
Abstract
Many patients infected with human immunodeficiency virus type-1 (HIV-1) suffer cognitive impairment ranging from mild to severe (HIV dementia), which may result from neuronal death in the basal ganglia, cerebral cortex and hippocampus. HIV-1 does not kill neurons by infecting them. Instead, viral proteins released from infected glial cells, macrophages and/or stem cells may directly kill neurons or may increase their vulnerability to other cell death stimuli. By binding to and/or indirectly activating cell surface receptors such as CXCR4 and the N-methyl-D-aspartate receptor, the HIV-1 proteins gp120 and Tat may trigger neuronal apoptosis and excitotoxicity as a result of oxidative stress, perturbed cellular calcium homeostasis and mitochondrial alterations. Membrane lipid metabolism and inflammation may also play important roles in determining whether neurons live or die in HIV-1-infected patients. Drugs and diets that target oxidative stress, excitotoxicity, inflammation and lipid metabolism are in development for the treatment of HIV-1 patients.
Similar content being viewed by others


Clinical Manifestations
Human immunodeficiency virus type-1 (HIV-1) infection is the commonest cause of dementia in adults less than 40 years of age. Prior to the availability of combination antiretroviral therapy, HIV dementia was noted in nearly 20% of patients with AIDS and often progressed rapidly over a period of 6 months.1, 2 These patients showed a subcortical dementia due to predominant involvement of the basal ganglia manifesting as psychomotor slowing.3 Some patients also developed Parkinsonism, cognitive difficulties and behavioral abnormalites including psychosis and depression. Due to involvement of the dopaminergic system, these patients have extreme sensitivity to side effects from antipsychotic agents.4, 5 Since the availability of combination antiretroviral therapy, the incidence of HIV dementia has decreased and the clinical manifestations are less severe. However, due to improved survival rates, the prevalence rates of HIV dementia continue to rise.6 In these milder forms of HIV dementia, cognitive dysfunction is more prevalent.
Our current understanding of the clinical features and pathophysiological mechanisms that underlie HIV dementia comes from the study of the clade B subtype of HIV-1, which is most prevalent in North America and Western Europe. However, there are nearly a dozen clades that have been identified and the most common HIV-1 clade worldwide is clade C. Epidemiological and pathological studies from these regions of the world, and experimental studies using these clades have not yet been performed. However, it is likely that important differences might exist among clades because viral sequences suggest that mutations may be present in regions of the virus implicated in the neuropathogenesis of HIV dementia.7, 8
Pathology
The pathological hallmark of HIV-1 infection of the brain is the presence of multinucleated giant cells which are formed by syncytia of HIV-1-infected macrophages.9 HIV-1 infects the perivascular macrophages, resident microglia10 and some astrocytes.11 Infection of other cell types may also occur but remains controversial. While the infection of macrophages and microglia is a productive infection with formation and release of viral particles,12 infection of astrocytes may result in viral latency and small amounts of virus are only released upon stimulation by cytokines.13 Prominent dendritic pruning14, loss of synapses15 and cell death16, 17 may occur in neurons. However, because neurons themselves are rarely infected, the damage to neurons is presumed to be indirect. Interestingly, while HIV-1 causes an immunodeficiency by loss of CD4 cells, the immune profiles within the brain suggest an immune activation involving mediators produced by microglia/macrophages and astrocytes.18 Thus, there is an increased production of cytokines such as TNF-α, interleukin (IL)-6 and IL-1β, and chemokines such as MCP-1.19, 20 Of these mediators, MCP-1 levels in the cerebrospinal fluid (CSF) correlate positively with HIV dementia.20
The HIV-1 Virus: Structure and Replication
HIV-1 belongs to the retrovirus family because these viruses have a unique enzyme called reverse transcriptase that converts viral RNA to DNA upon viral entry into the cell. Viral replication occurs after proviral DNA is integrated into host cell chromosomal DNA. The viral genome encodes two general classes of proteins, structural and regulatory.
The structural proteins form the envelope, the core and the matrix of the virus. Three regions within the HIV-1 genome, namely, env, pol and gag, encode all the structural proteins. The env gene codes for gp160 which is cleaved to form the two major envelope glycoproteins, gp120 and gp41. gp120 forms the surface spikes on the virion and gp41 is a transmembrane glycoprotein. The pol gene codes for reverse transcriptase, a protease that cleaves the polyproteins coded by the pol and gag genes into their active forms and an endonuclease that is responsible for viral integration into the host genome. The gag gene codes for all the core proteins. Regulatory proteins encoded by the viral genome control viral genome expression either at the level of proviral DNA or the viral mRNA. At least six genes (tat, rev, nef, vif, vpu and vpr) code for proteins that are involved in the regulation of viral replication. These regulatory proteins do not get incorporated into the viral particle, but regulate viral replication and release at multiple levels. For example, Tat, Rev and Nef are targeted to the nucleus of the cell. However, Nef can also be trapped within the cytoplasm of the cell (e.g. in astrocytes) and Tat may be actively released into the extracellular environment.21 Some of the structural and regulatory proteins have been shown to cause neuronal dysfunction and/or death and thus may be referred to as virotoxins (Table 1).
Neurotoxic Viral Proteins
There are several HIV-1 proteins that have been identified as neurotoxins. These include the structural proteins gp120 and gp41 and the nonstructural proteins Tat, Nef, Vpr and Rev. The HIV-1 coat protein gp120 is a potent neurotoxin with lethal effects on neurons (Figure 1) at concentrations in the picomolar range.22 Transgenic mice expressing gp120 under the control of a glial fibrillary acidic protein (GFAP; astrocyte-specific protein) promoter exhibited dendritic abnormalities in neurons, gliosis and age-related changes in long-term potentiation (LTP; considered a cellular basis of learning and memory), open field activity and spatial reference memory.23, 24, 25 The gp120 protein can directly modify neuronal function26, 27 and can indirectly damage neurons by disrupting glial functions. Gp120 can act on astrocytes to stimulate an inducible form of nitric oxide (NO) synthase,28 inhibit β-adrenergic function,29 induce tyrosine kinase activity,30 modify the expression of adhesion molecules31 and produce cytoskeletal changes.32 Astrocytes normally protect neurons from excitotoxic damage by buffering the excitatory amino-acid glutamate. By actions that stimulate Na+/H+ exchange, gp120 induces alkalization, leading to the inhibition of Na+-dependent glutamate influx in astrocytes.33, 34 The effects of gp120 on Na+/H+ exchangers, which can be blocked by amiloride, may lead to membrane depolarization.35, 36

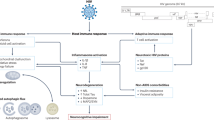
Mechanism by which HIV-1 may induce neuronal death in HIV-1 dementia. The virus infects monocytes and macrophages/microglia which enter the brain. The infected cells release cytotoxic HIV-1 proteins, most notably gp120 and Tat, which induce cell death cascades in neurons. The cell death cascade involves binding of gp120 and Tat to cell surface proteins such as CXCR4, overactivation of glutamate receptors (particularly the NMDA receptor), calcium influx, oxidative stress and release of cytotoxic membrane lipids such as 4-hydroxynonenal and ceramides. The latter events may trigger the intrinsic pathway of apoptosis, which involves mitochondrial membrane permeabilization, release of cytochrome c and activation of caspases and endonucleases, which finalize the cell death process. HIV-1 infection also induces monocytes and macrophages/microglia to produce proinflammatory cytokines which may potentiate neuronal cell death
Cytokine levels and COX-2 activity are increased in HIV-encephalitis and have been implicated as mediators of neurotoxic processes.37, 38 The activation of monocyte/macrophages by gp120 can promote oxidative damage and increase the release of neurotoxic cytokines, including TNF-α, IL-1β and prostaglandin E2 (PGE2; an arachidonic acid metabolite from the cyclooxygenase and lipoxygenase pathway).39, 40, 41, 42
The transmembrane protein gp41 that links gp120 to the envelope of the virion has been shown to be elevated in patients with severe HIV dementia. In vitro, gp41 is lethal to neurons in the low nanomolar range and requires the presence of glia, suggesting an indirect mechanism of cell death. Indeed, it has been demonstrated that astrocytes treated with the carboxy-terminus of gp41 have deficits in glutamate transport and release glutamate.43 Neuronal death induced by gp41 occurs by mechanisms that involve activation of inducible nitric oxide synthase (iNOS), NO formation, depletion of glutathione and disruption of mitochondrial function.44, 45, 46
The HIV trans-acting protein Tat is essential for viral replication. Accumulating evidence suggests that Tat is also an important mediator of neurotoxicity with lethal effects in the low nanamolar range.47, 48, 49, 50 Brain regions that are particularly susceptible to the toxic effects of Tat include the striatum, hippocampal dentate gyrus and the CA3 region of the hippocampus.16, 50, 51 Although neurons are rarely, if ever, infected with HIV, neurons can be exposed to Tat following active release from HIV-infected lymphoid cells and glial cells.52, 53, 54
In addition to the direct effect of Tat on neurons, Tat perturbs glial and monocyte/macrophage function, promoting the release of neurotoxic agents including matrix metalloproteinases (MMPs), IL-6, IL-8, RANTES, MCP-1 and TNF-α.55, 56, 57, 58, 59 Although the exact role of these agents in Tat-mediated neurotoxicity remains to be determined, Tat-induced TNF-α release has been implicated in neurotoxicity and MCP-1 levels in CSF have been shown to correlate with the severity of dementia in patients with HIV encephalitis.21, 60, 61, 62 MCP-1 and MMPs released by Tat may also increase the permeability of the blood–brain barrier and promote the transmigration of monocytes across the blood–brain barrier.63, 64 Together, these findings suggest that Tat may be an important mediator of the inflammatory response in the brain.
The nonstructural protein Nef is required for the proper budding of virions from HIV-infected cells. Abundant Nef expression has been detected in astrocytes of HIV-infected patients with pathological indications of neuronal damage.11, 65, 66 In vitro, Nef is lethal to neurons and glial cells in nanomolar concentrations and can increase the expression of MMPs, thus potentially modifying the permeability of the blood–brain barrier.67, 68 Nef has significant sequence homology with scorpion neurotoxins and like these venoms can cause the inactivation of a large-conductance potassium channels.69, 70, 71 Based on these findings, it has been suggested that Nef contributes to the neuronal degeneration seen in HIV encephalitis.
An important HIV accessory protein, Vpr, is thought to be important for effective viral replication in the early stages of infection. Vpr is important for transport of the pre-integration complex, nuclear localization, induction of cell cycle arrest in G2, transactivation of the HIV-1 LTR and other cellular promoters.72, 73, 74, 75, 76 Vpr is found in the serum and the CSF of HIV-1-infected patients.77 In HIV-infected patients with neurological dysfunction, CSF levels of Vpr are elevated, suggesting an involvement of Vpr in AIDS-related neurological disorders. In vitro, Vpr is lethal to neurons in micromolar concentrations when applied extracellularly by mechanisms that involve the formation of a large inward cation current and caspase-8 activation.78, 79 The N-terminal region of Vpr consisting of the first 40 amino acids has been found to be necessary and sufficient to form this sodium channel.80 Similar effects were found when Vpr was expressed intracellularly in terminally differentiated neurons.81 Thus, Vpr may be lethal to neurons by events triggered at the plasma membrane and intracellular associated processes.
The HIV phosphoprotein Rev is involved in the nuclear export of unspliced viral mRNAs. Rev contains a nuclear localization sequence and an RNA-binding domain in the N-terminal region that is critical for Rev function.82, 83 Extracellular Rev has neurotoxic properties. Intracerebroventricular injection of a synthetic peptide spanning the basic region of Rev was neurotoxic and lethal to rodents.84 The neurotoxic effects of Rev may involve an interaction with acidic phospholipids and the formation of α-helical conformations. This interaction and conformational change of Rev may disrupt membrane topology and produce toxic or lethal effects.84
Host Genetics
Genetic factors that may increase or decrease the susceptibility of HIV-infected patients to neurological complications include CCR5, MCP-1 and ApoE polymorphisms. A variation in the chemokine receptor CCR5wt-Delta32 is associated with delayed disease progression, while the CCR5-59029-A/A variant is associated with a more rapid disease progression and greater neurological impairment.85 Although rare in occurrence, the chemokine variant stromal cell-derived factor (SDF)1–3′-A/A has been associated with a more rapid disease progression and neurological deterioration in pediatric patients infected with HIV-1.85 Homozygosity for the MCP-1-2578G allele has been associated with a 50% reduction in the risk for HIV-1 infection. However, once a patient is infected with HIV-1, homozygosity for the MCP-1-2578G allele is associated with accelerated disease progression and a 4.5-fold increased risk of dementia.86
Although initial studies found no correlation between the risk of HIV dementia and ApoE genotype,87 later findings suggested that HIV-infected patients with the E4 isoform of ApoE are more likely to suffer neurological complications including dementia and peripheral neuropathy.88 The biophysical determinants of the E4 genotype that predispose neural tissue to malfunction and injury have not been determined, although a role for the regulation of redox balance by ApoE has been suggested.89, 90 Although the pathological consequences of cholesterol accumulation in brains of HIV patients with dementia remain to be determined, modification of cholesterol levels has been associated with poor cognitive function and may impair learning and memory.91, 92 Emerging evidence suggests that dysfunctions of sphingolipid and sterol metabolism in the setting of HIV dementia may have important pathological consequences.93
Viral Genetics
HIV-1 may enter the brain soon after infection using blood-derived macrophages as carriers (Figure 1).94 There is, in addition, evidence that HIV may also enter the brain by first infecting brain capillary endothelial cells.95 These findings suggest that macrophage and endothelial-tropic strains of HIV have a neuropathic potential. Consistent with this hypothesis, most viruses isolated from the brain are macrophage trophic and use CCR5 as the principal co-receptor for binding and entry into cells.96 Once in the brain, HIV-1 may infect resident microglia, and neural stem cells (Figure 2).

Infection of human neural progenitor cells by HIV-1. Using fluorescence-activated cell sorting, neural progenitor cells were isolated from dissociated fetal human cerebral cortical cells with a purity of 98%. In the presence of media containing epidermal growth factor (EGF) and basic fibroblast growth factor (bFGF), these cells were cultured as floating neurospheres for more than 30 days. (a) Cells were infected for 3 h at 37°C with HIV-1NL4–3 containing a YFP insert in the nef coding region (HIV-YFP; MOI=0.2). Cells were washed three times with PBS and lightly trypsinized (0.5 mg/ml trypsin for 10 min at 37°C). At 3 days after infection cells were fixed, and infection was visualized by immunofluorescent microscopy at × 100 magnification. (b) Free-floating neurosphere cultures were exposed to the HIV-1 isolates IIIB, Ba-L or Sim-br (RT activity of ∼106 cpm/ml) for 2 h at 37°C. To prevent carryover of contaminating viral DNA, viral inoculums were pretreated with RNAase-free DNAase (50 U/ml) for 30 m. HIV-1 Ba-L is a macrophage-tropic isolate that uses the CCR5 coreceptor and HIV-1 IIIB is a T-cell line-adapted isolate that uses the CXCR4 coreceptor; Sim-br is a primary brain isolate. After viral exposure, cells were washed, trypsin treated and the supernatant was sampled at 0, 3, 7 and 12 days, and p24 levels were measured by ELISA. In neurospheres, p24 levels increased in a linear manner over 12 days in cultures infected with HIV-1 IIIB and Sim-br, but only a small increase in viral production was detected in cultures infected with Ba-L. Data are mean±S.D. (ANOVA with Tukey post hoc comparisons)
There is a broad range of viral diversity in the brain with replication-competent HIV-1 genomes, complex mixtures of defective viral forms and chromosomally integrated provirus present.97 The hypervariable V3 domain of gp120 is an important determinant of neuropathogenesis, regardless of the HIV clade from which it was derived, and distinct HIV envelope sequences that are associated with the clinical expression of dementia have been isolated.98 Cloning and sequencing of the entire V3 region has provided evidence that genetically unique sequences exist in different brain regions.99 Viral variation in different brain compartments may be the product of selection pressures that result from variable access to antiretroviral drugs; or compartmentalized viral evolution. The influence of brain-specific actively replicating viral reservoirs may be important for the development of dementia. Indeed, it has been demonstrated that the CSF viral burden, but not the plasma viral burden, correlates with the presence and severity of dementia.100, 101 A positive correlation between CSF levels of β2-microglobulin and dementia has also been demonstrated, suggesting that immune activation is involved in neural dysfunction and degeneration in HIV dementia.100 Compared to HIV patients with no dementia, Tat sequences in the brains of HIV-1 patients with dementia showed greater nonsynonymous/synonymous substitution rates, with greatest sequence diversity in regions that influence viral replication and transport.7, 102 The functional significance of Tat molecular diversity in the brain remains to be determined.
Viral Effects on Neurogenesis
The brains of adult mammals contain small populations of cells that are capable of dividing and differentiating into neurons and glial cells.103 In the dentate gyrus, new neurons are functionally incorporated into the hippocampus104 and may participate in the formation of hippocampal-dependent memory.105 Neurogenesis is decreased during aging106 and by toxic stimuli, including stress, methamphetamine, opiates, alcohol and β-amyloid (a toxic form of amyloid found in the senile plaques of Alzheimer's patients).107, 108, 109 Neural progenitors express a variety of chemokine receptors including CXCR4 and CCR5,110 and thus have the potential to bind HIV-1. Based on the findings that HIV-1 infection in the brain is often associated with encephalitis and CNS inflammation, processes that have been shown to decrease neurogenesis,111, 112 it is possible that deficits of neurogenesis occur in HIV dementia. Consistent with this possibility, one study provided evidence for reduced numbers of proliferating cells in the hippocampus of HIV-1-infected patients,113 and HIV-1 can successfully infect and replicate in neural progenitor cells (Figure 2).
Neurotoxic HIV-1 proteins may also contribute to deficits of neurogenesis in HIV dementia. The second exon of Tat contains a conserved Arg–Gly–Asp cell adhesion motif that binds to integrin receptors. Additionally, the basic region of Tat in the first exon also contributes to its cell adhesive properties.114 It is thus possible that Tat may interfere with the normal development and migration of neurons, as well as remodeling after a traumatic insult to the accessory cells, by competing with extracellular matrix cellular proteins. Unpublished observations from our laboratory suggest that Tat interferes with the normal function of neural progenitor cells in vitro and in vivo. Although these initial findings are intriguing, further experiments are required to determine the pathogenic mechanisms of HIV-1 protein-induced neural progenitor cell dysfunction and the consequence of these interactions on cognitive function.
Cell Death Cascades
The biochemical and molecular cascades that result in the death of neurons in HIV dementia are complex, involving not only the virus and neurons, but also glial cells, macrophages and lymphocytes. In general, the mechanisms by which neurons become dysfunctional and die in HIV dementia are similar to those that occur in Alzheimer's disease.115 In both cases, apoptosis, excitotoxicity and inflammation are involved. In Alzheimer's disease, the β-amyloid protein may initiate these cascades, whereas in HIV dementia HIV-1 proteins (particularly gp120 and Tat) are likely initiating factors.
Apoptosis
Analyses of the brains of patients who died with HIV-1 dementia, together with experiments in cultured cells, suggest that many neurons undergo a form of programmed cell death called apoptosis. DNA fragmentation, caspase activation and mitochondrial alterations consistent with apoptosis have been documented in affected brain regions of HIV dementia patients.116 The tumor suppressor protein p53 may mediate apoptosis in HIV dementia because p53 levels are increased in affected brain regions of HIV dementia patients and cortical neurons lacking p53 are resistant to death induced by gp120 and Tat.117 Neural cell death induced by isolates from macrophages infected with HIV-1 was prevented by overexpression of Bcl-2, suggesting that the neurotoxic macrophage-derived factors activate an intrinsic (mitochondrial) apoptotic cascade.118 Prostate apoptosis response-4 (Par-4) is a leucine zipper protein that is thought to mediate apoptosis in several different cell types including neurons. Par-4 levels are increased in neurons in the hippocampus of patients with HIV encephalitis and in monkeys infected with a chimeric strain of HIV-1 and simian immunodeficiency virus. Par-4 production is rapidly increased in cultured hippocampal neurons after exposure to Tat, suppression of Par-4 production using antisense technology protects the neurons from being killed by Tat.119 These findings suggest that Par-4 may be a mediator of neuronal apoptosis in HIV dementia.
Brain-derived neurotrophic factor (BDNF), nerve growth factor and activity-dependent neurotrophic factor protected cultured cerebellar granule neurons from being killed by Tat.120 The neuroprotective effect of each trophic factor involved activation of the transcription factor NF-κB and upregulation of the expression of Bcl-2. The transcription factor CREB (cyclic AMP response element-binding protein) plays a key role in learning and memory, and mediates activity-dependent neuronal survival by upregulating the expression of BDNF.121 Long-term exposure (hours to days) of cultured PC12 cells to Tat resulted in decreased levels of CREB expression and activity which preceded apoptosis.122 Thus, in addition to activating proapoptotic cascades, neurotoxic HIV-1 proteins may inhibit pathways that promote cell survival.
Both infected and uninfected T cells undergo apoptosis during the course of HIV-1 infection. The mechanisms of apoptosis in T cells and neurons appear to be similar, involving death receptors (TNF receptors and Fas), some chemokines, calcium dysregulation, mitochondrial membrane permeabilization and caspase activation.123 Both gp120 and Tat may be key triggers of apoptosis in different cell types that die in HIV-1 AIDS. HIV-1-infected monocytes produce a protein called FLJ21908 that is cytotoxic to CD4(+) and CD8(+) T and B cells, as well as in neuroblastoma cells.124 FLJ21908 is present in the brain and lymph tissue of HIV dementia patients, suggesting a role for this protein in disease pathogenesis. Increased levels of the HIV-1 accessory protein Vpr have been detected in the CSF of AIDS dementia patients, and Vpr can kill cultured hippocampal neurons, apparently by forming ion-conducting pores in the plasma membrane.79 Vpr has been shown to induce apoptosis of cultured neurons by a mechanism involving caspase-8.78
Culture medium collected from HIV-1-infected T cells induces neuronal apoptosis only when virions are present in the medium, whereas the medium from infected macrophages is toxic whether or not virions are present.125 Several endogenous factors produced by macrophages may promote neuronal apoptosis, including excitatory amino acids, NO, MMPs and proinflammatory cytokines such as TNF-α and SDF-1.126, 127, 128 Many HIV-1-infected patients also use drugs of abuse including heroin. μ-Opioid receptors are widely expressed by astrocytes and astrocyte precursors, where their activation by opioids such as morphine can induce apoptosis. Prolonged exposure of cultured striatal cells to Tat and morphine resulted in the preferential death of astrocyte precursor cells and astrocytes.129 The available evidence suggests that interactive neurotoxic effects of viral infection and drugs of abuse accelerate the development and progression of HIV dementia.
Oxidative stress
Evidence for oxidative stress in HIV-1 dementia was obtained from analyses of brain tissue and CSF, including the presence of oxidized proteins.130, 131 Analyses of the brain tissue of HIV-1 dementia patients have revealed evidence for membrane-associated oxidative stress correlated with disease pathogenesis and cognitive impairment, including increased levels of the cytotoxic lipid peroxidation product 4-hydroxynonenal (HNE).93, 132 HNE is known to covalently modify numerous proteins on cysteine, lysine and histidine residues and, by this mechanism, can impair the function of membrane ion-motive ATPases and glucose and glutamate transporters.133, 134 Generation of HNE in response to gp120 and Tat may therefore play a major role in the neurotoxicity of these HIV-1 proteins. Indeed, HNE is known to mediate oxidative stress-induced apoptosis of cultured neurons135 and can damage neurons and cause cognitive dysfunction in vivo.136 Tat-induced apoptosis of cultured human cortical neurons was potentiated by TNF-α by a mechanism that involved increased oxidative stress.61 Several antioxidants, including L-deprenyl and ebeselen blocked the toxicity of the CSF, suggesting a key role for oxidative stress in the neuronal death that occurs in HIV-1 dementia.131
Excitotoxicity and cellular calcium overload
The bulk of the evidence that neurons may die as a result of overactivation of glutamate receptors (excitotoxicity) in HIV-1 dementia comes from studies of the neurotoxic actions of gp120 and Tat. Gp120 can promote the release of arachidonic acid from glial cells, which can modify the kinetics of NMDA receptors on neurons to increase the mean open probability and channel open time.137 The combined mechanisms of an increased pool of extracellular glutamate and deregulated NMDA receptor function can result in neuronal calcium overload and cellular dysfunction or death by mechanisms involving disruptions of redox balance and sphingolipid metabolism.39, 93, 138 The glutamate receptor antagonist memantine has been shown to inhibit gp120-evoked calcium changes in neurons and astrocytes and protects neurons from gp120-induced cell death.139, 140, 141, 142 A clinical trial using this drug is currently under way in patients with HIV dementia.
Tat can directly interact with neurons and has been demonstrated to depolarize neuronal membranes independent of Na+ flux.49 Tat-evoked responses are similar to those induced by the non-NMDA agonist kainate, with the distinction that repetitive applications of Tat do not desensitize non-NMDA receptors.49, 143 Based on evidence suggesting that the desensitization of glutamate receptors inversely predicts agonist toxicity, this unusual action of Tat on membrane polarization has been suggested to be an important mechanism for the toxic effects of Tat.144, 145, 146 As evidence, Tat-induced neuronal cell death can be significantly reduced by antagonists of excitatory amino-acid receptors.49 Tat potentiates excitatory amino-acid (glutamate and NMDA) triggered calcium flux and neurotoxicity in cultured neurons.147 Subtoxic concentrations of Tat combined with subtoxic concentrations of glutamate caused neuronal cell death. NMDA receptors play an important role in the neurotoxicities of both gp120 and Tat, which may explain the apparent efficacy of the noncompetitive NMDA receptor antagonist memantine in the treatment of HIV dementia patients.148 Excitotoxicity is likely exacerbated by proinflammatory cytokines such as IL-1β and TNF.149 While gp120 and Tat can render neurons vulnerable to excitotoxicity by enhancing activation of NMDA receptors, recent findings suggest an important role for the chemokine receptor CXCR4 in gp120-induced neuronal death.150 The latter study showed that a CXCR4 antagonist (AMD3100) can protect cultured neurons against gp120 toxicity.
Tat-triggered neuronal cell death is calcium dependent.138 When applied onto neurons, Tat evokes an initial burst of intracellular calcium release through IP3-sensitive pools, that is followed by glutamate receptor-mediated calcium influx.151, 152 The ensuing intracellular events of mitochondrial calcium uptake, disruption of redox balance and caspase activation result in neuronal death.119, 153 Consistent with these findings, inhibitors of IP3 receptors, agents that stabilize mitochondrial membrane permeability, antioxidants, NO synthase and caspase inhibitors protect neurons from the toxic effects of Tat.147, 151, 152 Calcium regulation in glial cells may also be perturbed during the course of HIV dementia. CSF from HIV dementia patients, but not CSF from HIV-1-infected but nondemented patients, disrupts cellular calcium homeostasis in cultured astrocytes.154 An antagonist of L-type calcium channels, nimodipine, has been shown to protect neurons from the toxic effects of gp120 in vitro, but failed in a clinical trial, possibly because of the inability of this drug to inhibit gp120-triggered disruptions of calcium homeostasis in glia.141, 155
Glial cell neurotoxins and inflammation
Infiltration of lymphocytes and macrophages, activation of microglia and astrocytes, and production of proinflammatory cytokines characterize the brains of HIV dementia patients. Examination of brain sections from HIV dementia and control brains with antibodies against iNOS, IL-1β and caspase-1 revealed that all the three markers of inflammation and oxidative stress are increased in areas of HIV-1 infection.156 The latter markers were increased in microglia and astrocytes, suggesting that these cells may be sites of production of reactive oxygen species. Levels of macrophage inflammatory protein-1 alpha (MIP-1α), MIP-1β, and regulated upon activation normal T cell expressed and secreted (RANTES) were altered in the CSF of demented HIV-1 patients compared to nondemented patients, suggesting roles for these chemokines in disease pathogenesis.157 Levels of the MMPs MMP-2, -7 and -9, and activities of MMP-2 and -9, are elevated in the CSF of HIV-1 dementia patients.158 The latter study further showed that brain-derived cells release MMP-2, -7, and -9, suggesting a mechanism for disruption of the blood–brain barrier and increased immune activation in the CNS in HIV dementia. Hippocampal CA3 and CA4 neurons from patients with HIV encephalitis express higher levels of the chemokine receptor CXCR4 and lower levels of CCR5 than do neurons in control cases or AIDS cases without encephalitis.159
Monkeys infected with simian immunodeficiency virus exhibit increased levels of vascular cell adhesion molecule-1, suggesting a mechanism for recruitment of lymphocytes into the central nervous system.160 Another possible mechanism for recruitment of monocytes into the brain involves HIV-1 protein-induced production of chemoattractant proteins by glial cells. For example, exposure of astrocytes to Tat induces them to produce MCP-1, and MCP-1 is elevated in the brain tissue and CSF of HIV-1 dementia patients.21 Inoculation of HIV-infected monocytes into the basal ganglia and cortex of severe combined immunodeficiency disease (SCID) mice results in pathological features similar to those of human HIV-1 dementia. In such SCID/HIV-1 mice, the expression of TNF-α, IL-6 and vascular endothelial growth factor (VEGF) was increased.161 The increased production of these proinflammatory proteins was associated with neuronal damage, as indicated by decreased levels of microtubule-associated protein-1 immunoreactivity. Pivotal roles for inflammatory processes in the neurodegenerative process in HIV-1 dementia are further suggested by studies showing that an inhibitor of TNF-α and MMPs, as well as a platelet-activating factor antagonist, reduce neuropathology in a mouse model of HIV-1 encephalitis.162
Lipid metabolism
Levels of cholesterol, ceramide and sphingomyelin are significantly increased in the brain tissues and CSF of HIV dementia patients.93 Membrane lipid peroxidation and increased ceramide production may be central to the death of neurons in HIV dementia because exposure of cultured neurons to gp120 and Tat resulted in increased cellular levels of HNE and ceramide. Moreover, the ceramide precursor palmitoyl-CoA sensitized neurons to Tat and gp120 toxicity, while an inhibitor of ceramide production reduced Tat and gp120-induced increases of ceramide and HNE and protected the neurons from Tat and gp120-induced death.93 HIV-1 infection may promote a lipid imbalance in neural cells, resulting in an overproduction of ceramide and consequent cellular dysfunction and death. Individuals with an apolipoprotein E4 allele are at an increased risk for Alzheimer's disease, and recent evidence suggests that the E4 allele also increases the risk of dementia in HIV-1-infected people.88 Cutler et al.132 found evidence of dysregulated lipid and sterol metabolism in HIV dementia patients with an E4 genotype. Levels of sphingomyelin, ceramide and cholesterol were significantly increased in the medial frontal cortex, parietal cortex and cerebellum of HIV dementia patients with an E3/4 or E4/4 genotype compared to patients with an E3/3 genotype.
Consistent with an important role for sphingomyelin hydrolysis and ceramide production in the pathogenic actions of neurotoxic HIV proteins, Jana and Pahan163 confirmed that gp120 and Tat induce ceramide production in cultured neurons. They further showed that gp120 and Tat induce sphingomyelinase activity by a mechanism involving induction of oxidative stress by CXCR4 activation. gp120 has a galactosylceramide-binding domain, and other proteins with such a domain regulate membrane trafficking from golgi to lipid rafts,164 suggesting a potential role for perturbed membrane cycling in HIV-1 dementia pathogenesis. Emerging findings suggest that membrane microdomains called lipid rafts play important roles in the pathogenesis of HIV dementia. Lipid rafts are regions of the plasma membrane that have high levels of cholesterol and sphingomyelin. Receptors for many different cytokines and growth factors are concentrated in lipid rafts. Lipid rafts are believed to be portals through which many different types of viruses, including HIV-1, enter cells165 and may also be regions where gp120 and Tat exert their neurotoxic actions.93, 132
Treatment with Antiretroviral Agents is not Sufficient
Within recent years, several antiretroviral drugs have become available that target either the reverse transcriptase or the HIV protease.166 Very recently, a drug that prevents HIV entry into cells has also been approved for use in HIV-1-infected patients.167 No drugs have yet been available against any of the other viral gene products. Nevertheless, treatments for AIDS patients, including the widely used nucleoside analogs and protease inhibitors, may impact on the cell death process induced by HIV-1 proteins.168 Nucleoside analogs have been shown to increase mitochondrial DNA damage and inhibit mitochondrial replication, which would be expected to exacerbate cell death. On the other hand, protease inhibitors can prevent apoptosis by protecting mitochondria.
It is clear that control of the virus in the peripheral blood can partially reverse HIV dementia, resulting in improvement in cognitive and psychomotor function.169, 170 Most antiretroviral drugs do not readily cross the blood–brain barrier. However, improvements in neurological function may be noticed within weeks to months, suggesting that viral products are an important driving force in this cascade. Indeed, viral proteins such as gp120171 and Tat172 can cross the blood–brain barrier. However, control of viral replication in the central nervous system is still important because in some patients cognitive decline continues despite excellent control of the virus in the periphery. These patients typically have elevated viral loads in the CSF, a phenomenon called CNS escape.173 This may occur due to poor penetration of the drugs across the blood–brain barrier and because of viral evolution in the brain. Further, once the cells in the brain are infected and viral integration has occurred, currently available antiretroviral drugs would have no effect on the production of nonstructural proteins such as Tat, which may still be released unchecked from infected microglia/macrophages and astrocytes. At this point of time, antiretroviral drugs alone are insufficient in the treatment of HIV dementia, and neuroprotective and anti-inflammatory approaches will likely be necessary. Finally, the evidence that lipid abnormalities and oxidative stress play important roles in the pathogenesis of HIV-1 dementia suggests that dietary modifications, including reductions in calories and cholesterol-elevating fats, may prove beneficial in the treatment of HIV-1-infected patients.163
Summary
The cellular and molecular alterations that result in synaptic dysfunction and neuronal degeneration in HIV dementia are beginning to be understood. In general, it appears that HIV-1 infects macrophages/microglia and possibly neural stem cells. The infected cells release neuotoxic HIV-1 proteins, such as Tat and gp120, as well as proinflammatory cytokines and excitotoxins. Neurons are damaged by HIV-1 and endogenous neurotoxins by mechanisms involving excessive calcium influx and oxidative stress. In addition to drugs that reduce viral load, therapeutic approaches to HIV dementia may include excitatory amino-acid receptor antagonists, antioxidants and calcium-stabilizing agents.
References
McArthur JC, Hoover DR, Bacellar H, Miller EN, Cohen BA, Becker JT, Graham NM, McArthur JH, Selnes OA and Jacobson LP (1993) Dementia in AIDS patients: incidence and risk factors. Multicenter AIDS Cohort Study. Neurology 43: 2245–2252
Navia BA, Jordan BD and Price RW (1986a) The AIDS dementia complex: I. Clinical features. Ann. Neurol. 19: 517–524
Price RW and Brew BJ (1988) The AIDS dementia complex. J. Infect. Dis. 158: 1079–1083
Koutsilieri E, Sopper S, Scheller C, ter Meulen V and Riederer P (2002) Parkinsonism in HIV dementia. J. Neural Transm. 109: 767–775
Mirsattari SM, Power C and Nath A (1998) Parkinsonism with HIV infection. Mov. Disord. 13: 684–689
Bouwman FH, Skolasky RL, Hes D, Selnes OA, Glass JD, Nance-Sproson TE, Royal W, Dal Pan GJ and McArthur JC (1998) Variable progression of HIV-associated dementia. Neurology 50: 1814–1820
Bratanich AC, Liu C, McArthur JC, Fudyk T, Glass JD, Mittoo S, Klassen GA and Power C (1998) Brain-derived HIV-1 tat sequences from AIDS patients with dementia show increased molecular heterogeneity. J. Neurovirol. 4: 387–393
Ranga U, Shankarappa R, Siddappa NB, Ramakrishna L, Nagendran R, Mahalingam M, Mahadevan A, Jayasuryan N, Satishchandra P, Shankar SK and Prasad VR (2004) Tat protein of human immunodeficiency virus type 1 subtype C strains is a defective chemokine. J. Virol. 78: 2586–2590
Navia BA, Cho ES, Petiio CK and Price RW (1986b) The AIDS dementia complex: II Neuropathology. Ann. Neurol. 19: 525–535
de la Monte SM, Ho DD, Schooley RT, Hirsch MS and Richardson EP (1987) Subacute encephalomyelitis of AIDS and its relation to HTLV-III infection. Neurology 37: 562–569
Ranki A, Nyberg M, Ovod V, Haltia M, Elovaara I, Raininko R, Haapasalo H and Krohn K (1995) Abundant expression of HIV Nef and Rev proteins in brain astrocytes in vivo is associated with dementia. AIDS 9: 1001–1008
Watkins BA, Dorn HH, Kelly WB, Armstong RC, Potts B, Micheals S, Kufta CV and Dubois-Dalcq ME (1990) Specific tropism of HIV-1 for microglia cells in primary human brain cultures. Science 249: 549–553
Tornatore C, Chandra R, Berger JR and Major EO (1994) HIV-1 infection of subcortical astrocytes in the pediatric central nervous system. Neurology 44: 481–487
Masliah E, Ge N, Morey M, DeTeresa R, Terry RD and Wiley CA (1992b) Cortical dendritic pathology in human deficiency virus encephalitis. Lab. Invest. 66: 285–291
Everall IP, Heaton RK, Marcotte TD, Ellis RJ, McCutchan JA, Atkinson JH, Grant I, Mallory M and Masliah E (1999) Cortical synaptic density is reduced in mild to moderate human immunodeficiency virus neurocognitive disorder. HNRC Group. HIV Neurobehavioral Research Center. Brain Pathol. 9: 209–217
Everall I, Barnes H, Spargo E and Lantos P (1995) Assessment of neuronal density in the putamen in human immunodeficiency virus (HIV) infection. Application of stereology and spatial analysis of quadrants. J. Neurovirol. 1: 126–129
Masliah E, Ge N, Achim CL, Hansen LA and Wiley CA (1992a) Selective neuronal vulnerability in HIV encephalitis. J. Neuropathol. Exp. Neurol. 51: 585–593
Wesselingh SL, Glass JD, McArthur JC, Griffin JW and Griffin DE (1994) Cytokine dysregulation in HIV-associated neurological disease. Adv. Neuroimmunol. 4: 199–206
Wesselingh SL, Power C, Glass JD, Tyor WR, McArthur JC, Farber JM, Griffin JW and Griffin DE (1993) Intracerebral cytokine messenger RNA expression in acquired immunodeficiency syndrome dementia. Ann. Neurol. 33: 576–582
Conant K, Garzino-Demo A, Nath A, McArthur JC, Halliday W, Power C, Gallo RC and Major EO (1998) Induction of monocyte chemoattractant protein-1 in HIV-1 Tat-stimulated astrocytes and elevation in AIDS dementia. Proc. Natl. Acad. Sci. USA 95: 3117–3121
van de Bovenkamp M, Nottet HS and Pereira CF (2002) Interactions of human immunodeficiency virus-1 proteins with neurons: possible role in the development of human immunodeficiency virus-1-associated dementia. Eur. J. Clin. Invest. 32: 619–627
Dreyer EB, Kaiser PK, Offermann JT and Lipton SA (1990) HIV-1 coat protein neurotoxicity prevented by calcium channel antagonists. Science 20: 364–367
D'hooge R, Franck F, Mucke L and De Deyn PP (1999) Age-related behavioural deficits in transgenic mice expressing the HIV-1 coat protein gp120. Eur. J. Neurosci. 11: 4398–4402
Krucker T, Toggas SM, Mucke L and Siggins GR (1998) Transgenic mice with cerebral expression of human immunodeficiency virus type-1 coat protein gp120 show divergent changes in short- and long-term potentiation in CA1 hippocampus. Neuroscience 83: 691–700
Toggas SM, Masliah E, Rockenstein EM, Rall GF, Abraham CR and Mucke L (1994) Central nervous system damage produced by expression of the HIV-1 coat protein gp120 in transgenic mice. Nature 367: 188–193
Lannuzel A, Lledo PM, Lamghitnia HO, Vincent JD and Tardieu M (1995) HIV-1 envelope proteins gp120 and gp160 potentiate NMDA-induced [Ca2+]i increase, alter [Ca2+]i homeostasis and induce neurotoxicity in human embryonic neurons. Eur. J. Neurosci. 7: 2285–2293
Vignoli AL, Martini I, Haglid KG, Silvestroni L, Augusti-Tocco G and Biagioni S (2000) Neuronal glycolytic pathway impairment induced by HIV envelope glycoprotein gp120. Mol. Cell. Biochem. 215: 73–80
Mollace V, Colasamto M, Persichini T, Bagetta G, lauro GM and Nistico G (1993) HIV gp120 glycoprotein stimulates the inducible isoform of NO synthase in human cultured astrocytoma cells. Biochem. Biophys. Res. Commun. 194: 439–445
Levi G, Patrizio M, Bernardo A, Petrucci TC and Agresti C (1993) Human immunodeficiency virus coat protein gp120 inhibits beta-adrenergic regulation of astroglial and microglial functions. Proc. Natl. Acad. Sci. 90: 1541–1545
Schneider-Schaulies J, Schneider-Schaulies S, Brinkmann R, Tas P, Halbrugge M, Walter U, Holmes HC and Ter Meulen V (1992) HIV-1 gp120 receptor on CD4-negative brain cells activates a tyrosine kinase. Virology 191: 765–772
Shrikant P, Benos DJ, Tang LP and Benveniste EN (1996) HIV glycoprotein 120 enhances intercellular adhesion molecule-1 gene expression in glial cells. Involvement of Janus kinase/signal transducer and activator of transcription and protein kinase C signaling pathways. J. Immunol. 156: 1307–1314
Pulliam L, West D, Haigwood N and Swanson RA (1993) HIV-1 envelope gp120 alters astrocytes in human brain cultures. AIDS Res. Hum. Retroviruses 9: 439–444
Drejer J, Meier E and Schousboe A (1983) Novel neuron-related regulatory mechanisms for astrocytic glutamate and GABA high affinity uptake. Neurosci. Lett. 37: 301–306
Patton HK, Zhou ZH, Bubien JK, Benveniste EN and Benos DJ (2000) gp120-induced alterations of human astrocyte function: Na(+)/H(+) exchange, K(+) conductance, and glutamate flux. Am. J. Physiol. 279: C700–C708
Bubien JK, Benveniste EN and Benos DJ (1995) HIV-gp120 activates large-conductance apamin-sensitive potassium channels in rat astrocytes. Am. J. Physiol. 268: C1440–C1449
Benos DJ, McPherson S, Hahn BH, Chaikin MA and Benveniste EN (1994) Cytokines and HIV envelope glycoprotein gp120 stimulate Na+/H+ exchange in astrocytes. J. Biol. Chem. 269: 13811–13816
Saha RN and Pahan K (2003) Tumor necrosis factor-alpha at the crossroads of neuronal life and death during HIV-associated dementia. J. Neurochem. 86: 1057–1071
Nottet HS (1999) Interactions between macrophages and brain microvascular endothelial cells: role in pathogenesis of HIV-1 infection and blood - brain barrier function. J. Neurovirol. 5: 659–669
Foga IO, Nath A, Hasinoff BB and Geiger JD (1997) Antioxidants and dipyridamole inhibit HIV-1 gp120-induced free radical-based oxidative damage to human monocytoid cells. J. Acquir. Immune Defic. Syndr. Hum. Retrovirol. 16: 223–229
Ilyin SE and Plata-Salaman CR (1997a) HIV-1 envelope glycoprotein 120 regulates brain IL-1beta system and TNF-alpha mRNAs in vivo. Brain Res. Bull. 44: 67–73
Merrill JE, Koyanagi Y, Zack J, Thomas L, Martin F and Chen IS (1992) Induction of interleukin-1 and tumor necrosis factor alpha in brain cultures by human immunodeficiency virus type 1. J. Virol. 66: 2217–2225
Wahl LM, Corcoran ML, Pyle SW, Arthur LO, Harel-Bellan A and Farrar WL (1989) Human immunodeficiency virus glycoprotein (gp120) induction of monocyte arachidonic acid metabolites and interleukin 1. Proc. Natl. Acad. Sci. USA 86: 621–625
Kort JJ (1998) Impairment of excitatory amino acid transport in astroglial cells infected with the human immunodeficiency virus type 1. AIDS Res. Hum. Retroviruses 14: 1329–1339
Sung JH, Shin SA, Park HK, Montelaro RC and Chong YH (2001) Protective effect of glutathione in HIV-1 lytic peptide 1-induced cell death in human neuronal cells. J. Neurovirol. 7: 454–465
Adamson DC, Kopnisky KL, Dawson TM and Dawson VL (1999) Mechanisms and structural determinants of HIV-1 coat protein, gp41-induced neurotoxicity. J. Neurosci. 19: 64–71
Adamson DC, Wildermann B, Sasaki MD, Glass JD, McArthur JC, Christov VI, Dawson TM and Dawson VL (1996) Immunologic NO synthase: elevation in severe AIDS dementia and induction by HIV-1 gp41. Science 274: 1917–1920
Hayman M, Arbuthnott G, Harkiss G, Brace H, Filippi P, Philippon V, Thomson D, Vigne R and Wright A (1993) Neurotoxicity of peptide analogues of the transactivating protein tat from Maedi-Visna virus and human immunodeficiency virus. Neuroscience 53: 1–6
Jones M, Olafson K, Del Bigio MR, Peeling J and Nath A (1998) Intraventricular injection of human immunodeficiency virus type 1 (HIV-1) Tat protein causes inflammation, gliosis, apoptosis, and ventricular enlargement. J. Neuropathol. Exp. Neurol. 57: 563–570
Magnuson DS, Knudsen BE, Geiger JD, Brownstone RM and Nath A (1995) Human immunodeficiency virus type 1 tat activates non-N-methyl-D-aspartate excitatory amino acid receptors and causes neurotoxicity. Ann. Neurol. 37: 373–380
Maragos WF, Tillman P, Jones M, Bruce-Keller AJ, Roth S, Bell JE and Nath A (2003) Neuronal injury in hippocampus with human immunodeficiency virus transactivating protein, Tat. Neuroscience 117: 43–53
Hayman M, Arbuthnott G, Harkiss G, Brace H, Filippi P, Philippon V, Thomson D, Vigne R and Wright A (1993) Neurotoxicity of peptide analogues of the transactivating protein tat from Maedi-Visna virus and human immunodeficiency virus. Neuroscience 53: 1–6
Chang HC, Samaniego F, Nair BC, Buonaguro L and Ensoli B (1997) HIV-1 tat protein exits from cells via a leaderless secretory pathway and binds to extracellular matrix-associated heparan sulfate proteoglycan through its basic region. AIDS 11: 1421–1431
Ensoli B, Buonaguro L, Barillari G, Fiorelli V, Gendelman R, Morgan R, Wingfield P and Gallo R (1993) Release, uptake, and effects of extracellular human immunodeficiency virus type-1 Tat protein on cell growth and viral replication. J. Virol. 67: 277–287
Tardieu M, Hery C, Peudenier S, Boespflug O and Montagnier L (1992) Human immunodeficiency virus type 1-infected monocytic cells can destroy human neural cells after cell-to-cell adhesion. Ann. Neurol. 32: 11–17
Kutsch O, Oh J, Nath A and Benveniste EN (2000) Induction of the chemokines interleukin-8 and IP-10 by human immunodeficiency virus type 1 tat in astrocytes. J. Virol. 74: 9214–9221
Chen P, Mayne M, Power C and Nath A (1997) The Tat protein of HIV-1 induces tumor necrosis factor-a production: implications for HIV associated neurological diseases. J. Biol. Chem. 272: 22385–22388
Nath A, Conant K, Chen P, Scott C and Major EO (1999) Transient exposure to HIV-1 Tat protein results in cytokine production in macrophages and astrocytes. A hit and run phenomenon. J Biol Chem 11: 17098–17102
Johnston JB, Zhang K, Silva C, Shalinsky DR, Conant K, Ni W, Corbett D, Yong VW and Power C (2001) HIV-1 Tat neurotoxicity is prevented by matrix metalloproteinase inhibitors. Ann. Neurol. 49: 230–241
Philippon V, Vellutini C, Gambarelli D, Harkiss G, Arbuthnott G, Metzger D, Roubin R and Filippi P (1994) The basic domain of the lentiviral Tat protein is responsible for damages in mouse brain: involvement of cytokines. Virology 205: 519–529
New DR, Maggirwar SB, Epstein LG, Dewhurst S and Gelbard HA (1998) HIV-1 Tat induces neuronal death via tumor necrosis factor-alpha and activation of non-N-methyl-D-aspartate receptors by a NFkappaB-independent mechanism. J. Biol. Chem. 273: 17852–17858
Shi B, Raina J, Lorenzo A, Busciglio J and Gabuzda D (1998) Neuronal apoptosis induced by HIV-1 Tat protein and TNF-alpha: potentiation of neurotoxicity mediated by oxidative stress and implications for HIV-1 dementia. J. Neurovirol. 4: 281–290
McManus CM, Weidenheim K, Woodman SE, Nunez J, Hesselgesser J, Nath A and Berman JW (2000) Chemokine and chemokine-receptor expression in human glial elements: induction by the HIV protein, Tat, and chemokine autoregulation. Am. J. Pathol. 156: 1441–1453
Weiss JM, Nath A, Major EO and Berman JW (1999) HIV-Tat induces MCP-1 mediated monocyte transmigration and upregulates CCR5 expression on human monocytes. J. Immunol. 163: 2953–2959
Ghorpade A, Persidskaia R, Suryadevara R, Che M, Liu XJ, Persidsky Y and Gendelman HE (2001) Mononuclear phagocyte differentiation, activation, and viral infection regulate matrix metalloproteinase expression: implications for human immunodeficiency virus type 1-associated dementia. J. Virol. 75: 6572–6583
Saito Y, Sharer LR, Epstein LG, Michaels J, Mintz M, Louder M, Golding K, Cvetkovich TA and Blumberg BM (1994) Overexpression of nef as a marker for restricted HIV-1 infection of astrocytes in postmortem pediatric central nervous tissues. Neurology 44: 474–481
Tornatore C, Meyers K, Atwood W, conant K and Major EO (1994) Temporal patterns of human immunodeficiency virus type 1 transcripts in human fetal astrocytes. J. Virol. 68: 93–102
Trillo-Pazos G, McFarlane-Abdulla E, Campbell IC, Pilkington GJ and Everall IP (2000) Recombinant nef HIV-IIIB protein is toxic to human neurons in culture. Brain Res. 864: 315–326
Sporer B, Koedel U, Paul R, Kohleisen B, Erfle V, Fontana A and Pfister HW (2000) HIV-1 Nef protein induces blood–brain barrier disruption in the rat: role of matrix metalloproteinase-9. J. Neuroimmunol. 102: 125–130
Garry RF, Kort JJ, Koch-Nolte F and Koch G (1991) Similarities of viral proteins to toxins that interact with monovalent cation channels. AIDS 5: 1381–1384
Kort JJ and Jalonen TO (1998) The nef protein of the human immunodeficiency virus type 1 (HIV-1) inhibits a large-conductance potassium channel in human glial cells. Neurosci. Lett. 251: 1–4
Werner T, Ferroni S, Saermark T, Brack-Werner R, Banati RB, Mager R, Steinaa L, Kreutzberg GW and Erfle V (1991) HIV-1 Nef protein exhibits structural and functional similarity to scorpion peptides interacting with potassium channels. AIDS 5: 1301–1308
Sherman MP, Schubert U, Williams SA, de Noronha CM, Kreisberg JF, Henklein P and Greene WC (2002) HIV-1 Vpr displays natural proteintransducing properties: implications for viral pathogenesis. Virology 302: 95–105
Tungaturthi PK, Sawaya BE, Singh SP, Tomkowicz B, Ayyavoo V, Khalili K, Collman RG, Amini S and Srinivasan A (2003) Role of HIV-1 Vpr in AIDS pathogenesis: relevance and implications of intravirion, intracellular and free Vpr. Biomed. Pharmacother. 57: 20–24
Zhang S, Feng Y, Narayan O and Zhao LJ (2001) Cytoplasmic retention of HIV-1 regulatory protein Vpr by protein-protein interaction with a novel human cytoplasmic protein VprBP. Gene 263: 131–140
Yao XJ, Lemay J, Rougeau N, Clement M, Kurtz S, Belhumeur P and Cohen EA (2002) Genetic selection of peptide inhibitors of human immunodeficiency virus type 1 Vpr. J. Biol. Chem. 277: 48816–48826
Philippon V, Matsuda Z and Essex M (1999) Transactivation is a conserved function among primate lentivirus Vpr proteins but is not shared by Vpx. J. Hum. Virol. 2: 167–174
Huang MB, Weeks O, Zhao LJ, Saltarelli M and Bond VC (2000) Effects of extracellular HIV-1 vpr protein in primary rat cortical cell cultures. J. Neurovirol. 6: 202–220
Patel CA, Mukhtar M and Pomerantz RJ (2000) HIV-1 Vpr induces apoptosis in human neuronal cells. J. Virol. 74: 9717–9726
Piller SC, Jans P, Gage PW and Jans DA (1998) Extracellular HIV-1 virus protein R causes a large inward current and cell death in cultured hippocampal neurons: implications for AIDS pathology. Proc. Natl. Acad. Sci. USA 95: 4595–4600
Piller SC, Ewart GD, Jans DA, Gage PW and Cox GB (1999) The amino-terminal region of Vpr from HIV-1 forms ion channels and kills neurons. J. Virol. 73: 4230–4238
Patel CA, Mukhtar M and Pomerantz RJ (2000) Human immunodeficiency virus type 1 Vpr induces apoptosis in human neuronal cells. J. Virol. 74: 9717–9726
Kubota S, Siomi H, Satoh T, Endo S, Maki M and Hatanaka M (1989) Functional similarity of HIV-I rev and HTLV-I rex proteins: identification of a new nucleolar-targeting signal in rev protein. Biochem. Biophys. Res. Commun. 162: 963–970
Bohnlein E, Berger J and Hauber J (1991) Functional mapping of the human immunodeficiency virus type 1 Rev RNA binding domain: new insights into the domain structure of Rev and Rex. J. Virol. 65: 7051–7055
Mabrouk K, Van Rietschoten J, Vives E, Darbon H, Rochat H and Sabatier JM (1991) Lethal neurotoxicity in mice of the basic domains of HIV and SIV Rev proteins. Study of these regions by circular dichroism. FEBS Lett. 289: 13–17
Singh KK, Barroga CF, Hughes MD, Chen J, Raskino C, McKinney RE and Spector SA (2003) Genetic influence of CCR5, CCR2, and SDF1 variants on human immunodeficiency virus 1 (HIV-1)-related disease progression and neurological impairment, in children with symptomatic HIV-1 infection. J. Infect. Dis. 188: 1461–1472
Gonzalez E, Rovin BH, Sen L, Cooke G, Dhanda R, Mummidi S, Kulkarni H, Bamshad MJ, Telles V, Anderson SA, Walter EA, Stephan KT, Deucher M, Mangano A, Bologna R, Ahuja SS, Dolan MJ and Ahuja SK (2002) HIV-1 infection and AIDS dementia are influenced by a mutant MCP-1 allele linked to increased monocyte infiltration of tissues and MCP-1 levels. Proc. Natl. Acad. Sci. USA 99: 13795–13800
Dunlop O, Goplen AK, Liestol K, Myrvang B, Rootwelt H, Christophersen B, Kvittingen EA and Maehlen J (1997) HIV dementia and apolipoprotein E. Acta Neurol. Scand. 95: 315–318
Corder EH, Robertson K, Lannfelt L, Bogdanovic N, Eggertsen G, Wilkins J and Hall C (1998) HIV-infected subjects with the E4 allele for APOE have excess dementia and peripheral neuropathy. Nat. Med. 4: 1182–1184
Kitagawa K, Matsumoto M, Kuwabara K, Takasawa K, Tanaka S, Sasaki T, Matsushita K, Ohtsuki T, Yanagihara T and Hori M (2002) Protective effect of apolipoprotein E against ischemic neuronal injury is mediated through antioxidant action. J. Neurosci. Res. 68: 226–232
Pedersen WA, Chan SL and Mattson MP (2000) A mechanism for the neuroprotective effect of apolipoprotein E: isoform-specific modification by the lipid peroxidation product 4-hydroxynonenal. J. Neurochem. 74: 1426–1433
Koudinov AR and Koudinova NV (2003) Cholesterol, synaptic function and Alzheimer's disease. Pharmacopsychiatry 36: S107–S112
Wagstaff LR, Mitton MW, Arvik BM and Doraiswamy PM (2003) Statin-associated memory loss: analysis of 60 case reports and review of the literature. Pharmacotherapy 23: 871–880
Haughey NJ, Cutler RG, Tamara A, McArthur JC, Vargas DL, Pardo CA, Turchan J, Nath A and Mattson MP (2004) Perturbation of sphingolipid metabolism and ceramide production in HIV-dementia. Ann. Neurol: 5: 257–267
Gartner S (2000) HIV infection and dementia. Science 28: 602–604
Moses AV, Stenglein SG, Strussenberg JG, Wehrly K, Chesebro B and Nelson JA (1996) Sequences regulating tropism of human immunodeficiency virus type 1 for brain capillary endothelial cells map to a unique region on the viral genome. J. Virol. 70: 3401–3406
Pierson TC and Doms RW (2003) HIV-1 entry and its inhibition. Curr. Top. Microbiol. Immunol. 281: 1–27
Li Y, Kappes JC, Conway JA, Price RW, Shaw GM and Hahn BH (1991) Molecular characterization of human immunodeficiency virus type 1 cloned directly from uncultured human brain tissue: identification of replication-competent and -defective viral genomes. J. Virol. 65: 3973–3985
Zhang K, Hawken M, Rana F, Welte FJ, Gartner S, Goldsmith MA and Power C (2001) Human immunodeficiency virus type 1 clade A and D neurotropism: molecular evolution, recombination, and coreceptor use. Virology 283: 19–30
Chang J, Jozwiak R, Wang B, Ng T, Ge YC, Bolton W, Dwyer DE, Randle C, Osborn R, Cunningham AL and Saksena NK (1998) Unique HIV type 1 V3 region sequences derived from six different regions of brain: region-specific evolution within host-determined quasispecies. AIDS Res. Hum. Retroviruses 14: 25–30
McArthur JC, McClernon DR, Cronin MF, Nance-Sproson TE, Saah AJ, St Clair M and Lanier ER (1997) Relationship between human immunodeficiency virus-associated dementia and viral load in cerebrospinal fluid and brain. Ann. Neurol. 42: 689–698
Robertson K, Fiscus S, Kapoor C, Robertson W, Schneider G, Shepard R, Howe L, Silva S and Hall C (1998) CSF, plasma viral load and HIV associated dementia. J. Neurovirol. 4: 90–94
Mayne M, Bratanich AC, Chen P, Rana F, Nath A and Power C (1998) HIV-1 tat molecular diversity and induction of TNF-alpha: implications for HIV-induced neurological disease. Neuroimmunomodulation 5: 184–192
Rao M and Mattson MP (2001) Stem cells and aging: expanding the possibilities. Mech. Ageing Dev. 122: 713–744
van Praag H, Schinder AF, Christie BR, Toni N, Palmer TD and Gage FH (2002) Functional neurogenesis in the adult hippocampus. Nature 28: 1030–1104
Shors TJ, Miesegaes G, Beylin A, Zhao M, Rydel T and Gould E (2001) Neurogenesis in the adult is involved in the formation of trace memories. Nature 410: 372–376
Kuhn HG, Dickinson-Anson H and Gage FH (1996) Neurogenesis in the dentate gyrus of the adult rat: age-related decrease of neuronal progenitor proliferation. J. Neurosci. 16: 2027–2033
Nishino H, Hida H, Takei N, Kumazaki M, Nakajima K and Baba H (2000) Mesencephalic neural stem (progenitor) cells develop to dopaminergic neurons more strongly in dopamine-depleted striatum than in intact striatum. Exp. Neurol. 164: 209–214
Haughey NJ, Nath A, Chan SL, Borchard AC, Rao MS and Mattson MP (2002) Disruption of neurogenesis by amyloid beta-peptide, and perturbed neural progenitor cell homeostasis, in models of Alzheimer's disease. J. Neurochem. 83: 1509–1524
Rice AC, Bullock MR and Shelton KL (2004) Chronic ethanol consumption transiently reduces adult neural progenitor cell proliferation. Brain Res. 11: 94–98
Tran PB, Ren D, Veldhouse TJ and Miller RJ (2004) Chemokine receptors are expressed widely by embryonic and adult neural progenitor cells. J. Neurosci. Res. 1: 20–34
Ekdahl CT, Claasen JH, Bonde S, Kokaia Z and Lindvall O (2003) Inflammation is detrimental for neurogenesis in adult brain. Proc. Natl. Acad. Sci. USA 11: 13632–13637
Monje ML, Toda H and Palmer TD (2003) Inflammatory blockade restores adult hippocampal neurogenesis. Science 302: 1760–1765
Krathwohl MD and Kaiser JL (2004) HIV-1 promotes quiescence in human neural progenitor cells. J. Infect. Dis. 15: 216–226
Benelli R, Mortarini R, Anichini A, Giunciuglio D, Noonan DM, Montalti S, Tacchetti C. and Albini A (1998) Monocyte-derived dendritic cells and monocytes migrate to HIV-Tat RGD and basic peptides. AIDS 12: 261–268
Mattson MP (2004) Pathways towards and away from Alzheimer's disease. Nature 430: 631–639
Petito CK and Roberts B (1995) Evidence of apoptotic cell death in HIV encephalitis (see comments). Am. J. Pathol. 146: 1121–1130
Garden GA, Guo W, Jayadev S, Tun C, Balcaitis S, Choi J, Montine TJ, Moller T and Morrison RS (2004) HIV associated neurodegeneration requires p53 in neurons and microglia. FASEB J. 18: 1141–1143
Chen W, Sulcove J, Frank I, Jaffer S, Ozdener H and Kolson DL (2002) Development of a human neuronal cell model for human immunodeficiency virus (HIV)-infected macrophage-induced neurotoxicity: apoptosis induced by HIV type 1 primary isolates and evidence for involvement of the Bcl-2/Bcl-xL-sensitive intrinsic apoptosis pathway. J. Virol. 76: 9407–9419
Kruman II, Nath A, Maragos WF, Chan SL, Jones M, Rangnekar VM, Jakel RJ and Mattson MP (1999) Evidence that Par-4 participates in the pathogenesis of AIDS dementia. Am. J. Pathol. 155: 39–46
Ramirez SH, Sanchez JF, Dimitri CA, Gelbard HA, Dewhurst S and Maggirwar SB (2001) Neurotrophins prevent HIV Tat-induced neuronal apoptosis via a nuclear factor-kappaB (NF-kappaB)-dependent mechanism. J. Neurochem. 78: 874–889
Mattson MP, Maudsley S and Martin B (2004) BDNF and 5-HT: a dynamic duo in age-related neuronal plasticity and neurodegenerative disorders. Trends Neurosci. 27: 589–594
Zauli G, Milani D, Mirandola P, Mazzoni M, Secchiero P, Miscia S and Capitani S (2001) HIV-1 Tat protein down-regulates CREB transcription factor expression in PC12 neuronal cells through a phosphatidylinositol 3-kinase/AKT/cyclic nucleoside phosphodiesterase pathway. FASEB J. 15: 483–491
Regulier EG, Reiss K, Khalili K, Amini S, Rappaport J, Zagury JF and Katsikis PD (2004) T-cell and neuronal apoptosis in HIV infection: implications for therapeutic intervention. Int. Rev. Immunol. 23: 25–59
Sperber K, Beuria P, Singha N, Gelman I, Cortes P, Chen H and Kraus T (2003) Induction of apoptosis by HIV-1-infected monocytic cells. J. Immunol. 170: 1566–1578
Xu Y, Kulkosky J, Acheampong E, Nunnari G, Sullivan J and Pomerantz RJ (2004) HIV-1-mediated apoptosis of neuronal cells: proximal molecular mechanisms of HIV-1-induced encephalopathy. Proc. Natl. Acad. Sci. USA 101: 7070–7075
Kaul M and Lipton SA (1999) Chemokines and activated macrophages in HIV gp120-induced neuronal apoptosis. Proc. Natl. Acad. Sci. USA 96: 8212–8216
Garden GA (2002) Microglia in human immunodeficiency virus-associated neurodegeneration. Glia 40: 240–251
Zhang K, McQuibban GA, Silva C, Butler GS, Johnston JB, Holden J, Clark-Lewis I, Overall CM and Power C (2003) HIV-induced metalloproteinase processing of the chemokine stromal cell derived factor-1 causes neurodegeneration. Nat. Neurosci. 6: 1064–1071
Khurdayan VK, Buch S, El-Hage N, Lutz SE, Goebel SM, Singh IN, Knapp PE, Turchan-Cholewo J, Nath A and Hauser KF (2004) Preferential vulnerability of astroglia and glial precursors to combined opioid and HIV-1 Tat exposure in vitro. Eur. J. Neurosci. 19: 3171–3182
Mollace V, Nottet HS, Clayette P, Turco MC, Muscoli C, Salvemini D and Perno CF (2001) Oxidative stress and neuroAIDS: triggers, modulators and novel antioxidants. Trends Neurosci. 24: 411–416
Turchan J, Pocernich CB, Gairola C, Chauhan A, Schifitto G, Butterfield DA, Buch S, Narayan O, Sinai A, Greiger J, Berger JR, Elford H and Nath A (2003) Oxidative stress in HIV demented patients and protection ex vivo with novel antioxidants. Neurology 28: 307–314
Cutler RG, Haughey NJ, Tammara A, McArthur JC, Nath A, Reid R, Vargas DL, Pardo CA and Mattson MP (2004) Dysregulation of sphingolipid and sterol metabolism by ApoE4 in HIV dementia. Neurology 63: 626–630
Keller JN, Pang Z, Geddes JW, Begley JG, Germeyer A, Waeg G and Mattson MP (1997) Impairment of glucose and glutamate transport and induction of mitochondrial oxidative stress and dysfunction in synaptosomes by amyloid beta-peptide: role of the lipid peroxidation product 4-hydroxynonenal. J. Neurochem. 69: 273–284
Mark RJ, Pang Z, Geddes JW, Uchida K and Mattson MP (1997) Amyloid beta-peptide impairs glucose transport in hippocampal and cortical neurons: involvement of membrane lipid peroxidation. J. Neurosci. 17: 1046–1054
Kruman I, Bruce-Keller AJ, Bredesen D, Waeg G and Mattson MP (1997) Evidence that 4-hydroxynonenal mediates oxidative stress-induced neuronal apoptosis. J. Neurosci. 17: 5089–5100
Bruce-Keller AJ, Li YJ, Lovell MA, Kraemer PJ, Gary DS, Brown RR, Markesbery WR and Mattson MP (1998) 4-Hydroxynonenal, a product of lipid peroxidation, damages cholinergic neurons and impairs visuospatial memory in rats. J. Neuropathol. Exp. Neurol. 57: 257–267
Lipton SA, Yeh M and Dreyer EB (1994) Update on current models of HIV-related neuronal injury: platelet-activating factor, arachidonic acid and nitric oxide. Adv. Neuroimmunol. 4: 181–188
Haughey NJ and Mattson MP (2002) Calcium dysregulation and neuronal apoptosis by the HIV-1 proteins Tat and gp120. J. Acquir. Immune Defic. Syndr. 1: S55–S61
Lipton SA (1992) Memantine prevents HIV coat protein-induced neuronal injury in vitro. Neurology 42: 1403–1405
Muller WE, Schroder HC, Ushijima H, Dapper J and Bormann J (1992) gp120 of HIV-1 induces apoptosis in rat cortical cell cultures: prevention by memantine. Eur. J. Pharmacol. 226: 209–214
Holden CP, Nath A, Haughey NJ and Geiger JD (1999) Involvement of Na+/H+ exchangers, Ca2+ channels, and excitatory amino acid receptors in intracellular Ca2+ responses to HIV-1 gp120 in cultured human fetal brain cells. Neuroscience 91: 1369–1378
Nath A, Haughey NJ, Jones M, Anderson C, Bell JE and Geiger JD (2000) Synergistic neurotoxicity by human immunodeficiency virus proteins Tat and gp120: protection by memantine. Ann. Neurol. 47: 186–194
Cheng J, Nath A, Knudsen B, Hochman S, Geiger JD, Ma M and Magnuson DSK (1998) Neuronal excitatory properties of human immunodeficiency virus type 1 tat protein. Neuroscience 82: 97–106
Brorson JR, Manzolillo PA, Gibbons SJ and Miller RJ (1995) AMPA receptor desensitization predicts the selective vulnerability of cerebellar Purkinje cells to excitotoxicity. J. Neurosci. 15: 4515–4524
Garthwaite J (1991) Glutamate, nitric oxide and cell–cell signalling in the nervous system. Trends Neurosci. 14: 60–67
Jonas P and Sakmann B (1992) Glutamate receptor channels in isolated patches from CA1 and CA3 pyramidal cells of rat hippocampal slices. J. Physiol. (London) 455: 143–171
Haughey NJ, Nath A, Mattson MP, Slevin JT and Geiger JD (2001) HIV-1 Tat through phosphorylation of NMDA receptors potentiates glutamate excitotoxicity. J. Neurochem. 78: 457–467
Jain KK (2000) Evaluation of memantine for neuroprotection in dementia. Expert Opin. Investig. Drugs 9: 1397–1406
Kaul M and Lipton SA (2004) Signaling pathways to neuronal damage and apoptosis in human immunodeficiency virus type 1-associated dementia: chemokine receptors, excitotoxicity, and beyond. J. Neurovirol. 10: S97–S101
Bachis A and Mocchetti I (2004) The chemokine receptor CXCR4 and not the N-methyl-D-aspartate receptor mediates gp120 neurotoxicity in cerebellar granule cells. J. Neurosci. Res. 75: 75–82
Haughey NJ, Holden CP, Nath A and Geiger JD (1999) Involvement of inositol 1, 4, 5-trisphosphate-regulated stores of intracellular calcium in calcium dysregulation and neuron cell death caused by HIV-1 protein tat. J. Neurochem. 73: 1363–1374
Kruman I, Nath A and Mattson MP (1998) HIV protein Tat induces apoptosis by a mechanism involving mitochondrial calcium overload and caspase activation. Exp. Neurol. 154: 276–288
New D, Ma M, Epstein LG, Nath A and Gelbard HA (1997) Human immunodeficiency virus type 1 Tat protein induces death by apoptosis in primary human neuron cultures. J. Neurovirol. 3: 168–173
Koller H, von Giesen HJ, Schaal H and Arendt G (2001) Soluble cerebrospinal fluid factors induce Ca2+ dysregulation in rat cultured cortical astrocytes in HIV-1-associated dementia complex. AIDS 15: 1789–1792
Navia BA, Dafni U, Simpson D, Tucker T, Singer E, McArthur JC, Yiannoutsos C, Zaborski L and Lipton SA (1998) A phase I/II trial of nimodipine for HIV-related neurologic complications. Neurology 51: 221–228
Zhao ML, Kim MO, Morgello S and Lee SC (2001) Expression of inducible nitric oxide synthase, interleukin-1 and caspase-1 in HIV-1 encephalitis. J. Neuroimmunol. 115: 182–191
Letendre SL, Lanier ER and McCutchan JA (1999) Cerebrospinal fluid beta chemokine concentrations in neurocognitively impaired individuals infected with human immunodeficiency virus type 1. J. Infect. Dis. 180: 310–319
Conant K, McArthur JC, Griffin DE, Sjulson L, Wahl LM and Irani DN (1999) Cerebrospinal fluid levels of MMP-2, 7, and 9 are elevated in association with human immunodeficiency virus dementia. Ann. Neurol. 46: 391–398
Petito CK, Roberts B, Cantando JD, Rabinstein A and Duncan R (2001) Hippocampal injury and alterations in neuronal chemokine co-receptor expression in patients with AIDS. J. Neuropathol. Exp. Neurol. 60: 377–385
Sasseville VG, Newman WA, Lackner AA, Smith MO, Lausen NC, Beall D and Ringler DJ (1992) Elevated vascular cell adhesion molecule-1 in AIDS encephalitis induced by simian immunodeficiency virus. Am. J. Pathol. 141: 1021–1030
Persidsky Y, Buttini M, Limoges J, Bock P and Gendelman HE (1997) An analysis of HIV-1-associated inflammatory products in brain tissue of humans and SCID mice with HIV-1 encephalitis. J. Neurovirol. 3: 401–416
Persidsky Y, Limoges J, Rasmussen J, Zheng J, Gearing A and Gendelman HE (2001) Reduction in glial immunity and neuropathology by a PAF antagonist and an MMP and TNFalpha inhibitor in SCID mice with HIV-1 encephalitis. J. Neuroimmunol. 114: 57–68
Mattson MP (2003) Gene-diet interactions in brain aging and neurodegenerative disorders. Ann. Intern. Med. 139: 441–444
Persaud-Sawin DA, McNamara 2nd JO, Rylova S, Vandongen A and Boustany RM. (2004) A galactosylceramide binding domain is involved in trafficking of CLN3 from golgi to rafts via recycling endosomes. Pediatr. Res. 56: 449–463
Fantini J, Garmy N, Mahfoud R and Yahi N (2002) Lipid rafts: structure, function and role in HIV, Alzheimers and prion diseases. Expert Rev. Mol. Med. 2002: 1–22
Martin AM, Nolan D, Gaudieri S, Phillips E and Mallal S (2004) Pharmacogenetics of antiretroviral therapy: genetic variation of response and toxicity. Pharmacogenomics 5: 643–655
Pierson TC, Doms RW and Pohlmann S (2004) Prospects of HIV-1 entry inhibitors as novel therapeutics. Rev. Med. Virol. 14: 255–270
Badley AD, Roumier T, Lum JJ and Kroemer G (2003) Mitochondrion-mediated apoptosis in HIV-1 infection. Trends Pharmacol Sci 24: 298–305
Sacktor N, Skolasky RL, Tarwater PM, McArthur JC, Selnes OA, Becker J, Cohen B, Visscher B, Miller EN and Multicenter AIDS Cohort Study (MACS) (2003) Response to systemic HIV viral load suppression correlates with psychomotor speed performance. Neurology 61: 567–569
Letendre SL, McCutchan JA, Childers ME, Woods SP, Lazzaretto D, Heaton RK, Grant I, Ellis RJ and HNRC Group (2004) Enhancing antiretroviral therapy for human immunodeficiency virus cognitive disorders. Ann. Neurol. 56: 416–423
Banks WA, Kastin AJ, Brennan JM and Vallance KL (1999) Adsorptive endocytosis of HIV-1gp120 by blood–brain barrier is enhanced by lipopolysaccharide. Exp. Neurol. 156: 165–171
Avraham HK, Jiang S, Lee TH, Prakash O and Avraham S (2004) HIV-1 Tat-mediated effects on focal adhesion assembly and permeability in brain microvascular endothelial cells. J. Immunol. 173: 6228–6233
von Giesen HJ, Hefter H, Jablonowski H and Arendt G (2000) HAART is neuroprophylactic in HIV-1 infection. J. Acquir. Immune Defic. Syndr. 23: 380–385
Author information
Authors and Affiliations
Corresponding author
Additional information
Edited by M Piacentini
Rights and permissions
About this article
Cite this article
Mattson, M., Haughey, N. & Nath, A. Cell death in HIV dementia. Cell Death Differ 12 (Suppl 1), 893–904 (2005). https://doi.org/10.1038/sj.cdd.4401577
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.cdd.4401577
Keywords
This article is cited by
-
Meta-Analysis on Nicotine's Modulation of HIV-Associated Dementia
Journal of Neuroimmune Pharmacology (2022)
-
Beneficial and Adverse Effects of cART Affect Neurocognitive Function in HIV-1 Infection: Balancing Viral Suppression against Neuronal Stress and Injury
Journal of Neuroimmune Pharmacology (2021)
-
A longitudinal study of brain volume changes in rhesus macaque model infected with SIV
Journal of NeuroVirology (2020)
-
HIV infection and latency induce a unique metabolic signature in human macrophages
Scientific Reports (2019)
-
HIV and Alzheimer’s disease: complex interactions of HIV-Tat with amyloid β peptide and Tau protein
Journal of NeuroVirology (2019)
