
Abstract
Accurate estimates of breast and ovarian cancer penetrance in BRCA1/2 mutation carriers are crucial in genetic counseling. Estimation is difficult because of the low frequency of mutated alleles and the often-uncertain mechanisms of family ascertainment. We estimated the penetrances of breast and ovarian cancers in carriers of BRCA1/2 mutations by maximizing the retrospective likelihood of the genetic model, given the observed test results, in 568 Italian families screened for germline mutations. The software BRCAPRO was used as a probability calculation tool in a Markov Chain Monte Carlo approach. Breast cancer penetrances were 27% (95% CI 20–34%) at age 50 years and 39% (27–52%) at age 70 in BRCA1 carriers, and 26% (0.18–0.34%) at age 50 and 44% (29–58%) at age 70 in BRCA2 carriers, and ovarian cancer penetrances were 14% (7–22%) at age 50 and 43% (21–66%) at age 70 in BRCA1 carriers and 3% (0–7%) at age 50 and 15% (4–26%) at age 70 in BRCA2 carriers. The new model gave a better fit than the current default in BRCAPRO, the likelihood being 70 log units greater; in addition, the observed numbers of mutations in families stratified by gene and by cancer profile were not significantly different from those expected. Our new penetrance functions are appropriate for predicting breast cancer risk, and for determining the probability of carrying BRCA1/2 mutations, in people who are presently referred to genetic counseling in Italy. Our approach could lead to country-customized versions of the BRCAPRO software by providing appropriate population-specific estimates.
Similar content being viewed by others


Introduction
Mutations in the two genes BRCA1 (MIM 113705) and BRCA2 (MIM 600185) account for 30–40% of the families with multiple breast cancer cases and for the vast majority of families with multiple breast and ovarian cancers cases.1, 2, 3 Taken together, these genes may be responsible for 5–10% of early-onset breast cancers4, 5, 6, 7 and about 10% of all ovarian cancers.8 Estimates of cancer risks among mutation carriers provide valuable opportunities to tailor cancer screening and prevention strategies and to refine clinical and behavioral interventions to reduce cancer risk.9, 10, 11, 12
Initial studies of multiple-case families with four or more cancer cases suggested that the lifetime risk of breast cancer in BRCA1 mutation carriers was 71–85%, and that of ovarian cancer was 42–63% at age 70.13, 14 Similarly, early-penetrance evaluations in BRCA2 led to an 84% risk of breast cancer and 27% for ovarian cancer at age 70.2 Later studies, based on extensive typing of incident cases, have provided lower risks,15, 16, 17 for example, 40% penetrance of breast cancer at age 7015 or 48% at age 80,17 though with large sampling errors. A recent combined analysis of several previous studies18 estimated the risk of breast cancer to be about 64% in BRCA1 and 39% in BRCA2 mutation carriers at age 70. Substantial uncertainty in the estimates remains. Issues of variation across different populations and across different mutations of the same gene need to be investigated in further detail.19
In the present work, we have addressed the question of estimating cancer- and gene-specific penetrances appropriate for the mutations segregating in a large sample of Italian multiple-case families screened for both genes. We used the software BRCAPRO,20, 21 a widely used program for predicting the presence of a germline mutation in a proband. Since the prediction made by this program is based on a specific genetic model (the set of parameter values, allele frequencies and penetrances that define the risk of disease in the population), it is possible to reverse the approach, that is, estimating some of the parameters from family data. This is accomplished by iteratively exploring new parameter values until the best genetic model is found, for which the prediction made by BRCAPRO is more accurate. In this approach, the likelihood of the genetic data (the observed test results) conditional on the phenotype is calculated. This is called the retrospective likelihood, and parameter estimates based on it remain unbiased even when the ascertainment method cannot be specified accurately, provided that ascertainment depends on phenotypes only.22 While use of the retrospective likelihood has precedents,2 our approach is novel in that it includes families negative at the genetic test, and accounts for test sensitivity.
Materials and methods
The families included in the present study have been described previously.23 Briefly, 568 Caucasian families ascertained in five clinical centers were submitted to genetic test for BRCA1 and/or BRCA2 (458 families were screened for both BRCA1 and BRCA2, 104 for BRCA1 only and eight for BRCA2 only). Among the families included in this study, 151 presented both breast and ovarian cancer, either in a single individual or in different relatives (hereditary breast-ovarian cancer (HBOC)), 357 included patients with breast cancer only (hereditary breast cancer (HBC)), 31 included patients with ovarian cancer only (hereditary ovarian cancer (HOC)), and 29 included at least one case of male breast cancer (MBC). In the present study, we estimate penetrance functions for females only, as the number of males affected with breast cancer is too small. Eligibility criteria for genetic testing varied across centers and, within centers, over time; families with multiple cases of breast/ovarian cancer or early-onset cancer cases were preferentially selected. Pedigree data were reported by family members to genetic counselors, and included information about breast and ovarian cancer in first- and second-degree relatives of probands. In total, 85 distinct germ-line mutations (46 in BRCA1 and 39 in BRCA2) were detected in 133 independently ascertained probands (26% of the screened families, 80 with mutation in BRCA1 and 53 in BRCA2, see Figure 1); 15 mutations of BRCA1 and seven mutations of BRCA2 were identified more than once, with a maximum of nine families with BRCA1*5382insC and four families with BRCA2*6696delTC and BRCA2*IVS16-2A>G. These mutation frequency spectra are typical of large, non-isolated populations. In mutation analysis, three centers used both direct automatic sequencing and PTT-SSCP, one center used both PTT-SSCP and fluorescence-assisted mutational analysis (FAMA) and the last center used PTT-SSCP only.

BRCA1 and BRCA2 maps of the mutations included in the present study, with indication of the number of families in which each mutation was identified.
The program BRCAPRO computes the probability that a proband is a carrier of a mutation in either BRCA1 or BRCA2, based on the family history of breast and ovarian cancer in her first- and second-degree relatives and on published estimates of cancer-specific penetrances and mutated-allele population frequencies; model parameter files are regularly updated following published studies.21, 24 BRCAPRO is also distributed as a part of the counseling package CaGene.25 In the present context, BRCAPRO is used as a probability calculation tool to obtain new parameter estimates via a Metropolis–Hastings26 Markov-Chain Monte Carlo (MCMC) method.27
The equations used to model the penetrance functions have the following form:

where fg,j(x) is the incidence at age x for the carriers of a mutation in gene g (g=1 or 2 for BRCA1 and BRCA2, respectively) for cancer type j (j=1 or 2 for breast and ovarian cancer, respectively), f0,j(x) are the corresponding incidences among non-carriers, and Y(x)=ϑ0+ϑ1x+ϑ2x2. Coefficients ϑ0, ϑ1, and ϑ2 are estimated. This form of the penetrance function allows for a parsimonious representation of the relationship between cancer, genotype and age. The density of cases in the general population, estimated using cancer registry data,21 is used as a baseline. The parameter ϑ0 allows for an increase in risk across all ages, while the parameters ϑ1 and ϑ2 allow to flexibly incorporate effects of genotype on age of onset. We also constrain the term Y(x) so that it is always greater than 0, to avoid the possibility of a lower penetrance in mutation carriers than in noncarriers. In addition to the penetrance functions, we also estimate from data the proportion m=p1/(p1+p2), where p1 and p2 are the population frequencies of the mutated alleles in BRCA1 and BRCA2, respectively; total prevalence of mutation carriers is thus approximately 2(p1+p2). We assume uniform prior distributions on all parameters.
At the kth iteration of the MCMC algorithm, a new set of parameters ϑs and m is formed, based on the parameter values at the (k−1)th iteration and a proposal drawn from a multivariate t distribution, and the corresponding penetrance functions are calculated; the carrier probabilities are then determined by BRCAPRO for all the families in the study, and the total log likelihood of the new parameter set, given the observed test results, is calculated as

where the zi's are indicator variables pointing to the carrier status of proband i (10=BRCA1, 01=BRCA2, 00=no mutation, 11=mutation in both genes), the πi's are the corresponding carrier probabilities, and β is the sensitivity of the genetic test, set to 0.7.21 In the families screened for one gene only, the log-likelihood function is modified by assuming that the determination of which gene to test is independent of genotype, as follows:

The proposed parameter set at each iteration k is accepted with probability=1 if Lk>Lk−1 and with probability=Lk/Lk−1 otherwise. Confidence intervals of parameters are obtained by examining their variation among the last 5000 iterations (after parameter values are stabilized); for each year of age, the values that excluded the 2.5% upper and lower tails of the penetrance distribution are taken as the 95% confidence intervals.
In our retrospective likelihood approach, it is not possible to estimate allele frequencies for the two genes separately along with penetrance parameters ϑs; therefore, the sum p=p1+p2 was initially set to 0.003.2, 14, 28, 29 Gene-specific allele frequencies are not being estimated in this work, only their ratio. The entire analysis was also repeated considering the P-values 0.001, 0.002, 0.004 and 0.005. We first considered the full ‘quadratic’ model including three parameters for each of four penetrance functions plus the proportion m of BRCA1 vs BRCA2 mutation in the population (total parameters 13). To check if the model could be simplified without affecting the overall fit, we also used the ‘linear’ function Y(x)=ϑ0+ϑ1x in equation (1); this led to a reduction in the number of estimated parameters from 13 to 9.
Results
In all calculations, the sample stabilized in less than 2000 iterations, though the chains were continued for 10 000 iterations. Acceptance rate was near 40%. Considering the model with P fixed to 0.003, we obtained a final log likelihood, evaluated at point estimates, of about −326 for both the quadratic and the linear models, and the four resulting penetrance curves were practically indistinguishable. Therefore, we accepted the linear model. Repeating the analysis with lower values of P (0.001 and 0.002) had the effect of decreasing the equilibrium values of the log likelihood to about −345 and −331, respectively (19 and 5 log unit difference). On the other hand, increasing P to 0.004 and 0.005 caused a slight increase in log likelihood (−324 and −322, respectively); however, the penetrance curves associated to these values were still lower. For example, breast cancer risk for BRCA1 carriers at age 70 decreased from 39% (P=0.003, see Table 1) to 35% (P=0.004) to 34% (P=0.005).
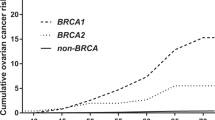
The four new penetrance curves of breast and ovarian cancer in BRCA1-2 mutation carriers corresponding to the model with p=0.003 are shown in Figure 2, together with 95% confidence limits. Dotted lines show, as a reference, the default penetrance curves distributed with BRCAPRO, used in our previous study.23 Breast cancer penetrance is lower, especially for BRCA1 carriers, whereas ovarian cancer penetrance in BRCA1 is higher (though the reference ovarian cancer penetrance curve is included in the 95% confidence limits of our estimate). The penetrance of ovarian cancer in BRCA2 is similar to the reference. Table 1 compares cancer penetrances at 50 and 70 years of age, as estimated by our model, to those of previous studies, whereas Table 2 shows the age-specific incidences at 10-year intervals in our estimates. Considering the frequency of the mutated alleles, the value of the parameter m was 0.57 (95% CI 0.44–0.69), leading to an estimate of 3.4 × 10−3 for the prevalence of BRCA1 mutation carriers (∼1:300 individuals) and 2.6 × 10−3 for that of BRCA2 mutation carriers (∼1:400 individuals) in our sample.

Breast and ovarian cancer penetrances in BRCA1 and BRCA2 mutation carriers estimated in the present work, with 95% confidence limits (dark and light continuous lines, respectively). Dotted lines: default penetrance functions in BRCAPRO.
Lastly, we used the estimated penetrances as input in BRCAPRO to predict the presence of a mutation in probands in our sample. As those are the same samples used in training the model, this exercise is a goodness-of-fit evaluation rather than a validation. In our previous evaluation study,23 it emerged that none of the currently available mutation-predicting models was able to accurately discriminate between the two genes, either because models did not address this (eg, Myriad Tables and Couch model) or because the discriminating ability was limited (BRCAPRO and IC models). However, difference in expressivity may be better exploited to distinguish between the two genes.30 We investigated this issue by stratifying our families in the four typical profiles (HBC, HBOC, HOC, and MBC), and computing the observed and expected number of mutations separately for the two genes (Table 3). While the estimated parameter values maximize the overall fit by construction, it is interesting to explore whether the fit varies by subgroup. We assessed calibration (the correspondence between observed and expected number of cases) by applying the χ2 test. The observed numbers of mutations in the four family profiles were not significantly different from those expected, both considering total data and data separated by gene. The highest χ2 contributions come from MBC families (penetrance of MBC was not estimated in the present work). The last column in Table 3 shows the difference between the log likelihoods computed with the penetrance functions estimated in this work and the default penetrance functions in BRCAPRO; the highest increases concern the HBC families in BRCA2 and the HBOC families in BRCA1. HBC families were disproportionately assigned to BRCA1 by the previous predictions.
Discussion
Determining the appropriate values of cancer risks in women who carry a mutation in BRCA1 or BRCA2 is crucial for establishing the most appropriate preventive measures. However, estimation of penetrance is a difficult task, being subjected to different sources of bias;31 considerable uncertainties remain about the ‘true’ values. A novel aspect of the present work is that it includes information from all tested families, both mutation-positive and mutation-negative; previously published estimates were based on information drawn from pedigrees of mutation carriers only.2, 16, 17, 18 Our approach consisted in determining the penetrance curves that optimize the goodness-of-fit of a Mendelian model that predicts the presence of a mutation in a proband, given its personal and family cancer history. An advantage of this methodology is that it can be applied to family collections whose ascertainment scheme is loose.
The most noticeable result of this work is that the breast cancer penetrance in BRCA1 carriers is lower than in previous studies. An important feature of our family data set is the high proportion of families with mild family history, which probably reflects the recent increase of genetic-test demand. From this point of view, our study sample may be considered intermediate between the multiple-case studies, based on highly selected families, and the population-based studies, in which typing is performed irrespective of family history of cancer; our penetrance estimates are more consistent with the latter studies. We have previously shown23 that all extant predictive models underestimated the probability of identifying mutations in the families with calculated prior probability <10%. If a persistent excess of mutations is identified as far as more and more low-risk families are typed, this supports the idea that cancer penetrances of these mutations were previously considered too high.
A complication in estimating a certain penetrance curve is that a single entity common to all populations may not exist;32 each population has its own mutation spectrum, with different prevalence of specific founder mutations, and different mutations may confer different breast and ovarian cancer risk. Consequently, each study estimates the average risk to carriers specifically for the population being studied. The population included in the present study is representative of the patient population that is referred for genetic testing for BRCA1 or BRCA2 mutations in Italy. Genetic counselors are interested in penetrance estimates relevant to the risks in the sort of families that are coming into counseling; thus, our estimated model is appropriate for use in determining the probability of carrying BRCA1/2 mutations in Italian families. We are implementing a version of BRCAPRO that can be distributed to genetic clinics in this country.
Another explanation for the finding of lower breast cancer risk accompanied by increased ovarian cancer risk in BRCA1 carriers could be the presence of a particular mutation spectrum in the study set, for example, if there was an unusually high proportion of mutations associated with early-onset ovarian cancer; however, this is not the case, as it was shown in our previous analysis of a similar data set,30 and it is also apparent by inspection of Figure 1. However, this issue should be investigated in greater details, as there may be substantial allelic heterogeneity in cancer risks in both BRCA1 and BRCA2.16, 17, 33 This issue has received little attention until recently, as it requires collecting large samples of families sharing the same mutations. We are investigating this issue in our data. It is possible that at least some of the mutations identified in families at relatively low cancer risk contribute to lower the average penetrance in comparison with the previous estimates, which were necessarily based on highly selected families, that is, those possibly harboring mutations with a relatively stronger phenotypic effect.
A disadvantage of the approach used here is that it is not possible to estimate simultaneously both the mutated allele frequencies and the penetrances in the same data set. Even in previous studies, either the prevalence of gene carriers assuming known penetrances were estimated,4, 17 or the penetrances assuming known prevalence were estimated.2, 13 We have chosen an initial value of 0.003 for the cumulative frequency of the mutated alleles based on an early segregation analysis study,28 in which all the parameters of the genetic model (gene frequency and penetrances) can be independently estimated; this requires, however, that the ascertainment method is clearly defined. We have tried to address the robustness of our penetrance estimates by repeating the entire analysis with different values of the cumulative allele frequency. Values lower than 0.003 caused a decrease in the total log likelihood, whereas higher values caused a small increase in the log likelihood, accompanied by a further decline of the penetrances. Therefore, the genetic model with p=0.003 may represent a combination of values (allele frequencies and penetrances) that gives a good representation of reality, though for an ultimate answer we probably must wait until technology will allow estimating the allele frequencies by direct allele count in large random samples of the general population.
A key element of the genetic counseling consultation is the identification of probands whose risk is higher than a certain threshold, such as to justify the genetic test. The ASCO has recently suggested that BRCA gene mutation screening should be limited to individuals whose probability for carrying a mutation is at least 10%.25, 34 Applying this criterion to our new model, we find 338 probands with carrier probability >10% (59.5% of all families), among which 119 mutations in either gene were identified, or a mutation detection rate of 35.2%. On the other hand, 14 mutations (seven in each gene) have been identified in the 230 probands (40.5%) with probability <10%, or a mutation detection rate of 6.1%. Considering the default penetrance files distributed with BRCAPRO, 263 families (46.3% of total sample) had carrier probability >10%, in which 101 mutations were detected (whereas 32 mutations were found in the low-risk group). Therefore, the 10% probability threshold requires testing 75 more families in the new model (an increase of 13.2%), but the proportion of mutations identified in the low risk group is lowered from 24.1 to 10.5%. Thus, not only the old model classified a higher number of families at low risk, but also the proportion of mutations identified among them was higher.
References
Martin A, Blackwood M, Antin-Ozerkis D et al: Germline mutations in BRCA1 and BRCA2 in breast-ovarian families from a breast cancer risk evaluation clinic. J Clin Oncol 2001; 19: 2247–2253.
Ford D, Easton DF, Stratton M et al: Genetic heterogeneity and penetrance analysis of the BRCA1 and BRCA2 genes in breast cancer families. The Breast Cancer Linkage Consortium. Am J Hum Genet 1998; 62: 676–689.
Szabo CI, King MC : Population genetics of BRCA1 and BRCA2. Am J Hum Genet 1997; 60: 1013–1020.
Peto J, Collins N, Barfoot R et al: Prevalence of BRCA1 and BRCA2 gene mutations in patients with early-onset breast cancer. J Natl Cancer Inst 1999; 91: 943–949.
Loman N, Johannsson O, Kristoffersson U, Olsson H, Borg A : Family history of breast and ovarian cancers and BRCA1 and BRCA2 mutations in a population-based series of early-onset breast cancer. J Natl Cancer Inst 2001; 93: 1215–1223.
Dite GS, Jenkins MA, Southey MC et al: Familial risks, early-onset breast cancer, and BRCA1 and BRCA2 germline mutations. J Natl Cancer Inst 2003; 95: 448–457.
de Sanjose S, Leone M, Berez V et al: Prevalence of BRCA1 and BRCA2 germline mutations in young breast cancer patients: a population-based study. Int J Cancer 2003; 106: 588–593.
Risch HA, McLaughlin JR, Cole DE et al: Prevalence and penetrance of germline BRCA1 and BRCA2 mutations in a population series of 649 women with ovarian cancer. Am J Hum Genet 2001; 68: 700–710.
Chang-Claude J, Becher H, Caligo M et al: Risk estimation as a decision-making tool for genetic analysis of the breast cancer susceptibility genes. EC Demonstration Project on Familial Breast Cancer. Dis Markers 1999; 15: 53–65.
Karp SE : Clinical management of BRCA1- and BRCA2-associated breast cancer. Semin Surg Oncol 2000; 18: 296–304.
Burke W, Daly M, Garber J et al: Recommendations for follow-up care of individuals with an inherited predisposition to cancer. II. BRCA1 and BRCA2. Cancer Genetics Studies Consortium. JAMA 1997; 277: 997–1003.
King M-C, Marx JH, Mandel JB : Breast and ovarian cancer risks due to inherited mutations in BRCA1 and BRCA2. Science 2003; 302: 643–646.
Easton DF, Ford D, Bishop DT : Breast and ovarian cancer incidence in BRCA1-mutation carriers. Breast Cancer Linkage Consortium. Am J Hum Genet 1995; 56: 265–271.
Narod SA, Ford D, Devilee P et al: An evaluation of genetic heterogeneity in 145 breast-ovarian cancer families. Breast Cancer Linkage Consortium. Am J Hum Genet 1995; 56: 254–264.
Hopper JL, Southey MC, Dite GS et al: Population-based estimate of the average age-specific cumulative risk of breast cancer for a defined set of protein-truncating mutations in BRCA1 and BRCA2. Australian Breast Cancer Family Study. Cancer Epidemiol Biomarkers Prev 1999; 8: 741–747.
Scott CL, Jenkins MA, Southey MC et al: Average age-specific cumulative risk of breast cancer according to type and site of germline mutations in BRCA1 and BRCA2 estimated from multiple-case breast cancer families attending Australian family cancer clinics. Hum Genet 2003; 112: 542–551.
Anglian_Breast_Cancer_Study_Group: Prevalence and penetrance of BRCA1 and BRCA2 mutations in a population-based series of breast cancer cases. Br J Cancer 2000; 83: 1301–1308.
Antoniou A, Pharoah PD, Narod S et al: Average risks of breast and ovarian cancer associated with BRCA1 or BRCA2 mutations detected in case series unselected for family history: a combined analysis of 22 studies. Am J Hum Genet 2003; 72: 1117–1130.
Hopper JL : Genetic epidemiology of female breast cancer. Semin Cancer Biol 2001; 11: 367–374.
Berry DA, Parmigiani G, Sanchez J, Schildkraut J, Winer E : Probability of carrying a mutation of breast-ovarian cancer gene BRCA1 based on family history. J Natl Cancer Inst 1997; 89: 227–238.
Parmigiani G, Berry D, Aguilar O : Determining carrier probabilities for breast cancer-susceptibility genes BRCA1 and BRCA2. Am J Hum Genet 1998; 62: 145–158.
Kraft P, Thomas DC : Bias and efficiency in family-based gene-characterization studies: conditional, prospective, retrospective, and joint likelihoods. Am J Hum Genet 2000; 66: 1119–1131.
Marroni F, Aretini P, D'Andrea E et al: Evaluation of widely used models for predicting BRCA1 and BRCA2 mutations. J Med Genet 2004; 41: 278–285.
Berry DA, Iversen Jr ES, Gudbjartsson DF et al: BRCAPRO validation, sensitivity of genetic testing of BRCA1/BRCA2, and prevalence of other breast cancer susceptibility genes. J Clin Oncol 2002; 20: 2701–2712.
Euhus DM, Smith KC, Robinson L et al: Pretest prediction of BRCA1 or BRCA2 mutation by risk counselors and the computer model BRCAPRO. J Natl Cancer Inst 2002; 94: 844–851.
Metropolis N, Rosenbluth AW, Rosenbluth MN, Teller AH, Teller E : Equations of state calculations by fast computing machine. J Chem Phys 1953; 21: 1087–1091.
Gilks WR, Richardson SJ : SD: Markov Chain Monte Carlo in Practice. London: Chapman & Hall, 1996.
Claus EB, Risch N, Thompson WD : Genetic analysis of breast cancer in the cancer and steroid hormone study. Am J Hum Genet 1991; 48: 232–242.
Easton DF, Bishop DT, Ford D, Crockford GP : Genetic linkage analysis in familial breast and ovarian cancer: results from 214 families. The Breast Cancer Linkage Consortium. Am J Hum Genet 1993; 52: 678–701.
Aretini P, D'Andrea E, Pasini B et al: Different expressivity of BRCA1 and BRCA2: analysis of 179 Italian pedigrees with identified mutation. Breast Cancer Res Treat 2003; 81: 71–79.
Begg CB : On the use of familial aggregation in population-based case probands for calculating penetrance. J Natl Cancer Inst 2002; 94: 1221–1226.
Pharoah PD, Antoniou A, Hopper J, Easton D : Re: On the use of familial aggregation in population-based case probands for calculating penetrance. J Natl Cancer Inst 2003; 95: 75–76, author reply 77–78.
Heimdal K, Maehle L, Apold J, Pedersen JC, Moller P : The Norwegian founder mutations in BRCA1: high penetrance confirmed in an incident cancer series and differences observed in the risk of ovarian cancer. Eur J Cancer 2003; 39: 2205–2213.
ASCO. Policy Statement: Genetic Testing for Cancer Susceptibility (approved February 20, 1996). Recommendations pertaining to clinical aspects of genetic testing for cancer susceptibility, ASCO: 2002.
Acknowledgements
We wish to thank the reviewers, whose constructive comments were very helpful in preparing the paper for publication. The Italian Consortium for Hereditary Breast and Ovarian Cancer is funded by the Italian Association for Cancer Research (AIRC). This work was in part supported by the National Research Council (CNR), and the Italian Ministry of University and Research (MIUR).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Marroni, F., Aretini, P., D'Andrea, E. et al. Penetrances of breast and ovarian cancer in a large series of families tested for BRCA1/2 mutations. Eur J Hum Genet 12, 899–906 (2004). https://doi.org/10.1038/sj.ejhg.5201256
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.ejhg.5201256
Keywords
This article is cited by
-
Risk reducing salpingectomy and delayed oophorectomy in high risk women: views of cancer geneticists, genetic counsellors and gynaecological oncologists in the UK
Familial Cancer (2015)
-
Novel and recurrent BRCA2 mutations in Italian breast/ovarian cancer families widen the ovarian cancer cluster region boundaries to exons 13 and 14
Breast Cancer Research and Treatment (2014)
-
Molecular and clinical characterization of an in frame deletion of uncertain clinical significance in the BRCA2 gene
Breast Cancer Research and Treatment (2012)
-
The BRCAPRO 5.0 model is a useful tool in genetic counseling and clinical management of male breast cancer cases
European Journal of Human Genetics (2010)
-
Personalized genomic medicine
Internal and Emergency Medicine (2010)
