
Summary:
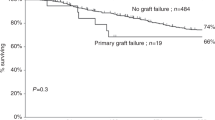
Over the last 15 years, we have performed a total of 30 haematopoietic stem cell transplants on 27 children suffering from Hurler's syndrome. These children were of median age 11 months at the time of diagnosis and 25 months at the time of transplantation. The phenotype was severe in 21 cases (78%). The donor was familial in 13 cases: nine genotypically identical, one phenotypically identical father and three HLA-mismatched donors. Unrelated donors were selected in 17 cases: four phenotypically identical and 13 with 1–4 HLA mismatches. The conditioning regimen generally consisted of busulphan 600 mg/m2 plus cyclophosphamide (Endoxan®) 260 mg/kg and cyclosporin with methotrexate for GvHD prophylaxis. Rabbit anti-thymocyte globulin (Thymoglobuline®) was given for all unrelated or familial mismatched transplantations. The median nucleated cell dose infused was 6.00 × 108 TNC/kg. No bone marrow (apart from one) was T cell depleted. For first transplants, engraftment was observed in 23/27 patients (pts) (85%). Primary graft failure was observed in 4/27 patients (16%), two were retransplanted from an unrelated donor, one with success. Four patients have died. The primary cause of death was infection in three cases (TRM : 11%) and disease progression in one case, after primary graft failure. Of the 23 living patients, two have disease progression after graft failure and 21 (78%) have functional grafts with a favourable long-term outcome after a median follow-up of 4.7 years, having either full or mixed chimaerism. Among surviving patients with functional grafts, 13 (62%) were transplanted from unrelated donors of whom 10 (77 %) had HLA disparities. There was a remarkably low incidence of GvHD. In our experience, haematopoietic stem cell transplantation using an HLA-matched familial donor or an HLA-matched or -mismatched unrelated donor without T cell depletion or irradiation can achieve a favourable outcome in Hurler's syndrome, with improved cognitive function, but with a limited effect on the corneas and skeleton.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
$259.00 per year
only $21.58 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout






Similar content being viewed by others

References
Hobbs JR, Hugh-Jones K, Barrett AJ et al. Reversal of clinical features of Hurler's disease and biochemical improvement after treatment by bone marrow transplantation. Lancet 1981; 2: 709–712.
Krivit W, Sung JH, Lockman LA, Shapiro EG . Central nervous system reconstitution after bone marrow transplantation for lysosomal and peroxisomal storage diseases. In: Rich RR, Fleisher TA, Schwartz BD, Shearer WT, Strober W (eds). Principles of Clinical Immunology, Vol 2. Mosby: St Louis, MO, 1995, p. 1852.
Whitley CB, Belani KG, Chang PN et al. Long-term outcome of Hurler syndrome following bone marrow transplantation. Am J Med Genet 1993; 46: 209–218.
Hoogerbrugge PM, Brouwer OF, Bordigoni P et al. A for the European group for Bone Marrow Transplantation. Allogeneic bone marrow transplantation for lysosomal storage diseases. Lancet 1995; 345: 1398–1402.
Peters C, Shapiro EG, Anderson J et al. Hurler Syndrome. II. Outcome of HLA-genotypically identical sibling and HLA-haploidentical related donor bone marrow transplantation in fifty-four children. Blood 1998; 91: 2601–2608.
Peters C, Balthazor M, Shapiro EG et al. Outcome of unrelated donor bone marrow transplantations in 40 children with Hurler syndrome. Blood 1996; 87: 4894–4902.
Guffon N, Souillet G, Maire I et al. Follow-up of nine patients with Hurler syndrome after bone marrow transplantation. J Pediatr 1998; 133: 119–125.
Piraud M, Boyer S, Mathieu M, Maire I . A diagnosis of mucopolysaccharidoses in a clinically selected population by urinary glycosaminoglycan analysis: a study of 2000 urine samples. Clin Chim Acta 1993; 221: 171–781.
Hall CW, Neufeld EF . α-L-iduronidase activity in cultured skin fibroblasts and amniotic fluid cells. Arch Biochem Biophys 1973; 158: 817–821.
Young EP . Prenatal diagnosis of Hurler disease by analysis of a-L-iduronidase in chorionic villi. J Inher Metab Dis 1992; 15: 224–230.
Gebuhrer L, Betuel H, Lambert J et al. A division of HLA-DQw1 associated with DR1, DR1x-DQw1, DR2, short, DRw10, and DRw14. I. Definition by alloantiserum LY1327. Human Immunol 1987; 18: 235–245.
Tiercy JM, Gorski J, Betuel H et al. DNA typing of DRw6 subtypes : correlation with DRB1 and DRB3 allelic sequences by hybridization with oligonucleotide probes. Human Immunol 1989; 24: 1–14.
Tiercy JM, Morel C, Freidel AC et al. Selection of unrelated donors for bone marrow transplantation is improved by HLA class II genotyping with oligonucleotide hybridization. Proc Natl Acad Sci USA 1991; 1988: 7121–7125.
Hobbs JR . The evolution of displacement bone marrow transplantation for inborn errors. In: Hobbs JR (ed.). Correction of Certain Genetic Diseases by Transplantation. The COGENT Fund: London, 1989, pp. 1–22.
Souillet G . Thymoglobulin: conditioning in children prior to bone marrow transplantation, a single center retrospective study. In: Thymoglobulin – anti-thymocyte globulin (rabbit). The role of Thymoglobulin in Alternative Donor Haematopoietic Stem Cell Transplantation. Clinical monograph, SangStat 2000, pp. 35–41.
Bleyzac N, Souillet G, Magron P et al. Improved clinical outcome of paediatric bone marrow recipients using a test dose and Bayesian pharmacokinetic individualization of busulfan dosage regimen. Bone Marrow Transplant 2001; 28: 743–751.
Glucksberg H, Storb R, Fefer A . Clinical manifestations of GVHD in human recipients of marrow from HLA matched sibling donors. Transplantation 1994; 18: 295–301.
Wong Z, Wilson V, Jeffreys AJ, Thein SL . Characterization of a panel of highly variable minisatellites cloned from human DNA. Ann Hum Genet 1996; 51: 269–288.
Souillet G, Rey S, Bertrand Y et al. Outcome of unrelated bone marrow donor search in 174 children resulting in 45 patients transplanted in HLA match and mismatch situation. Bone Marrow Transplant 2000; 26: 31–43.
Bunge S, Kleijer WJ, Steglich C et al. Mucopolysaccharidosis type I: identification of 8 novel mutations and determination of the frequency of the two common alpha-L-iduronidase mutations (W402X and Q70X) among European patients. Hum Mol Genet 1994; 3: 861–866.
Scott HS, Bunge S, Gal A et al. Molecular genetics of mucopolysaccharidosis type I: diagnostic, clinical, and biological implications. Hum Mutat 1995; 6: 288–302.
Beesley CE, Meaney CA, Greenland G et al. Mutational analysis of 85 mucopolysaccharidosis type I families: frequency of known mutations, identification of 17 novel mutations and in vitro expression of missense mutations. Hum Gen 2001; 109: 503–511.
Scott SH, Litjens T, Nelson PV et al. α-L-iduronidase mutations (Q70X and P533R) associate with a severe Hurler phenotype. Hum Mutat 1992; 1: 333–339.
Shapiro EG, Lockman LA, Balthazor M, Krivit W . Neuropsychological outcomes of several storage diseases with and without bone marrow transplantation. J Inherit Metab Dis 1995; 18: 41.
Fleming DR, Henslee-Downey PJ et al. The use of partially HLA mismatched donors for allogeneic transplantation in patients with mucopolysaccharidosis-I. Pediatr Transplant 1998; 2: 250–253.
Bolinger AM, Zangwill AB, Slattery JT et al. An evaluation of engraftment, toxicity and busulfan concentration in children receiving bone marrow transplantation for leukaemia or genetic disease. Bone Marrow Transplant 2000; 25: 925–930.
Vassal G, Deroussent A, Challine D et al. Is 600 mg/m2 appropriate dosage of busulfan in children undergoing bone marrow transplantation? Blood 1992; 79: 2475–2479.
Vassal G, Fisher A, Challine D et al. Busulfan disposition below the age of three: alteration in children with lysosomal storage disease. Blood 1993; 82: 1030–1034.
Vellodi A, Young EP, Cooper A . Bone marrow transplantation for mucopolysaccharidosis type I: experience of two British centres. Arch Dis Child 1997; 76: 92–99.
Field RE, Buchana JAF, Copplemans MGJ, Aichroth PM . Bone marrow transplantation in Hurler syndrome: effect on skeletal development. J Bone Joint Surg Br 1994; 76B: 975–981.
Masterson EL, Murphy PG, O'Meara A et al. Hip dysplasia in Hurler's syndrome : orthopaedic management after bone marrow transplantation. J Pediatr Orthop 1996; 16: 731–733.
Kakkis ED, Muenzer J, Tiller GE et al. Enzyme-replacement therapy in mucopolysaccharidosis I. N Engl J Med 2001; 344: 182–188.
Acknowledgements
We thank the assistant physicians, nurses and medical staff of the Immuno-haematology & Paediatric Bone Marrow Transplant Unit, Debrousse Hospital, for their support and commitment (Prof Philippe, Dr K Kebaili, Dr R Mardini) and the physicians and technicians of Histocompatibility Laboratory in Lyon, for their efforts and efficient collaboration (Dr A Eljaafari, Dr D Rigal, Dr JP Tremisi, JP Bourgeot, Ph Debost, S Rey). All of them have contributed to help the patients. We also address special thanks to Prof C Peters and Dr N Whitaker for their constructive and useful suggestions in reviewing the manuscript.
Author information
Authors and Affiliations
Rights and permissions
About this article
Cite this article
Souillet, G., Guffon, N., Maire, I. et al. Outcome of 27 patients with Hurler's syndrome transplanted from either related or unrelated haematopoietic stem cell sources. Bone Marrow Transplant 31, 1105–1117 (2003). https://doi.org/10.1038/sj.bmt.1704105
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.bmt.1704105
Keywords
This article is cited by
-
Long term disease burden post-transplantation: three decades of observations in 25 Hurler patients successfully treated with hematopoietic stem cell transplantation (HSCT)
Orphanet Journal of Rare Diseases (2021)
-
An online survey on burden of illness among families with post-stem cell transplant mucopolysaccharidosis type I children in the United States
Orphanet Journal of Rare Diseases (2019)
-
Enzyme replacement therapy for mucopolysaccharidoses; past, present, and future
Journal of Human Genetics (2019)
-
Spine challenges in mucopolysaccharidosis
International Orthopaedics (2019)
-
Treatment of thoracolumbar kyphosis in patients with mucopolysaccharidosis type I: results of an international consensus procedure
Orphanet Journal of Rare Diseases (2019)
