
Abstract
SNPs, abundant in human genome with lower mutation rate, are attractive to genetic application like forensic, anthropological and evolutionary studies. Universal SNPs showing little allelic frequency variation among populations while remaining highly informative for human identification were obtained from previous studies. However, genotyping tools target only dozens of markers simultaneously, limiting their applications. Here, 124 SNPs were simultaneous tested using Ampliseq technology with Ion Torrent PGM platform. Concordance study was performed with 2 reference samples of 9947A and 9948 between NGS and Sanger sequencing. Full concordance were obtained except genotype of rs576261 with 9947A. Parameter of FMAR (%) was introduced for NGS data analysis for the first time, evaluating allelic performance, sensitivity testing and mixture testing. FMAR values for accurate heterozygotes should be range from 50% to 60%, for homozygotes or Y-SNP should be above 90%. SNPs of rs7520386, rs4530059, rs214955, rs1523537, rs2342747, rs576261 and rs12997453 were recognized as poorly performing loci, either with allelic imbalance or with lower coverage. Sensitivity testing demonstrated that with DNA range from 10 ng-0.5 ng, all correct genotypes were obtained. For mixture testing, a clear linear correlation (R2 = 0.9429) between the excepted FMAR and observed FMAR values of mixtures was observed.
Similar content being viewed by others

Introduction
Single nucleotide polymorphisms (SNPs), essentially zero rate of recurrent mutation1, are likely in the near future to have a fundamental role in human identification and description. However, currently available dedicated SNP genotyping tools, usually based on the principle of single-base primer extension using commercially available SnapShot chemistry (Life Technologies, San Francisco, CA), only allow the parallel analysis of up to a few dozen SNPs2,3. Next generation sequencing (NGS), also named Massively parallel sequencing (MPS) and the barcoding system can obtain detailed sequence information of genetic markers and collect massive amounts of data from multiple samples simultaneously4,5,6,7,8. The libraries of targeted markers in this study were amplified via multi-PCR technology (called Ampliseq) and got sequenced and then aligned to human genome (Hg19). Currently, MiSeq (Illumina) and Ion Torrent Personal Genome Machine (Ion Torrent PGM) (Life Technologies), offer modest set-up and running costs for marker detection, are the most commonly applied for forensic and clinic application4,5,8,9. In this study, Ion Torrent PGM which exploits a sensitive semiconductor-based detection of H+ ion release during base incorporation onto short template sequences bound to micro-spheres9,10, was adopted as the studied NGS platform. Ion Torrent PGM is the first commercial sequencing machine that does not require fluorescence and camera scanning, resulting in higher speed, lower cost and smaller instrument size.
Since universal SNPs showing little allelic frequency variation among populations while remaining highly informative for human identification were obtained from previous studies11,12,13, Life Technologies delivered several beta assays. HID_SNP_v1.0 (containing 103 autosomal SNPs and 33 Y-SNPs), the first beta panel, was tested by Budowle et al.14. HID_SNP_ v2.2 (containing 136 autosomal SNPs and 33 Y-chromosome markers), the second beta panel for human identification, was first tested by Morling group15 and then inter-evaluated by six laboratories10. Based on these testing data, 34 upper Y-clade SNPs13 and 90 autosomal SNPs11,12 that have high heterozygosity and a low fixation index (FST), were include in the first commercially available panel named HID-Ion AmpliSeq™ SNP-124. Since no data of this panel has been published yet, we evaluated the panel and explored the application in Chinese HAN population this time.
Materials and Methods
The main experiments were conducted in Forensic Genetics Laboratory of Institute of Forensic Science, Ministry of Justice, P.R. China, which is an accredited laboratory by ISO 17025, in accordance with quality control measures. All the methods were carried out in accordance with the approved guidelines of Institute of Forensic Sciences, Ministry of Justice, P.R. China.
Sample preparation
Control DNA of 9947A (Life Technologies) and 9948 (Promega) were adopted as reference samples. Human blood samples involved for study were collected with the approval of Ethics Committee of Institute of Forensic Sciences, Ministry of Justice, P.R. China. Informed consent was obtained for each participant. DNA was extracted using QIAamp® DNA Blood Mini Kit (Qiagen, Hilden, Germany). The quantity of DNA was determined by Quantifiler Human DNA Quantification Kit (Life Technologies) with 7500 Real-time PCR System (Life Technologies).
Primer Pool for Library Preparation
Primers of 124 universal SNPs (90 autosomal SNPs and 34 upper Y-clade SNPs) were combined in one primer pool. For the 90 auto-SNPs, 43 Ken Kidd SNPs11 and 48 SNPforID12 (with 1 shared SNP) were included. The other 34 Y-SNPs designated the major haplogroups in the Y-chromosome parsimony tree13. The average PCR fragment was 132 bp for the 90 autosomal SNPs and 141 bp for the 34 upper Y-clade SNPs. SNP location information and primer sequences were listed in Supplementary Table S1.
Library Preparation, Quantification and Emulsion PCR (emPCR)
New technology of Ampliseq and according chemistry of Ion AmpliSeq Library Kit 2.0–96 LV (Life Technologies) were applied for library preparation. Ampliseq technology delivers simple and fast library construction for affordable targeted sequencing of genomic regions. The Ion AmpliSeq™ workflow is based on a transformative technology that simplifies ultrahigh-multiplex PCR amplification and library construction. Utilizing low input DNA, this single-tube workflow is as simple as setting up a PCR reaction and can avoid contamination. The library-PCR system contained 4 μL of 5X Ion AmpliSeq HiFi Master Mix and 10 μL of 2X HID-Ion AmpliSeq™ SNP-124 Panel. Except for the sensitivity and mixture testing of the panel, the initial DNA input was 10 ng for each library construction. The library-PCR parameters were as follows: 2 min at 99°, 18 cycles of 15 s at 99° and 4 min at 60° followed by a 10° hold. For sensitivity testing, 21 cycles were used when DNA amount lower than 1 ng in order to get sufficient amplification. The resulting amplicons were treated with 2 μL FuPa reagent (Life Technologies) to partially digest primers. All libraries were barcoded using Ion XpressTM Barcode Adapters (Life Technologies). After the ligation with barcodes, libraries were purified with Agencourt AMPure XP Reagents (Beckman Coulter, Brea, CA). Then qPCR methods with Ion Library Quantitation kit (Life Technologies) was adopted for accurate library quantification.
The accurately quantified and diluted pool library was then used to generate template positive Ion SphereTM Particles (ISP) containing clonally amplified DNA with emPCR technology, which performed on Ion OneTouch2 (OT2) (Life Technologies) by using Ion PGM Template OT2 200 Kit. Quality of emPCR products were evaluated with Ion SphereTM Quality Control Kit (Life Technologies). The optimal amount of library corresponds to the library dilution point that gives percent of template ISPs between 10–30%. The emPCR products were then enriched on the Ion OneTouchTM ES (Life Technologies).
Sequencing and Data analysis
NGS was performed on Ion Torrent PGM (Life Technologies) with Ion PGMTM Sequencing 200 Kit v2. Ion Chip types (314, 316 or 318) varied depending on the sample size. Assuming 80% chip loading and 60% usable chip, 8, 38 and 77 samples can been sequenced on 314, 316 and 318 chip. Raw sequencing data were collected as DAT files which were processed on the Ion Torrent Suite Server (v4.0.2). Signal processing, base-calling and barcode de-convolution were performed with Server v4.0.2. HID_SNP_Genotyper.42 (v4.2), Variant Caller (v4.0-r76860) and Coverage Analysis (v4.0-r77897) plug-ins were adopted for data analysis. The reference genome was Hg19.
Evaluation of HID-Ion AmpliSeq™ SNP-124 Panel
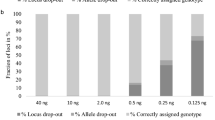
For the concordance and accuracy testing, female sample of 9947A (Life Technology) and male sample of 9948 (Promega) were applied as reference samples. 124 SNPs of the two reference samples were sequenced by NGS and Sanger technologies. Here, Sanger method was adopted for validation of NGS results. For the NGS fragment above 150 bp, same primer pair was used for Sanger sequencing; for the NGS fragment below 150 bp, different primer pair was designed for Sanger sequencing (listed in Supplementary Table S1). For the evaluation of the panel and forensic performance of the 124 SNPs, 45 unrelated healthy Chinese HAN individuals were involved in the study. For the sensitivity testing of the panel, serial dilutions of an In-house male control sample were performed to generate DNA concentrations of 10, 5, 2, 1, 0.5 and 0.2 ng/μL. And 1 μl of each concentration was added in the library PCR-setup system. In other words, the DNA input for sensitivity testing was ranged between 10 ng and 0.2 ng. The libraries of the 6 different concentrations were tested with 314 Ion chip twice. For the mixture study, mixture DNA from control samples of 9947A and 9948 were generated to give ratios of 100:1, 10:1, 5:1, 1:1, 1:5, 1:10 and 1:100. For the 1:1 ratio, 5 ng of each DNA was mixed together. For the ratios of 100:1 and 10:1, 50 pg, 500 pg of 9948 were added to 5 ng of 9947A. For the ratios of 1:10 and 1:100, 500 pg, 50 pg of 9947A were added to 5 ng of 9948. For the 5:1 ratio, 2.5 ng of 9947A was mixed with 500 pg of 9948. Therefore, the DNA input was ranged from 3 ng to 10 ng for library preparation and the 7 libraries of mixtures were tested with 314 Ion chip twice.
Results and Discussion
Concordance study
Control samples of 9947A (Life Technology) and 9948 (Promega) were chosen for concordance study. 124 SNPs (listed in Supplementary Table S1) of these samples were sequenced by NGS and Sanger technologies. HID_SNP_Genotyper.42 (v4.2) plug-in and Chromas were used for the genotyping analysis of NGS data and Sanger sequencing data, respectively. NGS technology has the property of ultra-high throughput but the read length is remarkably short compared to conventional Sanger sequencing. In this study, the shortest PCR length of targeted SNP for NGS is 77 bp and the longest is 244 bp. Thus, for the fragment below 150 bp, different primer pairs were designed for Sanger sequencing (Supplementary Table S1). Sequencing results of 9947A and 9948 by NGS and Sanger sequencing were listed as Supplementary Table S2-1 and Table S2-2, respectively. Except rs576261 (SNP No. 77) of control DNA 9947A (Supplementary Table S2-1), there was complete concordance between the results from NGS and Sanger sequencing of the two reference samples. For SNP rs576261, the NGS results of 9947A was ‘AC’ while the Sanger sequencing result was ‘C’ (Fig. 1). By analyzing BAM file with IGV software, the accurate genotyping at SNP rs576261 of sample 9947A should be ‘C’. The sequence context surrounding SNP rs576261 is TCTGTCACCA[A/C]CCCTGGCCTC. The SNP followed by a homopolymer stretch and a possible allele is identical to the stretch. Misalignment of reads and wrong call of alleles leads to wrong genotyping of NGS (Fig. 1A). And the reads for base A (193) and base C (1422) vary quite significantly.

NGS data analyzed with IGV software (A) and Sanger sequencing data analyzed with Chromas (B) at SNP of rs576261 with control sample 9947A.
A parameter of FMAR (Frequency of Major Allele Reads) was adopted here. Analysis with HID_SNP_Genotyper.42 (v4.2) plug-in can provide detail reads at each bases (A, C, G and T) for each SNP. FMAR was calculated as the biggest reads among the four bases dividing the total detected reads. For homozygotes, the optimal FMAR (%) should be equal to 100, while for heterozygotes, the optimal FMAR (%) should be 50. In previous study, Intra-locus balance (the lower peak height dividing the higher peak height for each locus) was applied to measure the balance of heterozygous alleles. According to Eduardoff et al., 50% is a ‘perfect balance’ and 40% threshold (60:40 heterozygote ratio) can give better equilibrium between gaining the highest proportion of reliable genotypes and balanced signals of SNPs. That means the FMAR (%) for accurate heterozygotes should be 50%–60%. And Intra-locus balance above 70% is desired to ensure accurate heterozygote genotyping and to facilitate mixture interpretation for STRs16. Here, if the same principle adopted for SNP calling, FMAR (%) for accurate heterozygotes should be ranged from 50% to 59% (1/(0.7 + 1)). Therefore, the boundaries of FMAR (%) was set as 50–60% for ideal allelic-balance of heterozygotes in this study. And according to Eduardoff et al., SNPs with major allelic reads frequencies of 90% or greater were deemed to be homozygous for that allele2, as the presence of other bases at a low proportion in the Ion Torrent PGM data arise from non-specific incorporation, but the proportion of a second allele must exceed 10% for Genotyper to call the genotype10. For this reason, when FMAR (%) reach >90%, samples cannot be mistyped as heterozygotes. In other words, FMAR (%) values above 90% were recognized as ideal for homozygotes. For the 34 Y-SNPs, only one allele can been detected for each locus. So the FMAR (%) value above 90% is also essential for accurate calling of Y-SNPs. Analyzing the FMAR (%) values of Supplementary Table S2-1, the average FMAR (%) for the detected 33 heterozygotes (except wrong genotype at rs576261) was 52.16. No values of FMAR (%) for heterozygotes was above 60%. While for the FMAR (%) data of Supplementary Table S2-2, three SNP loci (rs7520386, rs214955 and rs4530059) were detected with FMAR (%) values above 60%. For all the detected homozygotes and Y-SNPs, FMAR (%) values above 90%.
Typing performance of 124 SNPs
Above concordance study demonstrated the accuracy rate of SNP typing by PGM sequencing and also suggested several noticeable SNPs. The genotype calling for heterozygote with FMAR above 60% does not equal to wrong calling. To be fully evaluated the Panel, 10 ng initial DNA of 45 unrelated individuals with barcode ‘1–45′ were sequenced on 318 Ion chip twice for intensive study. Identical genotyping results were obtained from the two times of NGS sequencing except at rs7520386 and rs214955 (Supplementary Table S3). Sanger method was adopted for validation of NGS results also. For the wrong genotyping of rs7520386 of sample ‘78#’, abnormal value of FMAR (79.05%) was detected; and for the wrong genotyping of rs214955 of sample ‘A12_045′, lower coverage (<100) was observed. These suggested that attention should be paid to data with lower coverage or abnormal values of FMAR. By analyze all the NGS data of the 45 individuals, imbalance of heterozygotes were found at SNPs of rs7520386, rs4530059, rs214955, rs1523537, rs2342747 and rs576261 (Supplementary Table S4). Among the 6 SNPs, SNPs of rs7520386 and rs4530059 showed higher imbalance with mean FMAR (%) values above 60%. Except the 6 SNPs, all the detected FMAR (%) values of the 45 individuals were plotted in Fig. 2. The range of FMAR (%) values were 50%–60% for heterozygotes and 90%–100% for homozygotes and Y-SNPs. And a minimum threshold of 100× coverage was recommended in this study.

FMAR (%) values of the 124 SNPs among the 45 individuals except rs7520386, rs4530059, rs214955, rs1523537, rs2342747 and rs576261.
The SNP loci No. can been found in Supplementary Table S2. The top read line indicated the lowest FMAR (%) values for homozygotes (90%) and the bottom read line indicated the highest FMAR (%) values for heterozygotes (60%).
There is consistently high coverage with little variation between the samples. However, variation in coverage was observed among the SNPs and each SNP generally showed similarly high or low coverage across the samples. The lower coverage of auto-SNPs were always happened at SNP rs2342747 and rs12997453, which is also observed when genotyping of 9947A and 9948 (Supplementary Table S2-1 and Supplementary Table S2-2). The differences in coverage may primarily be related to differences in PCR amplification efficiency. Modifications of primer concentrations of SNP rs12997453 (lower coverage but ideal performance of heterozygotes) in the pool may provide more library yield. For the SNPs mentioned in Supplementary Table S4, modifications of the primers and/or primer concentrations may provide more balance and higher yield across the SNPs of the panel. Therefore, NGS results of heterozygote SNP with FMAR (%) values above 60% or total coverage below 100x analyzed with HID_SNP_Genotyper.42 (v4.2) plug-in should be checked or discarded. In this study, the 6 SNPs (rs7520386, rs4530059, rs214955, rs1523537, rs2342747 and rs576261) detected with abnormal values of FMAR and SNP of rs2342747, rs12997453 with lower coverage were recognized as poorly performing SNPs. SNP rs2342747 always detected with abnormal values of FMAR and lower coverage maybe should be deleted from the panel.
In the previous Inter-laboratory evaluation of the HID_SNP_ v2.2 with 169-markers for ancestry inference, discordant genotypes detected in 5 SNPs (rs1979255, rs1004357, rs938283, rs2032597 and rs2399332) indicate these loci should be excluded from the panel10. Two SNPs of them (rs1979255 and rs938283) were also included in this panel. For the 2 SNPs, the genotyping results of tested samples were correct and the FMAR (%) values were in the range for heterozygotes and homozygotes. Therefore, modification of the primers of ‘problematic SNPs’ may effectively improve the performance of new panel.
Sensitivity study
Serial dilutions of 10 ng-0.2 ng of a control male DNA sample were made and dilutions were sequenced on Ion 314 chip. Concordant genotyping results were obtained from all these samples except DNA of 0.2 ng at rs2342747 with ‘N/N’. The total reads at this locus was only 20.
As expected, the allelic balance of heterozygotes varied more in experiments with lower amounts of DNA. The FMAR (%) values of heterozygotes of the 90 auto-SNPs for each dilution were shown in Fig. 3 (except the 7 poorly performing SNPs: rs7520386, rs4530059, rs214955, rs1523537, rs2342747, rs576261 and rs12997453). DNA ranged from 2–10 ng are with optimal FMAR (%) values for heterozygotes. Some terrible FMAR data of heterozygotes were observed when DNA ranged from 0.2–1 ng, especially when DNA below 0.5 ng (Fig. 3). Although NGS data analyzed with correct genotypes by default setting of HID_SNP_Genotyper.42 plug-in for all the called samples, further analysis is essential when data with low coverage or abnormal FMAR values. Above results demonstrated that the optimal amount of DNA in the PCR seemed to be above 0.5 ng, comparable to current STR analysis requirements16,17. It seems likely that the sensitivity can be improved by further optimization of the primer pool or the PCR or by removing some poor performing SNPs from the panel.

FMAR (%) values of observed heterozygotes with different amount of DNA at the 90 auto-SNPs.
7 poorly performing SNPs (rs7520386, rs4530059, rs214955, rs1523537, rs2342747, rs576261 and rs12997453) were excluded for analysis.
Mixture study
In this study, mixtures of two reference samples (9947A and 9948) with ratios of 100:1, 10:1, 5:1, 1:1, 1:5, 1:10 and 1:100 were studied. Table 1 listed the theoretical FMAR values of mixtures with all possible genotypes except the two control samples with same genotypes. Figure 4 shows the theoretical FMAR and observed FMAR values of mixtures with genotypes mentioned in Table 1. 7 poorly performing auto-SNPs (rs7520386, rs4530059, rs214955, rs1523537, rs2342747, rs576261 and rs12997453) were excluded from this analysis. There was a clear linear correlation between the excepted and observed FMA values of all the mixtures (R2 = 0.9429), which indicated that the assay generated a loyal representation of DNA samples. Detection of mixtures with auto-SNPs is possible by analyzing the FMAR values with NGS data. NGS data can give balanced heterozygous genotypes, providing a more secure basis for analyzing mixtures. It is vital to reliably recognize SNP data as originating from a mixture and not a single profile with the commonly used SnapShot system.

Plotting profiles of values of theoretical FMAR and observed FMAR with different ratios of mixtures.
7 poorly performing SNPs (rs7520386, rs4530059, rs214955, rs1523537, rs2342747, rs576261 and rs12997453) were excluded from the analysis. The correction coefficient of R2 is 0.9429.
The genotyping results of 34 Y-SNPs of the mixtures were listed in Table S5. 14.71%, 97.06% and 100% of the Y-SNPs were detected in the 100:1, 10:1 and 5:1 mixture of 9947A/9948. For other mixture ratios, 100% of the Y-SNPs were detected.
Genetic analysis of 124 SNPs in Chinese HAN population
A total of 45 unrelated individuals (17 females and 28 males) of Chinese HAN population were sequenced with the HID-Ion SNP124 panel on Ion 318 chip twice. All the genotypes obtained at the 7 poorly performing SNPs were checked by Sanger sequencing.
Genetic analysis of these 124 SNPs was performed with SNP Analyzer Software18. No significant deviation from HWE expectations was detected in the distribution after Bonferroni correction among HAN population (N = 45) of the 90 auto-SNPs. The allelic frequencies and forensic parameters of the 90 auto-SNPs were listed in Table 2. Based on the data of auto-SNPs investigated among HAN individuals, LD analysis was explored. By pairwise LD calculation and Gabriel’s method18, the results (Supplementary Fig. S1) shows that no LD was existed among the 90 auto-SNPs. Therefore, the CDP (Cumulative Discrimination Power) was 1–5.2192−23 for the 90 auto-SNPs in Chinese HAN. For the Y-SNP analysis, 6 haplotypes were found in the 28 unrelated male individuals. These suggested that the HID-Ion SNP124 panel is suitable for personal identification of HAN population from China.
Conclusion
NGS plus Ampliseq technology have the capacity to sequence targeted regions of multiple DNA samples with high coverage simultaneously. Compared with Sanger sequencing, this technology also can reduce labor and cost on a per nucleotide bases and indeed on a per sample basis. In this study, with the commercially available SNP panel, high coverage and high throughput of 124 specified targets were detected. The parameter of FMAR (%) was applied for evaluating allelic performance, sensitivity testing and mixture testing, making the NGS data easy to interpret. Further modification of the panel can been explored based on the obtained data. This pilot study of the Ion Torrent PGM Sequencer has demonstrated considerable potential for SNP detection as a low to medium throughput NGS platform. And although capillary electrophoresis remains the gold standard and most cost-effective option for human identification with short tandem repeats (STRs)16,17, the PGM Sequencer System extends forensic analysis capabilities. SNPs regarding the bio-geographical ancestry (BGA) or externally visible characteristics (EVC) or STRs were explored with PGM also10,15. These features make markers typing on a NGS platform particularly appealing.
Additional Information
How to cite this article: Zhang, S. et al. Parallel Analysis of 124 Universal SNPs for Human Identification by Targeted Semiconductor Sequencing. Sci. Rep. 5, 18683; doi: 10.1038/srep18683 (2015).
References
Reich, D. E. et al. Human genome sequence variation and the influence of gene history, mutation and recombination. Nat Genet. 32, 135–42 (2002).
Daniel, R. et al. A SNaPshot of next generation sequencing for forensic SNP analysis. Forensic Sci Int Genet. 14, 50–60 (2015).
Ralf, A., van, O. M., Zhong, K. & Kayser, M. Simultaneous analysis of hundreds of Y-chromosomal SNPs for high-resolution paternal lineage classification using targeted semiconductor sequencing. Hum Mutat. 36, 151–9 (2015).
Flicek, P. & Birney, E. Sense from sequence reads: methods for alignment and assembly. Nature methods. 6, S6–S12 (2009).
Andersen, J. D. et al. Next-generation sequencing of multiple individuals per barcoded library by deconvolution of sequenced amplicons using endonuclease fragment analysis. Biotechniques. 57, 91–94 (2014).
Allegue, C. et al. Genetic Analysis of Arrhythmogenic Diseases in the Era of NGS: The Complexity of Clinical Decision-Making in Brugada Syndrome. PLoS One. 10, e0133037 (2015).
Børsting, C. & Morling, N. Next generation sequencing and its applications in forensic genetics. Forensic Sci Int Genet. 15, S1872–4973 (2015).
Geppert, M. et al. Identification of new SNPs in native South American populations by resequencing the Y chromosome. Forensic Sci Int Genet. 15, 111–114 (2015).
Merriman, B., Ion Torrent R & D Team & Rothberg, J. M. Progress in Ion Torrent semiconductor chip based sequencing. Electrophoresis. 33, 3397–3417 (2012).
Eduardoff, M. et al. Inter-laboratory evaluation of SNP-based forensic identification by massively parallel sequencing using the Ion PGMTM. Forensic Sci Int Genet. 17, 110–121 (2015).
Kidd, K. K. et al. Developing a SNP panel for forensic identification of individuals. Forensic Sci Int. 164, 20–32 (2006).
Musgrave-Brown, E. et al. Forensic validation of the SNPforID 52-plex assay. Forensic Sci Int Genet. 1, 186–90 (2007).
Karafet, T. M. et al. New binary polymorphisms reshape and increase resolution of the human Y chromosomal haplogroup tree. Genome Res. 18, 830–838 (2008).
Seo, S. B. et al. Single nucleotide polymorphism typing with massively parallel sequencing for human identification. Int J Legal Med. 127, 1079–1086 (2013).
Børsting, C., Fordyce, S. L., Olofsson, J., Mogensen, H. S. & Morling, N. Evaluation of the Ion Torrent™ HID SNP 169-plex: A SNP typing assay developed for human identification by second generation sequencing. Forensic Sci Int Genet. 12, 144–154 (2014).
Collins, P. J. et al. Developmental Validation of a Single-Tube Amplification of the 13 CODIS STR Loci, D2S1338, D19S433 and Amelogenin: The AmpFl STR Identifiler PCR Amplification Kit. J Forensic Sci. 49, 1265–77 (2004).
Mönich, U. J. et al. Probabilistic characterisation of baseline noise in STR profiles. Forensic Sci Int Genet. 19, 107–122 (2015).
Yoo, J., Lee, Y., Kim, Y., Rha, S. Y. & Kim, Y. SNPAnalyzer 2.0: a web-based integrated workbench for linkage disequilibrium analysis and association analysis. BMC Bioinformatics. 9, 290 (2008).
Acknowledgements
The authors gratefully acknowledge the technical support of Life Technologies. This study was supported by grants from the National Natural Science Foundation of China (No. 81330073, No. 81302620, No. 81222041) and National Key Technology Research & Development Program of the Ministry of Science and Technology of People’s Republic of China (2012BAK16B01) and the funders had no role in study design, data analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
S.Z. and Y.B. wrote the manuscript, Y.B., L.Z. and Z.W. conducted the experiment, S.Z., J.C., Y.G. and C.J. analyzed the results and modified the manuscript, Z.Z. and H.Z. provide the technical support, C.L. and Y.H. conceived the experiment. All authors reviewed the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Zhang, S., Bian, Y., Zhang, Z. et al. Parallel Analysis of 124 Universal SNPs for Human Identification by Targeted Semiconductor Sequencing. Sci Rep 5, 18683 (2015). https://doi.org/10.1038/srep18683
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep18683
This article is cited by
-
Comparison of three massively parallel sequencing platforms for single nucleotide polymorphism (SNP) genotyping in forensic genetics
International Journal of Legal Medicine (2023)
-
Forensic genetic analysis of single-nucleotide polymorphisms and microhaplotypes in Koreans through next-generation sequencing using precision ID identity panel
Genes & Genomics (2023)
-
Forensic nanopore sequencing of STRs and SNPs using Verogen’s ForenSeq DNA Signature Prep Kit and MinION
International Journal of Legal Medicine (2021)
-
A new strategy to confirm the identity of tumour tissues using single-nucleotide polymorphisms and next-generation sequencing
International Journal of Legal Medicine (2020)
-
A SNP panel for identification of DNA and RNA specimens
BMC Genomics (2018)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
