
Abstract
Erythropoietin (EPO) increases the number of circulating erythrocytes primarily by preventing apoptosis of erythroid progenitors. In addition to this proerythroid action, results of recent studies show that systemically administered EPO is protective in vivo, in several animal models of neuronal injury. In vitro, EPO prevents neuronal apoptosis induced by a variety of stimuli. This review summarizes the neuroprotective actions of EPO and discusses the underlying mechanisms in terms of signal transduction pathways involved. The understanding of these mechanisms will help differentiate the neuroprotective actions of EPO from its role in the bone marrow.
Similar content being viewed by others


Introduction
Erythropoietin (EPO) was named for its long-appreciated hormonal effect of maintaining the circulating erythrocyte mass. In recent years, however, it has been recognized that EPO is a member of the cytokine type I superfamily. Typical for cytokines, EPO has multiple functions outside of the bone marrow. Many of these effects parallel its action in hematopoiesis, where EPO functions to promote proerythroblast survival and maturation. Over the last 10 years, a prominent role for EPO has been defined in the nervous system and there is a growing interest in the potential therapeutic use of EPO for neuroprotection. In this review, we will outline the mechanism of action of EPO in erythropoiesis with a focus on inhibition of apoptosis. This concept will then be extended to examine the neuroprotective activities of EPO, which in addition to a critical role in antiapoptosis, also embody other beneficial actions.
Erythropoietin is a cytoprotective cytokine induced by the hypoxia inducible factor family
EPO is the hematopoietic factor responsible for the production of red blood cells and for this function is produced mainly by the adult kidney. EPO is induced by hypoxia via the hypoxia-inducible factor (HIF) family of transcription factors. This growing family mediates physiological adaptation to low tissue oxygen concentration (such as that encountered by high altitude or anemia) ultimately resulting in an increased number of erythrocytes within the circulation and therefore improved tissue oxygenation. Along the same line, HIF also regulates genes for neoangiogenesis (e.g., vascular endothelial growth factor, VEGF) as well as for vascular tone (e.g., nitric oxide (NO) synthase).
While erythropoiesis and angiogenesis represent adaptive responses to improve tissue oxygenation that require several days to develop (for instance, the time required for EPO-mediated appearance of mature erythrocytes in the circulation is about a week), HIF also mediates short-term adaptive responses to hypoxia, and several glucose transporters and glycolytic enzymes are transcriptionally regulated by HIF, as well as genes involved in cell growth and survival including insulin-like growth factor 2 and IGFBP (reviewed in: Semenza,1 Wiesener and Maxwell2 and Wenger3). The induction of glycolytic enzymes demonstrates a role for HIF in the metabolic, cell-autonomous adaptation to hypoxia represented by the switch of ATP generation from oxidative phosphorylation to glycolysis, the Pasteur effect.4 It has also been suggested that, because HIF is often constitutively expressed in tumors, it may mediate the high constitutive expression of glycolytic enzymes that is the basis of the Warburg effect.3, 5
The metabolic changes induced by hypoxia clearly play a role in ischemic preconditioning, a well-known phenomenon, reported for various organs including the heart, brain, kidney and liver, whereby pre-exposure to hypoxia (such as a minor ischemic event) protects from subsequent, severe ischemia.1 Activation of HIF and/or pre-exposure to hypoxia also protects neuronal cells from apoptosis induced by oxidative stress.6
However, HIF-mediated protection from apoptosis may also involve other hypoxia-inducible cytokines implicated in the physiological responses to hypoxia, such as EPO and VEGF. In fact, there is much evidence showing that EPO protects neuronal cells in vitro, an action independent of its erythropoietic activity. In particular, EPO not only protects neurons from cell death induced by hypoxia, but also from a variety of other agents including excitotoxins and glucose deprivation (Table 1). More recently, it was reported that VEGF is also a neuroprotective and trophic factor for neurons.7, 8, 9
The finding that systemically administered EPO, a widely used drug with a well-known safety profile, crosses the blood–brain barrier in rats and in humans10, 11 and has protective effects in various models of nervous system injury (Table 2), has greatly focused attention on the neuroprotective actions of EPO and the underlying mechanisms. The protective effects of EPO are not restricted to the CNS as it was recently shown that EPO has also antiapoptotic effects on cardiomyocytes in vitro and in vivo in a rat model of myocardial infarction, where it normalizes hemodynamic functions.12 All these data stress the importance of EPO as an antiapoptotic and cytoprotective, not only neuroprotective, cytokine.
Inhibition of apoptosis in the hematopoietic activity of EPO
Erythrocyte development proceeds from progenitor cells that require multiple growth factors to initiate cellular differentiation. Cells entering into development express receptors for EPO (EPOR) during specific phases in differentiation and unless EPO is present during the critical period, the progenitors will undergo apoptosis. EPO specifically augments the number of circulating erythrocytes by promoting the survival and therefore enabling the proliferation and differentiation of erythroid progenitor cells.13, 14 Although EPO, through the EPOR, can effectively support the proliferation of murine erythroid progenitor cells ex vivo15 and induce the entry of erythroid progenitors into cell cycle if dormant,16 promotion of survival due to prevention of apoptosis of late erythroid progenitors is thought to be a major mechanism in EPO action.17, 18
Using the EPO-dependent human erythroid progenitor cell line HCD-57, Silva et al.19 showed that EPO maintained their viability via repressing apoptosis by upregulating Bcl-xL, an antiapoptotic gene of the Bcl-2 family. When these cells are cultured in the absence of EPO, Bcl-2 and Bcl-xL are downregulated and the cells undergo apoptosis.19 To further support the importance of this pathway, Bcl-xL knockout mice exhibit fetal liver hematopoietic defects and severe anemia during embryogenesis.20
EPO exerts its erythrodifferentiating effect via the EPOR, which belongs to the hematopoietic growth factor receptor superfamily. Other members include receptors for IL-2 beta chain, IL-3, IL-4, IL-5, G-CSF, GM-CSF, thrombopoietin, growth hormone, prolactin and the receptors for several cytokines of the IL-6 family (IL-6, leukemia inhibitory factor (LIF), ciliary neurotrophic factor (CNTF) and oncostatin M).21, 22
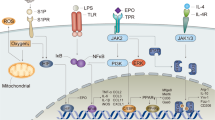
One possible signal transduction pathway involved in the antiapoptotic action of EPO in erythroid cells is outlined in Figure 1, based on the known signal transduction pathways activated by EPO (excellently reviewed in Wojchowski et al.23). Binding of EPO to the extracellular domain of preformed EPOR dimers24 initiates signaling that in turn results in recruitment and activation of Janus kinase 2 (JAK2) by the intracellular domain of EPOR. JAK2 then phosphorylates several proteins including STAT5. There is evidence in favor of a role of STAT5 in the antiapoptotic action of EPO on erythroid precursors. In particular, STAT5 appears to mediate the induction of Bcl-xL by EPO,25 and anemia and high levels of apoptosis in erythroid progenitors are observed in STAT5 knockout mice, possibly due to a disruption of the Bcl-xL response pathway.26 Further, EPOR activation activates voltage-sensitive Ca2+ channels, which in turn modulates neurotransmitter.27

Summary of demonstrated EPO-signaling pathways in neuronal protection, binding of EPO to its receptor (1) leads to phosphorylation of janus kinase 2 (2). This subsequently activates multiple cascades recruiting PI3-K, Stat5 and MAPKinase. Further, flux of Ca2+ through voltage sensitive channels is modulated (generally inhibited (3)), affecting neurotransmitter release and therefore neuronal excitability (4). NF-kB is reported to be dually activated by JAK-2 and by Akt (5). The net effect is a reduction in the proapoptotic protein BAD (6), and therefore the probability of mitochondrial leakage of cytochrome C (7), an increased production of antiapoptotic proteins of the Bcl-x family (8) and ultimate preservation of the DNA integrity (9). To the extent cytochrome C leakage is not prevented, caspase activation also occurs, inducing DNA degradation and the externalization of phosphatidyl serine on the cell membrane (10), promoting the activation of the inflammatory cells. Solid lines indicate activation; dashed indicate inhibition
Another pathway involves activation of phosphatidylinositol-3 kinase (PI3 K). In fact, PI(3)K is activated by EPO in the EPO-dependent UT-7 leukemia cell line, where it recruits Akt.28 This PI3K–Akt pathway also leads to upregulation of Bcl-xL and inhibition of apoptosis in Baf-3 cells.29 A further mechanism could be represented by NF-kB that is also a target of the PI3K-Akt pathway and mediates antiapoptotic signaling by platelet-derived growth factor.30
EPO as a neuroprotective agent
Study of experimental brain ischemia has shown that EPO and EPOR function as components of an innate response to metabolic stress. Multiple studies have shown that whereas normal brain expresses both EPO and EPOR in a highly restricted and limited manner, following ischemic and other stressors a marked induction of both proteins occurs in animal models31 as well as in human disease.32, 33 Presumably, this regulation is accomplished by specific proinflammatory cytokines, for example, TNF-alpha, IL-1 and IL-6.34 Temporally, EPOR upregulation occurs first by 12 h, primarily in neurons and endothelial cells of the microcirculation, followed hours later by an increase in EPO by both astrocytes and neurons.31 These effects are especially prominent in the penumbral (at risk) region surrounding injury. Study has shown that following the induction, this region is characterized by an increased resistance to subsequent stressors.
These activities in vivo appear to depend upon several processes. In many of these models, diverse pathogenic components play a role, including neuronal death, inflammation and, in the recovery phase, neurorepair. In some cases, the primary mechanism of action of EPO is represented by neuroprotection, while in other models (e.g., experimental autoimmune encephalitis) it could be due to reduced neuroinflammation. We have listed in Table 1 some of the in vitro models where EPO shows neuroprotective effects. It can be seen that, in most models, inhibition of cellular apoptosis was documented. In contrast to its antiapoptotic effect, EPO has not shown any activity in preventing necrosis in either the nervous system (Sinor and Greenberg35 and Mennini et al., unpublished) or the heart.12
The EPO/EPOR is highly prominent during fetal development,36 with the very high levels of expression found in many tissues diminishing rapidly after birth to the generally low levels found in the adult. Gene knockout experiments37 have suggested the importance of this system in development, as the embryos of EPOR −/− animals have abnormal brain development characterized by decreased neuronal progenitor cells as well as grossly decreased neuronal densities associated with massive neuronal apoptosis. Additionally, the heart is underdeveloped. On the other hand, erythroid-specific expression of EPOR in EPOR knockout mice rescues them from lethality suggesting that nonhematopoietic EPOR is not essential for development.38 It would be interesting to evaluate these mice in models of tissue injury.
EPO affects the interaction between apoptosis and inflammation
In many in vivo models of CNS disease for which EPO shows a protective effect, inflammation is also an important pathogenic component, induced by production of cytokines and chemokines followed by leukocyte infiltration and glial activation and proliferation. The complicated inter-related protective effects initiated by EPO administration have been examined in some detail in experimental stroke in the rat. Using a model of ischemia with reperfusion, it was shown that neurons within the penumbra would undergo apoptosis unless exposed to EPO within 3 h of injury.39 In addition to this antiapoptotic effect, EPO administration is also associated with a marked decrease in proinflammatory cytokines within the hemisphere undergoing infarction.40 This profoundly reduced the activation of astrocytes within the region (as indicated by increased glial fibrillary acidic protein (GFAP) expression) as well as grossly decreased influx of inflammatory microglia. The marked in vivo reduction of recruitment of microglia into the region of injury is likely explained by the reduced expression of cell surface markers of apoptosis, for example, phosphatidylserine.41 It should be noted that this proinflammatory role of phosphatidylserine conflicts with other works showing that it stimulates phagocytosis but not inflammation – rather anti-inflammation (for instance: Chan et al.,42 De Simone et al.43 and Brouckaert et al.44)).
Using an in vitro system of neurons cocultured with glia, Villa et al.40 have shown that the anti-inflammatory effects of EPO do not arise from a direct antagonism or a reduction of proinflammatory cytokines, but rather by an antiapoptotic effect on neurons. In fact, neurons exposed to trimethyltin, a toxin that induces neuronal apoptosis,45 release as yet unidentified factors that stimulate inflammatory TNF production by glial cells that, in turn, amplifies neurotoxicity.46 In this context, EPO inhibits TNF production by TMT-exposed neuron-glia co-cultures but not by pure glial preparations directly exposed to neuronal products or LPS.40 This pathway of ‘anti-inflammation by neuroprotection’ is outlined in Figure 2.

Interplay between the antiapoptotic action of EPO and neuroinflammation
According to a well-accepted scheme, apoptosis is a way of dying that triggers much less inflammation than by necrosis, and this is certainly true in many experimental models.47, 48 However, this model of ‘docking without shocking’,49 may not apply to all pathological conditions, and production of inflammatory cytokines by tissue macrophages during phagocytosis of apoptotic cells.50, 51 Similarly to what is observed in a model of cerebral ischemia with EPO,40 inhibition of apoptosis by lysophosphatidic acid or caspase inhibitors decreases inflammation in models of ischemic injury in the kidney and the brain.52, 53
It should be noted, however, that an anti-inflammatory effect of EPO was demonstrated in a model of experimental autoimmune encephalomyelitis, an inflammatory pathology of the CNS of autoimmune origin with marked inflammation in the almost complete absence of neuronal death, at least in the earliest times of the disease.54 Thus, it cannot be ruled out that EPO exerts an anti-inflammatory action on the CNS by different mechanisms than those depicted in Figure 2, although it is presently unclear what these mechanisms may be.
EPO signaling in nervous system cells
EPO has been shown to have multiple effects on neurons. Activation of the EPOR modulates Ca2+ influx55, 56 in vitro. Inhibition of Ca2+ influx upon depolarization directly reduces synaptic vesicle release of glutamate, which acts to reduce the magnitude of neuronal injury (Figure 1). In addition, it could explain the effects of EPO in reducing seizure severity10 as well as reported effects on learning and memory.57 Other indirect effects of EPO that reduce neuronal injury have also been delineated. For example, EPO treatment increases astrocyte production of glutathione peroxidase and in this manner ameliorates neuronal damage caused by excitotoxins.58 EPO also promotes the differentiation and maturation of astrocytes and oligodendrocytes and directly induces the proliferation of astrocytes,59 actions that can modify potential injury. Undoubtedly, other indirect neuronal protection functions remain to be described.
Several antiapoptotic pathways regulated by EPO in erythroid precursors are also activated in neurons and may play a role in prevention of neuronal apoptosis. Triggering NF-kB via activated Jak2 has been reported, and the antiapoptotic action of EPO on nitric oxide-induced cortical neuron apoptosis is blocked by inhibitors of NF-kB translocation as well as by overexpression of an IkBalpha super-repressor or of a dominant interfering form of Jak2.60 The antiapoptotic action of NF-kB in neurons also involves activation of Akt1 and Bad phosphorylation41, 61 as well as Bcl-xL upregulation.62, 63 Bcl-xL was also found induced by EPO in a study where DNA microarrays were used to identify modulated genes in PC12 neuronal-like cells64 and in vivo in a gerbil model of cerebral ischemia.62
Of note, Ruscher et al.,61 studying the protective effect of EPO against oxygen glucose deprivation-induced apoptosis of primary cortical neurons, confirmed that the protective effect requires Jak2, as shown by Digicaylioglu and Lipton,60 and of PI(3)Kinase, as shown by us,39 using specific inhibitors of these kinases. In particular, the MAPK inhibitor PD98059 and the PI(3)Kinase inhibitor LY984002 blocked the protective effect of EPO in hippocampal neurons exposed to hypoxia.39 On the other hand, unlike others reported in erythroid cells, the other pathway downstream of Jak2, activation of STATs, was not activated by EPO in this study, and no induction of mRNA for Bcl2, Bcl-xL, Bag-1, Bax or Bad, was observed, as measured by PCR.61
In addition to directly activating survival kinases such as ERK, EPO, while not activating Akt under normal conditions, prevents the decrease in Akt phosphorylation induced by a 15-h exposure of neurons to hypoxia.39 A similar pattern has been recently observed in vivo where administration of EPO, protects newborn rats from neuronal apoptosis induced by MK801 and prevents MK801-induced decrease in the phosphorylation level of ERK1/2 and Akt.65
EPO has profound effects in reducing injury and enhancing recovery in compression injuries of peripheral nerves.66, 67 Campana et al.66, 67 performed a crush injury of a spinal nerve root for which follow-up showed a robust effect by EPO to prevent apoptosis in dorsal root ganglion neurons which occurred by Jak2 signaling, as well as a corresponding improvement in pain behavior.
Clearly, the relevance of the pathways outlined above in the neuroprotection observed in vivo with EPO is far from being established. For instance, in a study of neonatal hypoxic ischemic injury in mice has reported that NF-kB-immunoreactivity in neurons in the injured areas was not decreased by EPO, neither was its subcellular distribution, despite a marked neuroprotective effect.68
Other tissue-protective activities of EPO
Capillary endothelial cells express EPOR on their intraluminal surfaces. EPO has been shown in vitro to antagonize the apoptosis of endothelial cells subjected to ischemic stressors.69 EPO thus plays a role in maintaining the integrity of the microvasculature. Additionally, EPO has also been shown to stimulate mitogenesis and support angiogenesis, both functioning to improve tissue oxygenation. The endothelial cell is additionally important as a conduit for systemically administered EPO to pass into organs with a barrier to the circulation. Recent in vitro studies have shown that EPO present on the apical (luminal) side is transported in a unidirectional manner through the endothelial cell by transcytosis.70 Further, EPO strengthens the tight junctions of endothelial cells and will antagonize the effects of VEGF, which is known to reduce tight junction proteins allowing leakage from the capillary around the endothelial cells into the brain parenchyma.70
EPO has been shown to be protective of myocardial ischemia either permanent or with reperfusion.12, 71 Further, cardiomyocytes losses were not completely prevented in EPO-treated animals, being reduced by ∼50%. In spite of tissue loss, maladaptive remodeling did not occur, resulting in a maintained normal cardiac function.12 Further, studies performed on isolated hearts have shown that EPO mediates ischemic preconditioning.72
The kidney is protected by EPO in the setting of both ischemic and toxic injuries.73, 74, 75 In ischemic injury with reperfusion, EPO prevents apoptosis of tubular epithelial cells as well as mediates intense mitotic activity of the surviving population of proximal tubular epithelial cells.73
The skin has also been shown to be protected by EPO in rodent flap models.76, 77 In these studies, the tissue-protective effects appear to depend strongly on the dose and treatment duration, suggesting that tissues may vary in their responses to EPO.
From the perspective of the evolving concept of EPO and EPOR as a endogenous local system of protection and the widespread expression of these molecules, it is likely that many other organs and tissues when examined for protective effects of EPO will demonstrate a positive role.
Therapeutic perspectives
Recombinant human EPO has been shown in general to be an exceedingly safe drug, as millions of patients have received it over the last decade for treatment of anemia. However, some side effects, including increased blood pressure and risk of thrombosis have been noted and these are of special concern for patients who are not anemic. EPO has been shown to mediate angiogenesis in the microcirculation, with a potency similar to VEGF.78 However, unlike VEGF, EPO does not reduce the tight junctions between endothelial cells, and therefore does not promote capillary leakage.70
The experimental models characterized by potential cellular barriers for the systemic administration of recombinant human EPO (e.g., brain, retina, spinal cord) have been found to require a minimum effective dose that is generally higher than the doses of EPO currently employed for treatment of anemia (but those used experimentally for bone marrow rescue have been much higher, as reviewed by Baron et al.79). Although acute (i.e. a few doses) administration of recombinant human EPO for treatment of tissue injury is not likely to be harmful, chronic dosing will at the least provoke increases in erythrocyte mass. This is undesirable from a rheologically perspective and could worsen injury. The recent Phase II trial of human stroke by EPO used three daily doses substantially higher than the clinical norm and, in these patients, EPO was shown to be safe and did not raise the hemoglobin concentration.80 Thus, this might represent a valuable therapeutic approach to acute CNS injury, such as brain and spinal cord trauma. On the other hand, use of EPO in chronic diseases associated with neuronal apoptosis or neuroinflammation, such as Parkinson's disease, Alzheimer disease or multiple sclerosis would be possible only if the erythropoietic effect was blocked.
The studies on the signaling mechanisms of the cytoprotective versus the hemopoietic actions of EPO, and the receptors involved, will help identifying molecules that retain the cytoprotective actions of EPO but not its hemopoietic effects. It was recently reported that desialylated EPO is a neuroprotective cytokine that does not induce erythrodifferentiation,81 indicating the feasibility of this approach.
In summary, EPO is a major modifier of apoptosis of multiple cell types in different tissues and organs in the setting of potential injury. However, EPO also exhibits many other actions that serve to protect cells either directly or indirectly. EPO should therefore be regarded also as a general tissue-protective cytokine.
Abbreviations
- AMPA:
-
amino-3-hydroxy-5-methylisoxazole-4-propionic acid
- CNS:
-
central nervous system
- EPO:
-
erythropoietin
- EPOR:
-
EPO receptor
- GFAP:
-
glial fibrillary acidic protein
- HIF:
-
hypoxia-inducible factor
- IGFBP:
-
insulin-like growth factor binding protein
- NMDA:
-
N-methyl-D-aspartate
- NO:
-
nitric oxide
- VEGF:
-
vascular endothelial growth factor
References
Semenza GL (2001) HIF-1 and mechanisms of hypoxia sensing. Curr. Opin. Cell Biol. 13: 167–171
Wiesener MS and Maxwell PH (2003) HIF and oxygen sensing; as important to life as the air we breathe? Ann. Med. 35: 183–190
Wenger RH (2002) Cellular adaptation to hypoxia: O2-sensing protein hydroxylases, hypoxia-inducible transcription factors, and O2-regulated gene expression. FASEB J. 16: 1151–1162
Seagroves TN, Ryan HE, Lu H, Wouters BG, Knapp M, Thibault P, Laderoute K and Johnson RS (2001) Transcription factor HIF-1 is a necessary mediator of the pasteur effect in mammalian cells. Mol. Cell Biol. 21: 3436–3444
Minchenko A, Leshchinsky I, Opentanova I, Sang N, Srinivas V, Armstead V and Caro J (2002) Hypoxia-inducible factor-1-mediated expression of the 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase-3 (PFKFB3) gene. Its possible role in the Warburg effect. J. Biol. Chem. 277: 6183–6187
Zaman K, Ryu H, Hall D, O'Donovan K, Lin KI, Miller MP, Marquis JC, Baraban JM, Semenza GL and Ratan RR (1999) Protection from oxidative stress-induced apoptosis in cortical neuronal cultures by iron chelators is associated with enhanced DNA binding of hypoxia-inducible factor-1 and ATF-1/CREB and increased expression of glycolytic enzymes, p21(waf1/cip1), and erythropoietin. J. Neurosci. 19: 9821–9830
Lambrechts D, Storkebaum E, Morimoto M, Del-Favero J, Desmet F, Marklund SL, Wyns S, Thijs V, Andersson J, van Marion I, Al-Chalabi A, Bornes S, Musson R, Hansen V, Beckman L, Adolfsson R, Pall HS, Prats H, Vermeire S, Rutgeerts P, Katayama S, Awata T, Leigh N, Lang-Lazdunski L, Dewerchin M, Shaw C, Moons L, Vlietinck R, Morrison KE, Robberecht W, Van Broeckhoven C, Collen D, Andersen PM and Carmeliet P (2003) VEGF is a modifier of amyotrophic lateral sclerosis in mice and humans and protects motoneurons against ischemic death. Nat. Genet. 34: 383–394
Oosthuyse B, Moons L, Storkebaum E, Beck H, Nuyens D, Brusselmans K, Van Dorpe J, Hellings P, Gorselink M, Heymans S, Theilmeier G, Dewerchin M, Laudenbach V, Vermylen P, Raat H, Acker T, Vleminckx V, Van Den Bosch L, Cashman N, Fujisawa H, Drost MR, Sciot R, Bruyninckx F, Hicklin DJ, Ince C, Gressens P, Lupu F, Plate KH, Robberecht W, Herbert JM, Collen D and Carmeliet P (2001) Deletion of the hypoxia-response element in the vascular endothelial growth factor promoter causes motor neuron degeneration. Nat. Genet. 28: 131–138
Sun Y, Jin K, Xie L, Childs J, Mao XO, Logvinova A and Greenberg DA (2003) VEGF-induced neuroprotection, neurogenesis, and angiogenesis after focal cerebral ischemia. J. Clin. Invest. 111: 1843–1851
Brines ML, Ghezzi P, Keenan S, Agnello D, de Lanerolle NC, Cerami C, Itri LM and Cerami A (2000) Erythropoietin crosses the blood-brain barrier to protect against experimental brain injury. Proc. Natl. Acad. Sci. USA 97: 10526–10531
Ehrenreich H, Degner D, Meller J, Brines M, Behe M, Hasselblatt M, Woldt H, Falkai P, Knerlich F, Jacob S, von Ahsen N, Maier W, Bruck W, Ruther E, Cerami A, Becker W and Siren AL (2004) Erythropoietin: a candidate compound for neuroprotection in schizophrenia. Mol. Psychiatry. 9: 42–54
Calvillo L, Latini R, Kajstura J, Leri A, Anversa P, Ghezzi P, Salio M, Cerami A and Brines M (2003) Recombinant human erythropoietin protects the myocardium from ischemia-reperfusion injury and promotes beneficial remodeling. Proc. Natl. Acad. Sci. USA 100: 4802–4806
Boyer SH, Bishop TR, Rogers OC, Noyes AN, Frelin LP and Hobbs S (1992) Roles of erythropoietin, insulin-like growth factor 1, and unidentified serum factors in promoting maturation of purified murine erythroid colony-forming units. Blood 80: 2503–2512
Koury MJ and Bondurant MC (1988) Maintenance by erythropoietin of viability and maturation of murine erythroid precursor cells. J. Cell Physiol. 137: 65–74
Miller CP, Liu ZY, Noguchi CT and Wojchowski DM (1999) A minimal cytoplasmic subdomain of the erythropoietin receptor mediates erythroid and megakaryocytic cell development. Blood 94: 3381–3387
Spivak JL, Pham T, Isaacs M and Hankins WD (1991) Erythropoietin is both a mitogen and a survival factor. Blood 77: 1228–1233
Yu H, Bauer B, Lipke GK, Phillips RL and Van Zant G (1993) Apoptosis and hematopoiesis in murine fetal liver. Blood 81: 373–384
Koury MJ and Bondurant MC (1990) Erythropoietin retards DNA breakdown and prevents programmed death in erythroid progenitor cells. Science 248: 378–381
Silva M, Grillot D, Benito A, Richard C, Nunez G and Fernandez-Luna JL (1996) Erythropoietin can promote erythroid progenitor survival by repressing apoptosis through Bcl-XL and Bcl-2. Blood 88: 1576–1582
Wagner KU, Claudio E, Rucker III EB, Riedlinger G, Broussard C, Schwartzberg PL, Siebenlist U and Hennighausen L (2000) Conditional deletion of the Bcl-x gene from erythroid cells results in hemolytic anemia and profound splenomegaly. Development 127: 4949–4958
Bazan JF (1989) A novel family of growth factor receptors: a common binding domain in the growth hormone, prolactin, erythropoietin and IL-6 receptors, and the p75 IL-2 receptor beta-chain. Biochem. Biophys. Res. Commun. 164: 788–795
Bazan JF (1990) Structural design and molecular evolution of a cytokine receptor superfamily. Proc. Natl. Acad. Sci. USA 87: 6934–6938
Wojchowski DM, Gregory RC, Miller CP, Pandit AK and Pircher TJ (1999) Signal transduction in the erythropoietin receptor system. Exp. Cell Res. 253: 143–156
Livnah O, Stura EA, Middleton SA, Johnson DL, Jolliffe LK and Wilson IA (1999) Crystallographic evidence for preformed dimers of erythropoietin receptor before ligand activation. Science 283: 987–990
Silva M, Benito A, Sanz C, Prosper F, Ekhterae D, Nunez G and Fernandez-Luna JL (1999) Erythropoietin can induce the expression of bcl-x(L) through Stat5 in erythropoietin-dependent progenitor cell lines. J. Biol. Chem. 274: 22165–22169
Socolovsky M, Fallon AE, Wang S, Brugnara C and Lodish HF (1999) Fetal anemia and apoptosis of red cell progenitors in Stat5a-/-5b-/- mice: a direct role for Stat5 in Bcl-X(L) induction. Cell 98: 181–191
Kawakami M, Sekiguchi M, Sato K, Kozaki S and Takahashi M (2001) Erythropoietin receptor-mediated inhibition of exocytotic glutamate release confers neuroprotection during chemical ischemia. J. Biol. Chem. 276: 39469–39475
Kashii Y, Uchida M, Kirito K, Tanaka M, Nishijima K, Toshima M, Ando T, Koizumi K, Endoh T, Sawada K, Momoi M, Miura Y, Ozawa K and Komatsu N (2000) A member of Forkhead family transcription factor, FKHRL1, is one of the downstream molecules of phosphatidylinositol 3-kinase-Akt activation pathway in erythropoietin signal transduction. Blood 96: 941–949
Leverrier Y, Thomas J, Mathieu AL, Low W, Blanquier B and Marvel J (1999) Role of PI3-kinase in Bcl-X induction and apoptosis inhibition mediated by IL-3 or IGF-1 in Baf-3 cells. Cell Death Differ. 6: 290–296
Romashkova JA and Makarov SS (1999) NF-kappaB is a target of AKT in anti-apoptotic PDGF signalling. Nature 401: 86–90
Bernaudin M, Marti HH, Roussel S, Divoux D, Nouvelot A, MacKenzie ET and Petit E (1999) A potential role for erythropoietin in focal permanent cerebral ischemia in mice. J. Cereb. Blood Flow Metab. 19: 643–651
Eid T, Brines ML, Cerami A, Spencer DD, Kim JH, Schweitzer JS, Ottersen OP and de Lanerolle NC (2004) Increased expression of erythropoietin receptor on blood vessels in the human epileptogenic hippocampus with sclerosis. J. Neuropathol. Exp. Neurol. 63: 73–83
Siren AL, Knerlich F, Poser W, Gleiter CH, Bruck W and Ehrenreich H (2001) Erythropoietin and erythropoietin receptor in human ischemic/hypoxic brain. Acta Neuropathol. (Berl.) 101: 271–276
Nagai A, Nakagawa E, Choi HB, Hatori K, Kobayashi S and Kim SU (2001) Erythropoietin and erythropoietin receptors in human CNS neurons, astrocytes, microglia, and oligodendrocytes grown in culture. J. Neuropathol. Exp. Neurol. 60: 386–392
Sinor AD and Greenberg DA (2000) Erythropoietin protects cultured cortical neurons, but not astroglia, from hypoxia and AMPA toxicity. Neurosci, Lett. 290: 213–215
Juul SE, Yachinis AT, Rojiani AM and Christensen RD (1999) Immunohistochemical Localization of Erythropoietin and Its Receptor in the Developing Human Brain. Pediat. Dev. Pathol. 2: 148–158
Yu X, Shacka JJ, Eells JB, Suarez-Quian C, Przygodzki RM, Beleslin-Cokic B, Lin CS, Nikodem VM, Hempstead B, Flanders KC, Costantini F and Noguchi CT (2002) Erythropoietin receptor signalling is required for normal brain development. Development 129: 505–516
Suzuki N, Ohneda O, Takahashi S, Higuchi M, Mukai HY, Nakahata T, Imagawa S and Yamamoto M (2002) Erythroid-specific expression of the erythropoietin receptor rescued its null mutant mice from lethality. Blood 100: 2279–2288
Siren AL, Fratelli M, Brines M, Goemans C, Casagrande S, Lewczuk P, Keenan S, Gleiter C, Pasquali C, Capobianco A, Mennini T, Heumann R, Cerami A, Ehrenreich H and Ghezzi P (2001) Erythropoietin prevents neuronal apoptosis after cerebral ischemia and metabolic stress. Proc. Natl. Acad. Sci. USA 98: 4044–4049
Villa P, Bigini P, Mennini T, Agnello D, Laragione T, Cagnotto A, Viviani B, Marinovich M, Cerami A, Coleman TR, Brines M and Ghezzi P (2003) Erythropoietin selectively attenuates cytokine production and inflammation in cerebral ischemia by targeting neuronal apoptosis. J. Exp. Med. 198: 971–975
Chong ZZ, Kang JQ and Maiese K (2003) Erythropoietin fosters both intrinsic and extrinsic neuronal protection through modulation of microglia, Akt1, Bad, and caspase-mediated pathways. Br. J. Pharmacol. 138: 1107–1118
Chan A, Seguin R, Magnus T, Papadimitriou C, Toyka KV, Antel JP and Gold R (2003) Phagocytosis of apoptotic inflammatory cells by microglia and its therapeutic implications: termination of CNS autoimmune inflammation and modulation by interferon-beta. Glia 43: 231–242
De Simone R, Ajmone-Cat MA, Tirassa P and Minghetti L (2003) Apoptotic PC12 cells exposing phosphatidylserine promote the production of anti-inflammatory and neuroprotective molecules by microglial cells. J. Neuropathol. Exp. Neurol. 62: 208–216
Brouckaert G, Kalai M, Krysko DV, Saelens X, Vercammen D, Ndlovu, Haegeman G, D'Herde K and Vandenabeele P (2004) Phagocytosis of necrotic cells by macrophages is phosphatidylserine dependent and does not induce inflammatory cytokine production. Mol. Biol. Cell. 15: 1089–1100
Viviani B, Rossi AD, Chow SC and Nicotera P (1995) Organotin compounds induce calcium overload and apoptosis in PC12 cells. Neurotoxicology 16: 19–25
Viviani B, Corsini E, Galli CL and Marinovich M (1998) Glia increase degeneration of hippocampal neurons through release of tumor necrosis factor-alpha. Toxicol. Appl. Pharmacol. 150: 271–276
Henson PM, Bratton DL and Fadok VA (2001) The phosphatidylserine receptor: a crucial molecular switch? Nat. Rev. Mol. Cell Biol. 2: 627–633
Devitt A, Moffatt OD, Raykundalia C, Capra JD, Simmons DL and Gregory CD (1998) Human CD14 mediates recognition and phagocytosis of apoptotic cells. Nature 392: 505–509
Savill J (1998) Apoptosis. Phagocytic docking without shocking. Nature 392: 442–443
Kurosaka K, Watanabe N and Kobayashi Y (2001) Production of proinflammatory cytokines by resident tissue macrophages after phagocytosis of apoptotic cells. Cell Immunol. 211: 1–7
Canbay A, Feldstein AE, Higuchi H, Werneburg N, Grambihler A, Bronk SF and Gores GJ (2003) Kupffer cell engulfment of apoptotic bodies stimulates death ligand and cytokine expression. Hepatology 38: 1188–1198
de Vries B, Matthijsen RA, van Bijnen AA, Wolfs TG and Buurman WA (2003) Lysophosphatidic acid prevents renal ischemia-reperfusion injury by inhibition of apoptosis and complement activation. Am. J. Pathol. 163: 47–56
Rabuffetti M, Sciorati C, Tarozzo G, Clementi E, Manfredi AA and Beltramo M (2000) Inhibition of caspase-1-like activity by Ac-Tyr-Val-Ala-Asp-chloromethyl ketone induces long-lasting neuroprotection in cerebral ischemia through apoptosis reduction and decrease of proinflammatory cytokines. J. Neurosci. 20: 4398–4404
Agnello D, Bigini P, Villa P, Mennini T, Cerami A, Brines ML and Ghezzi P (2002) Erythropoietin exerts an anti-inflammatory effect on the CNS in a model of experimental autoimmune encephalomyelitis. Brain Res. 952: 128–134
Kawakami M, Iwasaki S, Sato K and Takahashi M (2000) Erythropoietin inhibits calcium-induced neurotransmitter release from clonal neuronal cells. Biochem. Biophys. Res. Commun. 279: 293–297
Masuda S, Nagao M, Takahata K, Konishi Y, Gallyas Jr. F, Tabira T and Sasaki R (1993) Functional erythropoietin receptor of the cells with neural characteristics. Comparison with receptor properties of erythroid cells. J. Biol. Chem. 268: 11208–11216
Mogensen J, Miskowiak K, Sorensen TA, Lind CT, Olsen NV, Springborg JB and Mala H (2004) Erythropoietin improves place learning in fimbria-fornix-transected rats and modifies the search pattern of normal rats. Pharmacol. Biochem. Behav. 77: 381–390
Genc S, Akhisaroglu M, Kuralay F and Genc K (2002) Erythropoietin restores glutathione peroxidase activity in 1-methyl-4- phenyl-1,2,5,6-tetrahydropyridine-induced neurotoxicity in C57BL mice and stimulates murine astroglial glutathione peroxidase production in vitro. Neurosci. Lett. 321: 73–76
Sugawa M, Sakurai Y, Ishikawa-Ieda Y, Suzuki H and Asou H (2002) Effects of erythropoietin on glial cell development; oligodendrocyte maturation and astrocyte proliferation. Neurosci. Res. 44: 391–403
Digicaylioglu M and Lipton SA (2001) Erythropoietin-mediated neuroprotection involves cross-talk between Jak2 and NF-kappaB signalling cascades. Nature 412: 641–647
Ruscher K, Freyer D, Karsch M, Isaev N, Megow D, Sawitzki B, Priller J, Dirnagl U and Meisel A (2002) Erythropoietin is a paracrine mediator of ischemic tolerance in the brain: evidence from an in vitro model. J. Neurosci. 22: 10291–10301
Wen TC, Sadamoto Y, Tanaka J, Zhu PX, Nakata K, Ma YJ, Hata R and Sakanaka M (2002) Erythropoietin protects neurons against chemical hypoxia and cerebral ischemic injury by up-regulating Bcl-xL expression. J. Neurosci. Res. 67: 795–803
Chong ZZ, Kang JQ and Maiese K (2002) Hematopoietic factor erythropoietin fosters neuroprotection through novel signal transduction cascades. J. Cereb. Blood Flow Metab. 22: 503–514
Renzi MJ, Farrell FX, Bittner A, Galindo JE, Morton M, Trinh H and Jolliffe LK (2002) Erythropoietin induces changes in gene expression in PC-12 cells. Brain Res. Mol. Brain Res. 104: 86–95
Dzietko M, Felderhoff-Mueser U, Sifringer M, Krutz B, Bittigau P, Thor F, Heumann R, Bührer C, Ikonomidou C and Hansen HH (2004) Erythropoietin protects the developing brain against N-methyl–aspartate receptor antagonist neurotoxicity. Neurobiol. Dis. 15: 177–187
Sekiguchi Y, Kikuchi S, Myers RR and Campana WM (2003) ISSLS prize winner: Erythropoietin inhibits spinal neuronal apoptosis and pain following nerve root crush. Spine 28: 2577–2584
Campana WM and Myers RR (2003) Exogenous erythropoietin protects against dorsal root ganglion apoptosis and pain following peripheral nerve injury. Eur. J. Neurosci. 18: 1497–1506
Matsushita H, Johnston MV, Lange MS and Wilson MA (2003) Protective effect of erythropoietin in neonatal hypoxic ischemia in mice. Neuro Report 14: 1757–1761
Chong ZZ, Kang JQ and Maiese K (2002) Erythropoietin is a novel vascular protectant through activation of Akt1 and mitochondrial modulation of cysteine proteases. Circulation 106: 2973–2979
Martinez-Estrada OM, Rodriguez-Millan E, Gonzalez-De Vicente E, Reina M, Vilaro S and Fabre M (2003) Erythropoietin protects the in vitro blood–brain barrier against VEGF-induced permeability. Eur. J. Neurosci. 18: 2538–2544
Moon C, Krawczyk M, Ahn D, Ahmet I, Paik D, Lakatta EG and Talan MI (2003) Erythropoietin reduces myocardial infarction and left ventricular functional decline after coronary artery ligation in rats. Proc. Natl. Acad. Sci. USA 100: 11612–11617
Cai Z, Manalo DJ, Wei G, Rodriguez ER, Fox-Talbot K, Lu H, Zweier JL and Semenza GL (2003) Hearts from rodents exposed to intermittent hypoxia or erythropoietin are protected against ischemia–reperfusion injury. Circulation 108: 79–85
Vesey DA, Cheung C, Pat B, Endre Z, Gobe G and Johnson DW (2004) Erythropoietin protects against ischaemic acute renal injury. Nephrol. Dial. Transplant. 19: 348–355
Yang CW, Li C, Jung JY, Shin SJ, Choi BS, Lim SW, Sun BK, Kim YS, Kim J, Chang YS and Bang BK (2003) Preconditioning with erythropoietin protects against subsequent ischemia–reperfusion injury in rat kidney. FASEB J. 17: 1754–1755
Bagnis C, Beaufils H, Jacquiaud C, Adabra Y, Jouanneau C, Le Nahour G, Jaudon MC, Bourbouze R, Jacobs C and Deray G (2001) Erythropoietin enhances recovery after cisplatin-induced acute renal failure in the rat. Nephrol. Dial. Transplant. 16: 932–938
Buemi M, Vaccaro M, Sturiale A, Galeano MR, Sansotta C, Cavallari V, Floccari F, D'Amico D, Torre V, Calapai G, Frisina N, Guarneri F and Vermiglio G (2002) Recombinant human erythropoietin influences revascularization and healing in a rat model of random ischaemic flaps. Acta Derm. Venereol. 82: 411–417
Saray A, Ozakpinar R, Koc C, Serel S, Sen Z and Can Z (2003) Effect of chronic and short-term erythropoietin treatment on random flap survival in rats: an experimental study. Laryngoscope 113: 85–89
Jaquet K, Krause K, Tawakol-Khodai M, Geidel S and Kuck KH (2002) Erythropoietin and VEGF exhibit equal angiogenic potential. Microvasc. Res. 64: 326–333
Baron F, Frere P, Fillet G and Beguin Y (2003) Recombinant human erythropoietin therapy is very effective after an autologous peripheral blood stem cell transplant when started soon after engraftment. Clin. Cancer Res. 9: 5566–5572
Ehrenreich H, Hasselblatt M, Dembowski C, Cepek L, Lewczuk P, Stiefel M, Rustenbeck HH, Breiter N, Jacob S, Knerlich F, Bohn M, Poser W, Ruther E, Kochen M, Gefeller O, Gleiter C, Wessel TC, De Ryck M, Itri L, Prange H, Cerami A, Brines M and Siren AL (2002) Erythropoietin therapy for acute stroke is both safe and beneficial. Mol. Med. 8: 495–505
Erbayraktar S, Grasso G, Sfacteria A, Xie QW, Coleman T, Kreilgaard M, Torup L, Sager T, Erbayraktar Z, Gokmen N, Yilmaz O, Ghezzi P, Villa P, Fratelli M, Casagrande S, Leist M, Helboe L, Gerwein J, Christensen S, Geist MA, Pedersen LO, Cerami-Hand C, Wuerth JP, Cerami A and Brines M (2003) Asialoerythropoietin is a nonerythropoietic cytokine with broad neuroprotective activity in vivo. Proc. Natl. Acad. Sci. USA 100: 6741–6746
Koshimura K, Murakami Y, Sohmiya M, Tanaka J and Kato Y (1999) Effects of erythropoietin on neuronal activity. J. Neurochem. 72: 2565–2572
Morishita E, Masuda S, Nagao M, Yasuda Y and Sasaki R (1997) Erythropoietin receptor is expressed in rat hippocampal and cerebral cortical neurons, and erythropoietin prevents in vitro glutamate-induced neuronal death. Neuroscience 76: 105–116
Sakanaka M, Wen TC, Matsuda S, Masuda S, Morishita E, Nagao M and Sasaki R (1998) In vivo evidence that erythropoietin protects neurons from ischemic damage. Proc. Natl. Acad. Sci. USA 95: 4635–4640
Grasso G (2001) Neuroprotective effect of recombinant human erythropoietin in experimental subarachnoid hemorrhage. J. Neurosurg. Sci. 45: 7–14
Kumral A, Ozer E, Yilmaz O, Akhisaroglu M, Gokmen N, Duman N, Ulukus C, Genc S and Ozkan H (2003) Neuroprotective effect of erythropoietin on hypoxic–ischemic brain injury in neonatal rats. Biol. Neonate 83: 224–228
Romsi P, Ronka E, Kiviluoma K, Vainionpaa V, Hirvonen J, Mennander A, Pokela M, Biancari F, Rimpilainen J and Juvonen T (2002) Potential neuroprotective benefits of erythropoietin during experimental hypothermic circulatory arrest. J. Thorac. Cardiovasc. Surg. 124: 714–723
Genc S, Kuralay F, Genc K, Akhisaroglu M, Fadiloglu S, Yorukoglu K, Fadiloglu M and Gure A (2001) Erythropoietin exerts neuroprotection in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-treated C57/BL mice via increasing nitric oxide production. Neurosci. Lett. 298: 139–141
Junk AK, Mammis A, Savitz SI, Singh M, Roth S, Malhotra S, Rosenbaum PS, Cerami A, Brines M and Rosenbaum DM (2002) Erythropoietin administration protects retinal neurons from acute ischemia–reperfusion injury. Proc. Natl. Acad. Sci. USA 99: 10659–10664
Celik M, Gokmen N, Erbayraktar S, Akhisaroglu M, Konakc S, Ulukus C, Genc S, Genc K, Sagiroglu E, Cerami A and Brines M (2002) Erythropoietin prevents motor neuron apoptosis and neurologic disability in experimental spinal cord ischemic injury. Proc. Natl. Acad. Sci. USA 99: 2258–2263
Gorio A, Gokmen N, Erbayraktar S, Yilmaz O, Madaschi L, Cichetti C, Di Giulio AM, Vardar E, Cerami A and Brines M (2002) Recombinant human erythropoietin counteracts secondary injury and markedly enhances neurological recovery from experimental spinal cord trauma. Proc. Natl. Acad. Sci. USA 99: 9450–9455
Bianchi R, Buyukakilli B, Brines M, Savino C, Cavaletti G, Oggioni N, Lauria G, Borgna M, Lombardi R, Cimen B, Comelekoglu U, Kanik A, Tataroglu C, Cerami A and hezzi P (2004) Erythropoietin both protects from and reverses experimental diabetic neuropathy. Proc. Natl. Acad. Sci. USA 101: 823–828
Akisu M, Kullahcioglu Girgin F, Baka M, Husseyinov A and Kultursay N (2001) The role of recombinant human erythropoietin in lipid peroxidation and platelet-activating factor generation in a rat model of necrotizing enterocolitis. Eur. J. Pediatr. Surg. 11: 167–172
Akisu M, Tuzun S, Arslanoglu S and Yalaz M and Kultursay N (2001) Effect of recombinant human erythropoietin administration on lipid peroxidation and antioxidant enzyme(s) activities in preterm infants. Acta. Med. Okayama 55: 357–362
Author information
Authors and Affiliations
Corresponding author
Additional information
Edited by Dr G Melino
Rights and permissions
About this article
Cite this article
Ghezzi, P., Brines, M. Erythropoietin as an antiapoptotic, tissue-protective cytokine. Cell Death Differ 11 (Suppl 1), S37–S44 (2004). https://doi.org/10.1038/sj.cdd.4401450
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.cdd.4401450
Keywords
This article is cited by
-
Nanoengineered injectable hydrogels derived from layered double hydroxides and alginate for sustained release of protein therapeutics in tissue engineering applications
Journal of Nanobiotechnology (2023)
-
Erythropoietin regulates developmental myelination in the brain stimulating postnatal oligodendrocyte maturation
Scientific Reports (2023)
-
Cross-platform transcriptomic profiling of the response to recombinant human erythropoietin
Scientific Reports (2021)
-
Erythropoietin promotes functional recovery via anti-apoptotic mechanisms in mouse unilateral ureteral obstruction
Cell Stress and Chaperones (2020)
-
Combination of intravitreal bevacizumab and erythropoietin versus intravitreal bevacizumab alone for refractory diabetic macular edema: a randomized double-blind clinical trial
Graefe's Archive for Clinical and Experimental Ophthalmology (2019)
