
Abstract
Adipose tissue plays a central role in maintaining metabolic homeostasis under normal conditions. Metabolic diseases such as obesity and type 2 diabetes are often accompanied by chronic inflammation and adipose tissue dysfunction. In this study, we observed that endoplasmic reticulum (ER) stress and the inflammatory response occurred in adipose tissue of mice fed a high-fat diet for a period of 16 weeks. After 16 weeks of feeding, ER stress markers increased and chronic inflammation occurred in adipose tissue. We found that ER stress is induced by free fatty acid (FFA)-mediated reactive oxygen species (ROS) generation and up-regulated gene expression of inflammatory cytokines in 3T3-L1 adipocytes. Oral administration to obese mice of chemical chaperons, which alleviate ER stress, improved chronic inflammation in adipose tissue, followed by the suppression of increased body weight and improved insulin signaling. These results indicate that ER stress plays important pathophysiological roles in obesity-induced adipose tissue dysfunction.
Similar content being viewed by others


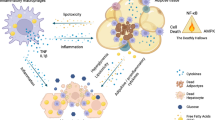
Introduction
The endoplasmic reticulum (ER) is a central cellular organelle in which transmembrane and secretory proteins are synthesized, folded and matured1,2. Various genetic and environmental insults lead to the accumulation of unfolded proteins in the ER lumen, causing ER stress. Excessive ER stress ultimately leads to apoptotic cell death. Eukaryotic cells have a system to mitigate ER stress, known as the unfolded protein response (UPR)3,4,5. Three major transducers of the UPR have been identified: PKR-like ER kinase (PERK), inositol-requiring enzyme 1 (IRE1) and activating transcription factor 6 (ATF6). These factors transmit signals from the ER to the cytoplasm or nucleus and activate three pathways: i) suppression of protein translation to avoid the generation of more unfolded proteins6; ii) induction of genes encoding ER molecular chaperones to facilitate protein folding7,8; and iii) activation of ER-associated degradation (ERAD) to reduce unfolded protein accumulation in the ER9,10. If these strategies fail, the cells are unable to maintain ER homeostasis and undergo apoptosis11,12.
Inflammation is a reaction to host tissue or cell damage caused by stimuli such as infection, chemicals, or physical injury. Inflammation functions as an attempt to eliminate these causal factors and maintain homeostasis. Acute inflammation is a temporary response activated to remove the stimulus and repair the tissue. By contrast, chronic inflammation is a prolonged response that is intended to eliminate the causal factors and/or repair the damaged tissues. Chronic inflammation was proposed as an underlying pathology of various diseases, including heart disease, cancer, inflammatory bowel disease and metabolic diseases13,14,15,16.
Obesity, which is defined as abnormal or excessive fat accumulation in adipose tissues, is a chronic inflammation disease17. Obese adipose tissue is characterized by enhanced hypertrophy and hyperplasia of adipocytes, as well as chronic inflammation involving inflammatory cell infiltration and activation of the cytokine network18,19,20. Although the features of chronic inflammation in obese adipose tissue are clearly defined, the signals and mechanisms that trigger chronic inflammation are not well understood.
It has previously been reported that obese adipose tissue are exposed to stressful conditions, including hypoxia, oxidant stress and ER stress21,22,23. Recently, ob/ob genetic diabetes mice were reported to reveal up-regulation of ER stress markers such as BiP, phosphorylated PERK and phosphorylated α-subunit of eukaryotic translational initiating factor 2 (eIF2α) in adipose tissue and the liver23. Interestingly, several studies have demonstrated that free fatty acids (FFAs), which are elevated in obesity, have the potential to induce ER stress in various cells, including adipocytes24,25. However, little is known about the mechanisms for induction of ER stress by obesity in adipose tissue, or the relationship between ER stress and chronic inflammation. Therefore, we investigated the roles of ER stress and its stress response in adipose tissue of obese mice. In the present study, we showed that FFA-mediated ROS generation causes ER stress in adipocytes. Furthermore, we found that alleviation of ER stress using chemical chaperones suppressed the inflammatory response, including the expression of inflammatory cytokines in adipose tissue and improved insulin signaling.
Results
High-fat diet induced-obesity causes ER stress and chronic inflammation in adipose tissue
To examine whether obesity induces ER stress in adipose tissue, we fed male C57BL/6 mice a high-fat diet for 16 weeks. The mouse model of diet-induced obesity is the most important tool for understanding the development of obesity26. Body weight significantly increased in obese mice throughout the study period compared with control mice fed a regular diet (Figure 1a). We conducted RT-PCR analysis of Bip using mRNA isolated from epididymal adipose tissue at various times after starting the high-fat diet. The expression levels of Bip were slightly up-regulated after 12 weeks and were markedly elevated after 16 weeks of feeding (Figure 1b). We also determined the expression levels of other ER stress markers, including c/EBP-homologous protein (Chop), activating transcription factor 4 (Atf4), ER degradation enhancer mannosidase (Edem), ER DnaJ homolog 4 (Erdj4) and protein disulfide isomerase (Pdi) in adipose tissue of mice fed a high-fat diet for 16 weeks. The expression levels of these ER stress markers were significantly up-regulated in obese mice (Figure 1c). Taken together, these results indicate that the persistence of obesity gradually induces ER stress and is followed by activation of the UPR signaling pathway in adipocytes.

ER stress and chronic inflammation occurs in adipose tissue of mice fed a high-fat diet.
Mice were fed a regular diet or high-fat diet for 16 weeks. A, Body weight. Mice fed a high-fat diet had higher weights compared with mice fed a regular diet. Values are mean ± SD (n = 12). RD, regular diet; HFD, high-fat diet. B, Bip mRNA expression in adipose tissue of mice fed a regular diet or a high-fat diet. Bip mRNA expression increased from 12 weeks in mice fed a high-fat diet. The expression of Bip is shown relative to Gapdh. Values are mean ± SD (n = 4). **p < 0.01 (unpaired Student's t-test). C, mRNA expression levels of ER stress markers in adipose tissue. mRNA expression levels of each ER stress marker were significantly up-regulated in mice fed a high-fat diet for 16 weeks. Values are mean ± SD (n = 4). *p < 0.05 and **p < 0.01 (unpaired Student's t-test). D, mRNA expression levels of inflammatory cytokines in adipose tissue. mRNA expression levels of inflammatory cytokines were significantly up-regulated in mice fed a high-fat diet for 16 weeks. Values are mean ± SD (n = 4). *p < 0.05 and ***p < 0.001 (unpaired Student's t-test). E, H&E staining of adipose tissue. Macrophage infiltration and adipocyte hypertrophy were observed in mice fed a high-fat diet for 16 weeks.
To examine the relationship between obesity-induced ER stress and chronic inflammation, we determined the mRNA expression levels of inflammatory genes in adipose tissue of mice fed a high-fat diet for 16 weeks. The expression levels of Tnf-α, a key inflammatory cytokine, were dramatically increased in obese mice (Figure 1d). The gene expression levels of other inflammatory cytokines, including Ifn-γ, Il-1β, Il-6 and monocyte chemoattractant protein-1 (Mcp-1) were also up-regulated. The gene expression levels of the mouse macrophage-specific marker F4/80 were also significantly elevated. Furthermore, adipocytes from obese mice exhibited hypertrophy and hyperplasia (Figure 1e). Infiltration of macrophages and lymphocytes was also observed in adipose tissue. These findings provide evidence of chronic inflammation occurring in obese adipose tissue, which is a consequence of ER stress.
ROS inhibitors suppress FFA-induced ER stress in 3T3-L1 adipocytes
To investigate the mechanisms for the triggers of obesity-induced ER stress in adipose tissue, in vitro experiments using cultured adipocytes were performed. FFAs were recently reported to cause ER stress in adipocytes25,27,28,29. To confirm this, 3T3-L1 adipocytes were exposed to 50 μg/ml FFA for 4 hours. RT-PCR showed that expression levels of the spliced form of Xbp-1 (sXBP-1) were up-regulated in 3T3-L1 adipocytes treated with FFA (Figure 2a). We then determined the expression levels of ER stress markers, Chop, Atf4, Edem, ERdj4 and Pdi in 3T3-L1 adipocytes treated with FFA and observed that they were significantly increased (Figure 2b). These results confirmed that FFAs induce ER stress in 3T3-L1 adipocytes.

Effects of ROS inhibitors on FFA-induced ER stress in 3T3-L1 adipocytes.
A, Treatment with FFA up-regulated sXbp-1 mRNA in 3T3-L1 adipocytes. 3T3-L1 adipocytes were treated with 50 μg/ml FFA/BSA for 4 hours. uXbp-1, unspliced forms of Xbp-1 mRNA; sXbp-1, spliced forms of Xbp-1 mRNA. B, mRNA expression levels of ER stress markers in 3T3-L1 adipocytes treated with FFA. 3T3-L1 adipocytes were treated with 50 μg/ml FFA/BSA for 4 hours. mRNA expression levels ER stress markers were significantly increased in treatment with FFA. Values are mean ± SD (n = 3). *p < 0.05 and **p < 0.01 (unpaired Student's t-test). C, Pretreatment with the ROS inhibitors NAC or ebselen prevented FFA-induced up-regulation of sXbp-1 mRNA in 3T3-L1 adipocytes. 3T3-L1 adipocytes were treated with 1 mM NAC or 20 μM ebselen for 30 minutes and added to 50 μg/ml FFA/BSA for 4 hours. Pretreatment with NAC and ebselen inhibited the up-regulation of sXBP-1 induced by FFA. D and E, Effects of NAC or ebselen on mRNA expression of the ER stress markers Bip (D) and Chop (E) in 3T3L1 adipocytes. 3T3-L1 adipocytes were treated with 1 mM NAC or 20 μM ebselen for 30 minutes and then treated with 50 μg/ml FFA/BSA for 4 hours. Pretreatment with NAC or ebselen suppressed the up-regulation of Bip and Chop induced by FFA. Values are mean ± SD (n = 3). **p < 0.01 (unpaired Student's t-test).
It has been reported that FFAs induce ROS generation in adipocytes22. To investigate upstream events that potentially lead to induction of ER stress we used ROS inhibitors. To examine whether FFA-induced ROS generation causes ER stress, 3T3-L1 adipocytes were pretreated with the ROS inhibitors NAC or ebselen for 30 minutes and then with FFA for 4 hours. FFA-induced Xbp-1 splicing was completely blocked by NAC and ebselen pretreatment (Figure 2c). Pretreatment with NAC or ebselen also significantly decreased FFA-induced up-regulation of Bip and Chop (Figure 2d, e), indicating that FFA-mediated ROS generation causes ER stress in 3T3-L1 adipocytes.
Chemical chaperones prevent the up-regulation of inflammatory cytokines in 3T3-L1 adipocytes by alleviating ER stress
We then focused on downstream events of ER stress and UPR signaling. Our findings also suggested that ER stress and chronic inflammation occur in adipose tissue of obese mice. Chronic inflammation in adipose tissue plays a crucial role in aggravating the consequences of obesity18. Therefore, to investigate the roles of ER stress in chronic inflammation, we examined the effects of ER stress on the expression of inflammatory cytokines in cultured adipocytes by administering them to chemical chaperones that alleviate ER stress. To achieve this, we stimulated 3T3-L1 adipocytes with 0.6 μg/ml tunicamycin, which causes ER stress by inhibiting N-linked glycosylation. This treatment significantly up-regulated mRNA expression of sXbp-1 in 3T3-L1 adipocytes (Figure 3a), confirming the induction of severe ER stress. Pretreatment with the chemical chaperones 4-PBA or TUDCA30,31 prevented tunicamycin-induced up-regulation of sXbp-1 mRNA expression (Figure 3a). 4-PBA and TUDCA also inhibited tunicamycin-induced up-regulation of Bip and Chop (Figure 3b, c), suggesting that these chemical chaperones reduce ER stress in 3T3-L1 adipocytes.

Effects of chemical chaperones on ER stress and the expression of inflammatory cytokines in 3T3-L1 adipocytes.
A, Pretreatment with the chemical chaperones 4-PBA or TUDCA inhibited tunicamycin-induced up-regulation of sXbp-1 mRNA in 3T3-L1 adipocytes. 3T3-L1 adipocytes were treated with 7.5 mM 4-PBA or 0.5 mg/ml TUDCA for 14 hours and then treated with 0.6 µg/ml tunicamycin for 6 hours. uXbp-1, unspliced forms of Xbp-1 mRNA; sXbp-1, spliced forms of Xbp-1 mRNA. B and C, Effects of 4-PBA or TUDCA on the mRNA expression of the ER stress markers Bip (B) and Chop (C) in 3T3-L1 adipocytes. 3T3-L1 adipocytes were treated with 7.5 mM 4-PBA or 0.5 mg/ml TUDCA for 14 hours and then treated with 0.6 μg/ml tunicamycin for 12 hours. Pretreatment with 4-PBA and TUDCA prevented the up-regulation of Bip and Chop induced by tunicamycin. Values are mean ± SD (n = 3). *p < 0.05 (unpaired Student's t-test). D and E, Effects of 4-PBA or TUDCA on mRNA expression of the inflammatory cytokines Tnf-α (D) and Il-6 (E) in 3T3L1 adipocytes. 3T3-L1 adipocytes were treated with 7.5 mM 4-PBA or 0.5 mg/ml TUDCA for 14 hours and then treated with 0.6 μg/ml tunicamycin for 12 hours. Pretreatment with 4-PBA or TUDCA suppressed the up-regulation of Tnf-α and Il-6 induced by tunicamycin. Values are mean ± SD (n = 3). *p < 0.05 (unpaired Student's t-test).
We then performed RT-PCR to examine whether ER stress influences gene expression of inflammatory cytokines in 3T3-L1 adipocytes. Tunicamycin up-regulated the expression of ER stress markers and the inflammatory cytokines Tnf-α and Il-6 in 3T3-L1 adipocytes (Figure 3d, e). Pretreatment with 4-PBA or TUDCA inhibited ER stress-induced up-regulation of inflammatory cytokine expression (Figure 3d, e). Taken together, these findings indicate that ER stress and UPR signaling directly regulate the expression of inflammatory cytokines in 3T3-L1 adipocytes.
Chemical chaperones suppress the gene expression levels of inflammatory cytokines in adipose tissue of mice fed a high-fat diet
Based on our observations in vitro, we hypothesized that obesity-induced ER stress could contribute to chronic inflammation in adipose tissue in vivo. To examine whether chemical chaperones improve chronic inflammation in adipose tissue of obese mice, obese mice were orally administered 4-PBA or TUDCA for 4 weeks. RT-PCR showed that gene expression of the ER stress markers Bip, Chop, Atf4, Edem, Erdj4 and Pdi was significantly down-regulated in epididymal adipose tissue of mice treated with 4-PBA or TUDCA (Figure 4a), indicating that these compounds could inhibit obesity-induced ER stress in vivo.

ER stress up-regulates the expression of inflammatory cytokines in adipose tissue of mice fed a high-fat diet.
Mice were fed a high-fat diet for 16 weeks and then treated with 4-PBA or TUDCA for 4 weeks at a dose of 1 g/kg/day. A, mRNA expression levels of ER stress markers in adipose tissue. mRNA expression levels of ER stress markers were significantly down-regulated by treatment with 4-PBA or TUDCA. Values are mean ± SD (n = 5). *p < 0.05 (unpaired Student's t-test). B, mRNA expression levels of inflammatory cytokines in adipose tissue. The expression levels of inflammatory cytokines were down-regulated by treatment with 4-PBA or TUDCA. Values are mean ± SD (n = 5). *p < 0.05 (unpaired Student's t-test).
We then examined the effect of chemical chaperones on the expression of inflammatory cytokines in adipose tissue. Treatment with 4-PBA or TUDCA also significantly down-regulated the gene expression levels of the inflammatory cytokines Tnf-α, Ifn-γ, Il-1 β. Il-6 and Mcp-1 in adipose tissue of mice fed a high-fat diet (Figure 4b). These results suggest that chemical chaperones prevent the inflammatory response in adipose tissue by alleviating ER stress.
Alleviation of ER stress improves the symptoms of obesity
We examined the effects of chemical chaperones on the symptoms of obesity. A significant feature of adipocytes from obese mice is hypertrophy of the cell bodies. In mice treated with 4-PBA or TUDCA, the adipocytes were smaller compared with those in vehicle-treated mice (Figure 5a, b). Also, the number of infiltrating macrophages and lymphocytes in adipose tissue decreased (Figure 5a). Additionally, mice treated with 4-PBA or TUDCA exhibited a slight but significant decrease in body weight compared with the control mice (Figure 5c).

ER stress contributes to symptoms of obesity in adipose tissue of mice fed a high-fat diet.
Mice were fed a high-fat diet for 16 weeks and then treated with 4-PBA or TUDCA for 4 weeks at a dose of 1 g/kg/day. A, H&E staining of adipose tissue. Treatment with 4-PBA or TUDCA inhibited macrophage infiltration into adipose tissue and slightly decreased the size of the adipocytes. B, Quantitative analysis of the size of adipocytes treated with 4-PBA or TUDCA. Values are mean ± SD (n = 5). *p < 0.05 (unpaired Student's t-test). C, Effects of 4-PBA or TUDCA on body weights of mice fed a high-fat diet. The body weights of obese mice treated with 4-PBA (n = 7) or TUDCA (n = 8) were significantly lower than those of vehicle-treated obese mice (n = 8). *p < 0.05 (unpaired Student's t-test). D, Western blot analysis of insulin signaling components in adipose tissue. Adipose tissue lysates were subjected to immunoprecipitation followed by immunoblotting. Treatment with 4-PBA or TUDCA down-regulated the expression of phospho-IRS-1 at Ser307 (IRS-1-pS) and up-regulated the expression of phospho-Akt at Ser473 (Akt-pS) (Left panel). Mice were fed regular diet or a high fat diet for 20 weeks (Right panel). E, Quantitative analysis of phospho-IRS-1 and phospho-Akt expression levels in adipose tissue. Values are mean ± SD (n = 5). *p < 0.05 (unpaired Student's t-test).
As obesity-induced inflammation promotes metabolic dysfunction followed by impaired insulin signaling32, we conducted immunoblotting to assess the insulin signaling status in adipose tissue. Mice treated with vehicle showed a significant increase in levels of insulin receptor substrate (IRS)-1 phosphorylated at Ser-307 (IRS-1-pS) (Figure 5d, e). Phosphorylation of IRS-1 at Ser-307 negatively regulates insulin signaling33. The levels of phosphorylated Akt at Ser-473 (Akt-pS), which is the distal event in the insulin signaling pathway34, decreased (Figure 5d, e). Treatment with 4PBA or TUDCA greatly reduced the elevated expression of phospho-IRS-1 (Ser307) and increased the expression of phospho-Akt (Ser473) to normal levels, indicating that chemical chaperones prevented insulin resistance in obese adipose tissue. Taken together, our results suggest that chemical chaperones can improve obesity-induced chronic inflammation followed by metabolic disorders in adipose tissue. We therefore concluded that ER stress plays important pathophysiological roles in obesity-induced adipose tissue dysfunction.
Discussion
In this study, activation of the inflammatory response and metabolic disorders were observed in adipose tissue of mice fed a high-fat diet. These pathophysiologic events were alleviated by treatment with chemical chaperones that can suppress ER stress. Obesity is a risk factor which can lead to lipid metabolism disorders, hypertension, arteriosclerosis and ischemic heart disease; as well as metabolic diseases, including insulin resistance and type 2 diabetes. Although the incidence of obesity has increased worldwide, current pharmacotherapeutic options for treating obesity and obesity-related diseases remain limited and ineffective35. It is therefore necessary to understand the molecular mechanisms in the development of obesity related problems. Our results provide evidence that ER stress and its stress responses in adipose tissue is a key factor in the aggravation of obesity related problems. Therefore, ER stress is a potential therapeutic target for the treatment for obesity.
We showed that high-fat diet-induced obesity causes ER stress and activates UPR signaling in adipose tissue. However, the molecular mechanisms of obesity-induced ER stress in adipocytes are not yet fully understood. The previous report showed that production of ROS increased selectively in adipose tissue of obese mice and elevated levels of FFA caused increase of ROS in cultured adipocytes22 In this study, we also observed FFA-mediated ROS generation in adipocytes. ROS oxidizes nascent proteins and increases malfolded and unfolded proteins in the ER36. Further, ROS acts on calcium channels in the ER membrane, followed by stimulation of calcium release from the ER. Decreased concentration of the total calcium in the ER lumen ultimately impairs protein folding of nascent proteins37. Thus, FFA-mediated ROS generation could induce ER stress in adipose tissue of obese mice. Additionally, it is conceivable that inflammatory response contributes to ER stress in adipose tissue; The inflammatory cytokine TNF-α can induce ROS generation in the cytosol via activation of NADPH oxidase38,39. Another inflammatory cytokine, IL-1β, depletes calcium stores in the ER by up-regulating the expression of inducible nitric oxide synthase (iNOS)40. It is already established that nitric oxide inhibits the ER calcium pump and induces ER stress41. All of these events could act in a coordinated manner and induce ER stress in adipocytes in obese mice.
Mice fed a high-fat diet showed severe inflammation involving increased expression of inflammatory cytokines. The three major UPR signaling pathways (mediated by PERK, IRE1 and ATF6) that are activated by ER stress are known to stimulate the expression of inflammatory cytokines in several cell types42. PERK signaling activates NF-κB, a transcriptional regulator that plays a central role in mediating the responses to inflammatory signaling. The PERK arm of UPR mediates inhibition of protein translation via phosphorylation of eIF2α. Translation of IκBα, the main negative regulator of NF-κB, is known to be inhibited by phosphorylation of eIF2α43. A decrease in the translation of IκBα results in removal of inhibition of NF-κB activity and promotes the translocation of NF-κB from the cytoplasm to the nucleus. Down-regulation of this inhibitory control allows the induction of its downstream inflammatory genes. ATF6 also regulates the NF-κB pathway44. ATF6 translocates to the golgi in response to ER stress, where it is cleaved into an active amino-terminal form. This activated N-terminus ATF6 then translocates to the nucleus. Although little is known about the precise mechanism for how the ATF6 signaling pathway activates NF-κB, ATF6 probably up-regulates the transcription of genes, which activate the NF-κB pathway. The third canonical branch of the UPR is the IRE1 signaling pathway. It was reported that the IRE1 signaling pathway directly activates c-Jun N-terminal kinase (JNK)12,45, an important inflammatory signaling mediator. JNK up-regulates the expression of inflammatory cytokines by activating the activator protein 1 (AP-1) transcription factor complex. In the present study, we demonstrated that ER stress markers were markedly up-regulated and the inflammatory response was successfully suppressed by chemical chaperones that alleviate ER stress in adipose tissue of mice fed a high-fat diet. Inflammation in adipose tissue of high-fat diet-induced obese mice could be caused by the activation of UPR signaling pathways from these ER stress sensors induced by ER stress.
One of the most common physiological consequences of obesity is the development of insulin resistance. A recently report showed that the IRE1–JNK signaling pathway directly inhibits cytoplasmic insulin signaling in ob/ob mice because activated JNK phosphorylates IRS-1 at Ser30723. It is possible that ER stress in adipocytes could disrupt insulin signaling through the activation of IRE1. Our findings showed that alleviation of ER stress improved insulin signaling via down-regulation of phosphorylation of IRS-1 at Ser307 in adipose tissue of mice fed a high-fat diet although it is unclear whether the mechanisms are dependent on suppressing the IRE1–JNK signaling pathway. The inflammatory cytokines secreted from adipocytes and macrophages in obese adipose tissue can also activate JNK pathways46. In addition, inflammatory cytokines can disrupt insulin signaling by interfering with IRS-1–insulin receptor binding and promoting IRS-1 degradation47. The effect of ER stress on insulin signaling requires further comprehensive study to elucidate the detailed mechanisms underlying the development of insulin resistance in obese adipose tissue.
The present study revealed the possibility that inhibition of ER stress may be an effective approach to reduce the risk of obesity and its complications. In other words, ER stress and its stress response (UPR) offer novel drug targets for obesity. However, ER stress has various physiologic roles, including escape from apoptosis in cells with unfolded protein in the ER11,12, regulation of secretory cell differentiation or maturation30,48,49,50 and maintenance of cellular homeostasis51. Consequently, complete elimination of ER stress by agents that prevent ER stress could cause disadvantage for living cells and biological regulation. To develop the agents targeting ER stress in clinical, further studies are now needed to characterize the functional changes in cells dependent on ER stress.
Methods
Antibodies and reagents
Antibodies against Akt, phospho-Akt (Ser473), insulin receptor substrate (IRS)-1 and phospho-IRS-1 (Ser307) were purchased from Cell Signaling Technology. Tauroursodeoxycholic acid (TUDCA), 3-isobutyl-1-methylxanthine (IBMX) and dexamethasone (DEX) were purchased from Wako. Insulin and N-acetylcysteine (NAC) were purchased from Sigma. 4-phenylbutyrate (4-PBA) and ebselen were purchased from Enzo Lifesciences. FFA was purchased from SRL.inc (Tokyo, Japan).
Mouse studies
4-week-old male C57BL/6 (Charles River Laboratory) were housed under a 12-h light/12-h dark cycle and followed free access to regular diet (6% fat; Oriental Yeast) or a high-fat diet (34.9%; Research Diets Inc.) to induce obesity. After 16 weeks on the diet, mice were orally administered with 4-PBA or TUDCA twice-daily in two divided doses (500 mg/kg at 7:30 am and 7:30 pm; total 1 g/kg/day). Mice in the control groups received the same volume of vehicle by oral gavage. The experimental procedures and housing conditions were approved by the Committee of Animal Experimentation, Hiroshima University.
Cell culture and treatment
3T3-L1 preadipocytes were purchased from JCRB (Osaka, Japan) and cultured in DMEM (Invitrogen) supplemented with 10% FBS (Hyclone), 2 mM L-glutamine, 50 U/ml penicillin and 50 µg/ml streptomycin. For experiments with chemical chaperones, 3T3-L1 adipocytes were treated with 7.5 mM 4-PBA or 0.5 mg/ml TUDCA for 14 h, followed by treatment with 0.6 μg/ml tunicamycin for 6 or 12 h. For ROS inhibitor studies, 3T3-L1 adipocytes were pretreated with 1 mM N-acetylcysteine (NAC) or 20 μM ebselen for 30 min and then stimulated with 50 μg/ml FFA in the presence of 5% BSA (Sigma) for 4 h.
Adipocyte differentiation
3T3-L1 preadipocytes were allowed to reach confluence (Day 0) and then cultured with stimulation/differentiation medium consisting of growth medium supplemented with 0.25 mM IBMX, 1 mM dexamethasone and 1 μg/ml insulin. Two days after stimulation, the cells were placed in post-stimulation medium containing DMEM, 10% FBS and 1 μg/ml insulin. The medium was replaced every 2 days for 8 days.
Protein extraction
For protein extraction, epididymal adipose tissue samples were placed in 600 μl of cell extraction buffer containing 25 mM Tris-HCl pH 7.6, 150 mM NaCl, 1% NP-40, 1% sodium deoxycholate, 0.1% SDS and protease inhibitor cocktail (MBL International). After homogenization on ice, the tissue lysates were incubated on ice for 45 min. After centrifugation at 15,000 rpm for 15 min at 4°C, the supernatants were collected. One milligram of total tissue protein was used for immunoprecipitation and immunoblotting.
Immunoprecipitation
Tissue lysates were incubated with anti-IRS-1 antibody on ice for 1.5 h and then rotated with protein G-agarose (Invitrogen) at 4°C for 1.5 h. The agarose beads were washed three times with cold lysis buffer. Next, protein G agarose beads were suspended in SDS sample buffer and incubated at 100°C for 5 min.
Immunoblotting
Tissue lysates or immunoprecipitates were resolved by SDS-PAGE and transferred to an Immobilon-P nylon membrane (Millipore). The membrane was treated with 5% skimmed milk overnight at 4°C and probed with the primary antibodies described above. The antibodies were detected using alkaline phosphatase-conjugated anti-rabbit IgG (Sigma) or anti-mouse IgG (Enzo Lifesciences). The density of each band was quantified using Adobe Photoshop CS software (Adobe Systems Incorporated).
RNA isolation and RT-PCR
Total RNA was isolated from epididymal adipose tissue or 3T3-L1 adipocytes using ISOGEN (Wako). First-strand cDNA was synthesized in a 20 μl reaction volume using random primers (Takara) and Moloney murine leukemia virus reverse transcriptase (Invitrogen). PCR was performed using specific primer sets in a total volume of 20 μl containing 0.5 μM of each primer, 0.15 mM dNTPs, 2 U Taq polymerase and 10× PCR buffer (Agilent). The primer sequences are summarized in Supplementary Information, Table S1. The PCR products were resolved by electrophoresis on a 4.8% acrylamide gel. The density of each band was quantified using Adobe Photoshop CS software.
Histological analysis
Epididymal adipose tissue samples were fixed overnight in 10% neutral buffered formalin. Samples were then dehydrated with ethanol, embedded in paraffin and sectioned (5 μm). H&E staining was performed using standard protocols.
Analysis on cell size of adipocyte
At 20× magnification, five representative images of each slide were captured. We counted the number of adipocytes and the mean cell size of adipocytes was determined by dividing the total area of the image by the number of adipocytes.
Statistical analysis
Statistical comparisons were made using unpaired Student's t-test. Differences between groups were considered statistically significant at values of p < 0.05. All results are presented as means ± standard deviation (SD).
References
Gething, M. J. & Sambrook, J. Protein folding in the cell. Nature 355, 33–45 (1992).
Ellgaard, L., Molinari, M. & Helenius, A. Setting the standards: quality control in the secretory pathway. Science 286, 1882–1888 (1999).
Schröder, M. & Kaufman, R. J. ER stress and the unfolded protein response. Mutat. Res. 569, 29–63 (2005).
Ron, D. Translational control in the endoplasmic reticulum stress response. J. Clin. Invest. 110, 1383–1388 (2002).
Kaufman, R. J. Orchestrating the unfolded protein response in health and disease. J. Clin. Invest. 110, 1389–1398 (2002).
Harding, H. et al. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol. Cell. 6, 1099–1108 (2000).
Li, M. et al. ATF6 as a transcription activator of the endoplasmic reticulum stress element. Thapsigargin stress-induced changes and synergistic interactions with NF-Y and YY1. Mol. Cell. Biol. 20, 5096–5106 (2000).
Yoshida, H., Haze, K., Yanagi, H., Yura, T. & Mori, K. Identification of the cis-acting endoplasmic reticulum stress response element responsible for transcriptional induction of mammalian glucose-regulated proteins. Involvement of basic leucine zipper transcription factors. J. Biol. Chem. 273, 33741–33749 (1998).
Ng, D. T., Spear, E. D. & Walter, P. The unfolded protein response regulates multiple aspects of secretory and membrane protein biogenesis and endoplasmic reticulum quality control. J. Cell Biol. 150, 77–88 (2000).
Travers, K. J. et al. Functional and genomic analyses reveal an essential coordination between the unfolded protein response and ER-associated degradation. Cell 101, 249–258 (2000).
Nakagawa, T. et al. Caspase-12 mediates endoplasmic-reticulum-specific apoptosis & cytotoxicity by amyloid-β. Nature 403, 98–103 (2000).
Urano, F. et al. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science 287, 664–666 (2000).
Manabe, I. Chronic inflammation links cardiovascular metabolic and renal diseases. Circ. J. 75, 2739–2748 (2011).
Coussens, L. M. & Werb, Z. Inflammation and cancer. Nature 420, 860–867 (2002).
Kevin, J. M. & Fiona, P. Intestinal homeostasis and its breakdown in inflammatory bowel disease. Nature 474, 298–306 (2011).
Hotamisligil, G. S. Inflammation and metabolic disorders. Nature 444, 860–867 (2006).
Weisberg, S. P. et al. Obesity is associated with macrophage accumulation in adipose tissue. J. Clin. Invest. 112, 1796–1808 (2003).
Wellen, K. E. & Hotamisligil, G. S. Obesity-induced inflammatory changes in adipose tissue. J. Clin. Invest. 112, 1785–1788 (2003).
Nishimura, S. et al. In vivo imaging in mice reveals local cell dynamics and inflammation in obese adipose tissue. J. Clin. Invest. 118, 710–721 (2008).
Xu, H. et al. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J. Clin. Invest. 112, 1821–1830 (2003).
Hosogai, N. et al. Adipose tissue hypoxia in obesity and its impact on adipocytokine dysregulation. Diabetes 56, 901–911 (2007).
Furukawa, S. et al. I. Increased oxidative stress in obesity and its impact on metabolic syndrome. J. Clin. Invest. 114, 1752–1761 (2004).
Ozcan, U. et al. Endoplasmic reticulum stress links obesity insulin action and type 2 diabetes. Science 306, 457–461 (2004).
Cnop, M., Foufelle, F. & Velloso, L. A. Endoplasmic reticulum stress obesity and diabetes. Trends. Mol. Med. 18, 59–68 (2012).
Jiao, P. et al. FFA-induced adipocyte inflammation and insulin resistance: involvement of ER stress and IKKβ pathways. Obesity (Silver Spring) 19, 483–491 (2011).
Van Heek, M. et al. Diet-induced obese mice develop peripheral, but not central, resistance to leptin. J. Clin. Invest. 99, 385–390 (1997).
Wang, D., Wei, Y. & Pagliassotti, M. J. Saturated fatty acids promote endoplasmic reticulum stress and liver injury in rats with hepatic steatosis. Endocrinology. 147, 943–951 (2006).
Wei, Y., Wang, D. & Pagliassotti, M. J. Saturated fatty acid-mediated endoplasmic reticulum stress and apoptosis are augmented by trans-10, cis-12-conjugated linoleic acid in liver cells. Mol. Cell. Biochem. 303, 105–113 (2007).
Gentile, C. L. & Pagliassotti, M. J. The role of fatty acids in the development and progression of nonalcoholic fatty liver disease. J. Nutr. Biochem. 19, 567–576 (2008).
Basseri, S., Lhoták, S., Sharma, A. M. & Austin, R. C. The chemical chaperone 4-phenylbutyrate inhibits adipogenesis by modulating the unfolded protein response. J. Lipid. Res. 50, 2486–2501 (2009).
Berger, E. & Haller, D. Structure-function analysis of the tertiary bile acid TUDCA for the resolution of endoplasmic reticulum stress in intestinal epithelial cells. Biochem. Biophys. Res. Commun. 409, 610–615 (2011).
Shen SW., Reaven, GM. & Farquhar, JW. Comparison of impedance to insulin-mediated glucose uptake in normal subjects and in subjects with latent diabetes. J. Clin. Invest. 49, 2151–2160 (1970).
Aguirre, V., Uchida, T., Yenush, L., Davis, R. & White, M. F. The c-Jun NH(2)-terminal kinase promotes insulin resistance during association with insulin receptor substrate-1 and phosphorylation of Ser(307). J. Biol. Chem. 275, 9047–9054 (2000).
Inoue, G., Cheatham, B., Emkey, R. & Kahn, C. R. Dynamics of insulin signaling in 3T3-L1 adipocytes. Differential compartmentalization and trafficking of insulin receptor substrate (IRS)-1 and IRS-2. J. Biol. Chem. 273, 11548–11555 (1998).
Cao, Y. Adipose tissue angiogenesis as a therapeutic target for obesity and metabolic diseases. Nat. Rev. Drug. Discov. 9, 107–115 (2010).
Malhotra, J. D. & Kaufman, R. J. Endoplasmic reticulum stress and oxidative stress: a vicious cycle or a double-edged sword? Antioxid. Redox. Signal. 9, 2277–2293 (2007).
Zhang, K. & Kaufman, R. J. From endoplasmic-reticulum stress to the inflammatory response. Nature. 454, 455–462 (2008)
Griendling, K. K., Sorescu, D. & Ushio-Fukai, M. NAD(P)H oxidase: role in cardiovascular biology and disease. Circ. Res. 86, 494–501 (2000).
Xue, X. et al. Tumor necrosis factor alpha (TNFalpha) induces the unfolded protein response (UPR) in a reactive oxygen species (ROS)-dependent fashion and the UPR counteracts ROS accumulation by TNFalpha. J. Biol. Chem. 280, 33917–33925 (2005).
Tsujino, M. et al. Induction of nitric oxide synthase gene by interleukin-1 beta in cultured rat cardiocytes. Circulation 90, 375–383 (1994).
Li, W. W., Alexandre, S., Cao, X. & Lee, A. S. Transactivation of the grp78 promoter by Ca2+ depletion. A comparative analysis with A23187 and the endoplasmic reticulum Ca(2+)-ATPase inhibitor thapsigargin. J. Biol. Chem. 268, 12003–12009 (1993).
Hotamisligil, G. S. Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell 140, 900–917 (2010).
Deng, J. et al. Translational repression mediates activation of nuclear factor kappa B by phosphorylated translation initiation factor 2. Mol. Cell. Biol. 24, 10161–10168 (2004).
Yamazaki, H. et al. Activation of the Akt-NF-kappaB pathway by subtilase cytotoxin through the ATF6 branch of the unfolded protein response. J. Immunol. 183, 1480–1487 (2009).
Hu, P., Han, Z., Couvillon, A. D., Kaufman, R. J. & Exton, J. H. Autocrine tumor necrosis factor alpha links endoplasmic reticulum stress to the membrane death receptor pathway through IRE1alpha-mediated NF-kappaB activation and down-regulation of TRAF2 expression. Mol. Cell. Biol. 26, 3071–3084 (2006).
Kyriakis, J. M. & Avruch, J. Mammalian mitogen-activated protein kinase signal transduction pathways activated by stress and inflammation. Physiol. Rev. 81, 807–869 (2001).
Shoelson, S. E., Lee, J. & Goldfine, A. B. Inflammation and insulin resistance. J. Clin. Invest. 116, 1793–1801 (2006).
Murakami, T. et al. Signalling mediated by the endoplasmic reticulum stress transducer OASIS is involved in bone formation. Nat. Cell. Biol. 11, 1205–1211 (2009).
Asada, R. et al. The endoplasmic reticulum stress transducer OASIS is involved in the terminal differentiation of goblet cells in the large intestine. J. Biol. Chem. 287, 8144–8153 (2012).
Kondo, S., Saito, A., Asada, R., Kanemoto, S. & Imaizumi, K. Physiological unfolded protein response regulated by OASIS family members, transmembrane bZIP transcription factors. IUBMB. Life. 63, 233–239 (2011).
Rutkowski, D. T. & Hegde, R. S. Regulation of basal cellular physiology by the homeostatic unfolded protein response. J. Cell. Biol. 189, 783–794 (2010).
Acknowledgements
We thank M. Harada, S. Nakagawa and K. Takedachi for technical supports. This work was partly supported by grants from the Japan Society for the Promotion of Science KAKENHI (#22020030, #22800049), Sumitomo Foundation, Mochida Memorial Foundation for Medical and Pharmaceutical Research, Astellas Foundation for Research on Metabolic Disorders, Takeda Science Foundation, The Pharmacological Research Foundation Tokyo, Daiichi-Sankyo Foundation of Life Science and The Naito Foundation.
Author information
Authors and Affiliations
Contributions
NK and KI designed the experiments. NK and RA did the experiments in Figures 1, 4, 5. NK, AS and SK did the experiments in Figures 2, 3. NK and KI wrote the manuscript. KI supervised the project. All authors reviewed the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
SUPPLEMENTARY INFORMATION
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareALike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Kawasaki, N., Asada, R., Saito, A. et al. Obesity-induced endoplasmic reticulum stress causes chronic inflammation in adipose tissue. Sci Rep 2, 799 (2012). https://doi.org/10.1038/srep00799
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep00799
This article is cited by
-
Chronic endoplasmic reticulum stress in myotonic dystrophy type 2 promotes autoimmunity via mitochondrial DNA release
Nature Communications (2024)
-
Role of obesity related inflammation in pathogenesis of peripheral artery disease in patients of type 2 diabetes mellitus
Journal of Diabetes & Metabolic Disorders (2023)
-
Panax notoginseng extract and total saponin suppress diet-induced obesity and endoplasmic reticulum stress in epididymal white adipose tissue in mice
Chinese Medicine (2022)
-
Thioredoxin interacting protein protects mice from fasting induced liver steatosis by activating ER stress and its downstream signaling pathways
Scientific Reports (2022)
-
Pro-inflammatory effects of DEHP in SGBS-derived adipocytes and THP-1 macrophages
Scientific Reports (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
