenic pathways in head and neck squamous cell carcinoma - are elderly a different subcategory?
Cellular Oncology (2022)
-
TERT promoter hotspot mutations and their relationship with TERT levels and telomere erosion in patients with head and neck squamous cell carcinoma
Journal of Cancer Research and Clinical Oncology (2020)
-
Relationship of telomere length in colorectal cancer patients with cancer phenotype and patient prognosis
British Journal of Cancer (2019)
