
Abstract
Idiopathic orbital inflammation (IOI) is a well-recognised pathological process usually confined to the orbit. It is an orbital mass lesion characterised by infiltration of soft tissues by inflammatory cells and fibrous tissue. It is essentially a diagnosis of exclusion. Extraorbital extension (EOE) is rare and, to the best of the authors' knowledge, inclusive of our series, there have been 22 cases of IOI with EOE reported in the literature. We describe four patients with IOI with EOE—their presentation, histopathological findings and management strategies. Histopathology of three patients revealed sclerosing and one had a nonspecific variant of IOI. In our series, all four patients required additional immunosuppression. Hence, corticosteroid therapy often needs to be supplemented by radiotherapy and/or immunosuppressive therapy.
Similar content being viewed by others

Introduction
Idiopathic orbital inflammation (IOI) is usually confined to the orbit. In total, 60% of lacrimal gland-located IOI respond to corticosteroids and up to 80% of extraorbital gland orbital IOI show resolution with this treatment. The first case of IOI was described in 1903 by Busse and Hochheim and later in 1905 by Birch-Hirschfeld, but the earliest report of IOI with intracranial extension was published in 1958 by H Jackson.1 There have been 18 cases of IOI with extraorbital extension (EOE) described in the literature, the majority being sclerosing inflammation.2, 3, 4, 5, 6, 7, 8, 9 We report a further four cases and review the published literature.
Case 1
A 45-year-old Caucasian female developed left retro-orbital pain. She had no visual symptoms and orbital examination was unremarkable. Computerised tomography (CT) scans revealed left orbital mass. Provisional diagnosis of idiopathic orbital inflammation was made and the patient was treated with oral prednisolone 50 mg daily, tapered to 5 mg daily over a 6-week period.
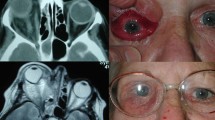
After 6 months, her left-sided headache recurred and was accompanied by facial numbness in the distribution of the maxillary division of left trigeminal nerve. Orbital examination remained unchanged. Magnetic resonance imaging (MRI) scan showed a mass at the lateral wall of left orbit extending into the infratemporal fossa, pterygopalatine fossa, along the lateral cavernous sinus to Meckel's cave, and involving the dura around the temporal lobe (Figure 1, 2). All additional investigations, including an autoimmune screen: ANA, ANCA, ACE, Rheumatoid factor, and lupus antigen, were unremarkable. A transantral biopsy of the pterygopalatine fossa mass was performed. The histopathology revealed fibrous tissue with a patchy infiltrate of lymphoplasmacytic cells with prominent sclerosis and no malignant potential (Figure 3). Immunoperoxidase stains confirmed polyclonality.

Coronal MRI scan showing a mass extending through the inferior orbital fissure and the pterygopalatine fissure into the left infratemporal fossa, lateral wall of the cavernous sinus to Meckel's cave and dura around the temporal lobe (Case 1).

Sagittal MRI scan showing a mass extending through the inferior orbital fissure and the pterygopalatine fissure into the left infratemporal fossa, lateral wall of cavernous sinus to Meckel's cave and dura around the temporal lobe (Case 1).

Transantral biopsy specimen showing fibrous tissue with a patchy infiltrate of lymphoplasmacytic cells (Case 1) (H & E, magnification × 80, courtesy of Professor Newland).
In addition to prednisolone 50 mg daily, the patient required oxycodone hydrochloride 10 mg four times daily to control her pain. Azathioprine 25 mg daily increased to 100 mg daily was added to her treatment regime. The patient's symptoms slowly regressed and she was able to taper and then cease her corticosteroid and opioid therapy. At 1 year after the initial presentation, a repeat MRI scan revealed small orbital lesion, 30% of its original size, and reduced enhancement of its extension to infratemporal fossa (Figure 4, 5). Azathioprine was finally discontinued and the patient remains symptom-free 2 years later.

Coronal MRI scan showing regression of the mass from infratemporal fossa, lateral wall of cavernous sinus and intracranial fossa (Case 1).

Sagittal MRI scan showing regression of the mass from infratemporal fossa, lateral wall of cavernous sinus and intracranial fossa (Case 1).
Case 2
A 30-year-old Chinese woman developed right periorbital pain. On examination her vision was 6/6 in both eyes, with a chemotic and injected right eye. A diagnosis of nodular scleritis was made and she was commenced on oral prednisolone 50 mg daily and hourly topical prednisolone acetate 1% to the right eye. Initial improvement lasted 4 weeks before her symptoms recurred associated with deterioration of vision in the right eye. Examination revealed nodular scleritis, vitritis, superior-temporal uveal effusion and corrected visual acuity of 6/12 in the right eye. There was also a painless 50 mm in diameter, subcutaneous lump on her left arm. Systemic investigations, including full blood count, autoimmune screen, VRDL, Lyme antibodies, and Leishmania antibodies, were negative. Treatment with prednisolone 50 mg daily, sulphamethoxazole 800 mg, trimethoprim 160 mg twice daily, and oxycodone hydrochloride 10 mg 4–6 hourly, was commenced, but with no improvement. Follow-up examination revealed 4 mm of nonaxial proptosis, right upper lid ptosis, and diffuse scleritis. Her visual acuity in the right eye deteriorated to count fingers. MRI scan revealed thickening of the right superior-temporal sclera and an adjacent lacrimal gland mass. Tissue biopsies were obtained from the lacrimal gland and the left arm mass. Histopathological findings of both lesions confirmed a chronic inflammatory process with plasmacytosis and lymphoid follicles. Subsequently, the patient developed right eye paresis. Repeat MRI scan showed an extension of the lesion to the lateral rectus, all around the globe and into the inferior orbital fissure. Further episcleral biopsy confirmed chronic inflammation. Mixed infiltrate with reactive lymphocytes and polymorphs, occasional eosinophils and histiocytic cells, were detected. Fungal and bacterial cultures, along with Gram and PAS stains did not reveal any organisms. Immunoperoxidase and IgH gene rearrangement studies showed polyclonal infiltrate. A diagnosis of idiopathic orbital inflammation with extraorbital extension and ocular involvement was made.
Radiotherapy to the right orbit, total of 30 Gy in 3 Gy fractions, brought gradual symptom relief over a 3-week period. The right visual acuity remained at count fingers only. Follow-up MRI scan revealed moderate decrease in the size of the lacrimal mass with minimal inferior extension. Abnormal tissue around the globe persisted. After 1 month, her pain recurred and she was started on azathioprine 75 mg daily, prednisolone 30 mg daily, and oxycodone hydrochloride 10 mg four times daily. Over a period of 6 weeks she was able to cease her medications and remained pain-free on 6 monthly reviews for 2 years. Her vision remained at count fingers in the right eye and 6/6 in the left. Proptosis regressed, but the right eye did not regain any movement.
Case 3
A 32-year-old Pacific Islander man from New Caledonia developed left retro-orbital pain associated with a rapid deterioration of vision in his left eye. He had a history of right anisometropic amblyopia and the corrected visual acuity in both eyes was 6/60. On examination he had 3 mm of nonaxial proptosis of the left eye with 2 mm of superior displacement. There was no afferent pupillary defect. CT scan demonstrated an orbital floor mass with areas of bony destruction. The mass extended to the pterygopalatine fossa, parasellar region of the left middle cranial fossa, sphenoid sinus and the adjacent medial aspect of the temporal lobe. A biopsy via an antrostomy revealed a chronic inflammatory process with prominent fibrosis, but no malignant cells or infective organisms. Immunophenotyping by flow cytometry showed polyclonal lymphocytes. He was started on intravenous methylprednisolone 500 mg daily for 3 days. His left eye visual acuity improved to 6/9 within 2 weeks. After 1 month, he developed left lateral rectus paresis. Repeated CT scan revealed a significant increase in the mass in the infratemporal fossa, left cavernous sinus, encircling the internal carotid artery without compression. The size of the orbital mass was unchanged. He continued on monthly pulsed methylprednisolone on his visits to Australia. At 6 months following initiation of therapy, the left eye remained paretic with a visual acuity of 6/9, despite the fact that the follow-up CT scan showed further extension into the pituitary fossa and along the free edge of the tentorium. Intravenous cyclophosphamide 1.5 mg and dexamethasone 8 mg every 3 weeks were administered.
His visual acuity remained at 6/9; paresis of left rectus muscle resolved and pain subsided. A repeat CT scan showed 70% regression of the mass size. After 2 months his therapy was tapered and discontinued. He remains symptom-free and clinical signs are unchanged at 2 years follow-up.
Case 4
A 48-year-old Caucasian woman presented with a 4-month history of progressive diplopia associated with right ptosis, mild periorbital swelling, and retrobulbar pain. On examination, she had 4 mm of right nonaxial proptosis with 2 mm of medial globe displacement, vision of 6/12 OD and 6/6 OS with a right relative afferent pupillary defect. There was marked limitation of right abduction, supraduction, and mild limitation of adduction. CT scan demonstrated a homogenously enhancing, infiltrative mass with obliteration of the lateral and superior recti, lacrimal gland, and the lateral and superior intraconal fat. There was extension through the superior orbital fissure into the middle cranial fossa and anterior cavernous sinus. This was associated with erosion of the posterior portion of the greater wing of sphenoid and early erosion of the orbital roof (Figure 6, 7). Biopsy of the mass via an anterior orbitotomy revealed sclerosing orbital inflammation with dense fibrosis and a paucicellular inflammatory infiltrate of lymphocytes and histiocytes. Systemic review by a rheumatologist, in addition to CT scans of the chest and abdomen, failed to reveal the presence of multifocal fibrosclerosis. She was treated with an aggressive immunosuppressive regime of pulsed steroids, cyclosporine, and cyclophosphamide over a 6-month period. There was complete resolution of all orbital symptoms and signs within 4 months and a repeat CT scan at 9 months showed no residual mass. At last follow-up 2 years later, there was no clinical evidence of recurrence.

Axial MRI scan showing large orbital mass extending into the intracranial cavity (Case 4).

Coronal MRI scan showing large orbital mass extending into the intracranial cavity (Case 4).
Discussion
IOI or nonspecific orbital inflammation is a space-occupying orbital lesion characterised by infiltration of soft tissues by inflammatory cells and is essentially a diagnosis of exclusion. There are three main histopathological subtypes of IOI: lymphocytic, granulomatous, and sclerosing. Alternate classifications subdivide IOI according to anatomical location into: myositis, dacryoadenitis, perineuritis, periscleritis, and perivasculitis, anterior, posterior, apical, and diffuse.1
EOE is rare and, to the best of the authors' knowledge, inclusive of our series, there have been 22 cases of IOI with EOE reported in the literature (Table 1). The mean age was 48 (range 16–86) with 12 male and 10 female cases. The most common symptoms were pain (77%), proptosis (68%), decreased visual acuity (59%), and diplopia (22%). Ophthalmoplegia was present in 55% and optic disc changes were noted in 46% of patients. With regard to imaging, all cases demonstrated homogenous infiltrative masses with mild-moderate enhancement on CT and 36% showed bone changes such as erosion, destruction, sclerosis, or remodelling. EOE of the inflammatory process was most commonly through the superior orbital fissure (55%), into the cavernous sinus in 46% and the intracranial space in 22%. Spread through the inferior orbital fissure into the pterygopalatine fossa occurred in 18% and 18% showed involvement of the infratemporal fossa.2, 3, 4, 5, 6, 7, 8, 9
Accurate data on histopathology were available in 14 cases with sclerosing inflammation the most frequently reported entity (13 patients). Although extraorbital involvement is occasionally seen in specific types of orbital inflammation such as Wegener's granulomatosis,10 it is distinctly rare in idiopathic cases with sclerosing inflammation appearing to have a greater propensity to extend outside the confines of the orbit than other subtypes of IOI. This may be a manifestation of the generally progressive nature of the fibrosing process seen in this condition.
Sclerosing inflammation was initially thought to be the sequelae of a long-standing IOI. Recent studies describe it as a specific subtype and a separate entity. It is characterised by a dense, fibrous replacement of orbital tissues with less pronounced inflammatory component.11 It can be associated with systemic sclerosing inflammatory conditions including: retroperitoneal fibrosis, mediastinal fibrosis, Riedel's thyroiditis, and sclerosing cholangitis.12, 13
Case 2 in our series is unusual in both its presentation and histology insofar as an idiopathic nonspecific inflammatory mass without sclerosing features extending outside the orbit has, to our knowledge, not previously been described. However, the skin involvement is somewhat suggestive of an underlying systemic disorder that investigation has thus far been unable to define.
The management of IOI with EOE can be challenging with only 36% of all cases being responsive to systemic corticosteroids alone. Radiotherapy was successful in 23%, and 18% responded to the use of immunosuppressive agents such as azathioprine, cyclophosphamide, and cyclosporine. Surgical debulking was not utilised in any of the reported cases. However, cases unresponsive to maximal medical treatment and showing inexorable intracranial progression have required exenteration.14, 15, 16 Our experience, wherein all four cases required additional immunosuppression in the form of azathioprine, cyclophosphamide and/or cyclosporine for partial or complete disease resolution reflects the previously published reports.
Mean follow-up for all cases was 24 months (range 1.5 months to 25 years) 23% were left with permanent disability, which was most commonly unilateral blindness or other neurologic deficits.
Conclusion
IOI with EOE is a rare condition that may lead to permanent visual disability if treatment is delayed or inadequate. The majority of cases appear to be the sclerosing variant of IOI and may need a more aggressive approach to management than simply steroid treatment alone. Hence, corticosteroid therapy often needs to be supplemented by radiotherapy and/or immunosuppressive therapy.
References
Clifton A, Borgstein R, Moseley I, Kendall B, Shaw P . Intracranial extension of orbital pseudotumour. Clin Radiol 1992; 45: 23–26.
De Jesus O, Inserni J, Gonzalez A, Colon L . Idiopathic orbital inflammation with intracranial extension. Case report. J Neurosurg 1996; 85: 510–513.
Cruz A, Akaishi P, Chahud F, Elias J . Sclerosing inflammation in the orbit and in the pterygopalatine and infratemporal fossae. Ophthalmic Plastic Reconstr Surg 2003; 19 (3): 201–206.
Norman E, Charles C, Turbin R . Bilateral sclerosing orbital pseudotumour with intracranial spread. Arch Ophthalmol 2003; 121 (3): 412–413.
Kaye A, Hahn J, Crociun A, Hanson M, Berlin J, Tubbs R . Intracranial extension of inflammatory pseudotumour of the orbit. Case report. J Neurosurg 1984; 60: 625–629.
Bencherif B, Zouaoui A, Chedid G, Kujas M, Van Effenterre R, Marsault C . Intracranial extension of the idiopathic orbital inflammatory pseudotumour. Am J Neuroradiol 1993; 14: 181–184.
Soares D, Crandon I, Char D, Carpenter R . Orbital pseudotmour with intracranial extension. A case report. West Indian Med J 1998; 47 (2): 68–71.
Noble S, Chandler W, Lloyd R . Intracranial extension of orbital pseudotumor: A case report. Neurosurgery 1986; 18 (6): 798–801.
Frohman L, Kupersmith M, Lnag J, Reede D, Bergeron T, Aleksic S et al. Intracranial extension and bone destruction in orbital pseudotumour. Arch Ophthalmol 1986; 104: 380–384.
Satorre J, Antle M, O'Sullivan R, White V, Nugent K, Rootman J . Orbital lesions with granulomatous inflammation. Can J Ophthalmol 1991; 26 (4): 174–195.
Weissler M, Miller E, Fortune M . Sclerosing orbital pseudotumor: a unique clinicopathologic entity. Ann Otol Rhinol Laryngol 1989; 98: 496–501.
Aylward GW, Sullivan TJ, Garner I, Moseley I, Wright JE . Orbital involvement in multifocal fibrosclerosis. Br J Ophthamol 1995; 79: 246–249.
Richards A, Skalka H, Roberts F, Flint A . Pseudotumor of the orbit and retroperitoneal fibrosis. Arch Ophthalmol 1980; 98: 1617–1620.
Rootman J . Why ‘orbital pseudotumour’ is no longer a useful concept. Br J Ophthalmol 1998; 82: 339–340.
Mombaerts I, Goldschmeding R, Schlingemann RO, Koornneef L . What is orbital pseudotumor? Surv Ophthalmol 1996; 41 (1): 66–78 (Review) (139 refs).
Eagle K, King A, Fisher C, Souhami R . Cyclophosphamide induced remission in relapsed, progressive idiopathic orbital inflammation (‘Pseudotumour’). Clin Oncol 1995; 7: 402–404.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Zborowska, B., Ghabrial, R., Selva, D. et al. Idiopathic orbital inflammation with extraorbital extension: case series and review. Eye 20, 107–113 (2006). https://doi.org/10.1038/sj.eye.6701780
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.eye.6701780
Keywords
This article is cited by
-
Orbital inflammatory pseudotumor: new advances in diagnosis, pathogenesis, and treatment
European Journal of Medical Research (2023)
-
Intracranial extension of orbital inflammatory pseudotumor: a case report and literature review
BMC Neurology (2016)
-
Suspected idiopathic sclerosing orbital inflammation presenting as immunoglobulin G4-related disease: a case report
Journal of Medical Case Reports (2011)
-
Orbital myositis: Diagnosis and management
Current Allergy and Asthma Reports (2009)
-
Orbital inflammatory disease: a diagnostic and therapeutic challenge
Eye (2006)
