
Abstract
HCV genetic diversity is high and impacts disease progression, treatment and drug resistance. HCV subtype 1a is divided in two clades (I and II), and the 80 K natural polymorphism in the viral NS3 protease is prevalent in clade I. Paradoxically, countries dominated by this clade have contrasting frequencies of 80 K. Over 2,000 HCV 1a NS3 sequences were retrieved from public databases representing Europe, Oceania and the Americas. Sequences were aligned with HCV reference sequences and subjected to phylogenetic analysis to investigate the relative presence of different subtype 1a clades and NS3 protease mutations. HCV-1a sequences split into clades I and II. Clade I was further structured into three subclades, IA to C. Sub-clade IA prevailed in the U.S., while subclade IC was major in Brazil. The NS3 80 K polymorphism was associated with subclade IA, but nearly absent in subclades IB and IC, a pattern similarly seen for the 91S/T compensatory mutation. Three HCV-1a-I sub-clades have been identified, with different frequencies in distinct regions. The 80 K and 91A/S mutations were associated with subclade IA, which provide an explanation for the disparities seen in simeprevir resistance profiles of countries dominated by HCV 1a-I, like the U.S. and Brazil.
Similar content being viewed by others

Introduction
Since its discovery in 1989 as the causative agent of non-A non-B hepatitis, the hepatitis C virus (HCV), a member of Flaviviridae family, accounts for the infection of approximately 1.6% of the human population worldwide1. This virus is characterized by a great genetic diversity that permits its classification into seven genetically distinct genotypes – gen - (1 through 7) and 67 subtypes (a-x)2. In adults, HCV gen1 represents almost half (46%) of the total infections, followed by gen3 (22%), 2 and 4 (13% each)1.
HCV genetic variability is known to impact disease progression, cancer development, treatment and acquisition of drug resistance. Patients infected with HCV gen1 and 4 have the lowest rates of sustained virological response (SVR) to traditional pegylated interferon and ribavirin when compared to gen2, 3, 5 and 63,4, and to overcome that limitation a new generation of direct-acting antivirals like the NS3 protease inhibitors have been developed5. Within HCV gen1, subtype 1b has a higher genetic barrier to develop NS3 protease inhibitor (PI) resistance than subtype 1a and, consequently, responds better to PI-based therapy6. In 2013, the new PI simeprevir was approved for treating HCV gen 1 and 4 infections. Again, the success therapeutic response of subtype 1a was lower than that of subtype 1b, a phenomenon attributed to the presence of the NS3 polymorphism 80 K in the former7.
Recent evidence suggests that HCV subtype 1a can be classified into two distinct genetic clades (I and II) with a non-homogenous geographic distribution8,9,10. Both clades seem to co-circulate at approximately similar prevalence in European countries, while clade I accounts for nearly 75% of the circulating HCV 1a strains in the United States (US)11 and >95% of the strains from Brazil9,12. A disparate prevalence of the NS3 polymorphism 80 K was also observed across different HCV subtype 1a clades. The 80 K polymorphism is present in around 50% of clade I isolates, but is very low prevalent (<3%) in clade II9. This may explain the disproportionate occurrence of 80 K in the US when compared to Europe and to other regions of the world11. In fact, the 80 K polymorphism has been recently suggested to have arisen in the US13.
The association between the HCV subtype 1a clade I and 80 K seems, however, to be more complex than originally envisaged. Despite HCV 1a strains from Brazil are mostly classified within clade I, a very low prevalence of 80 K polymorphism (<7%) has been described in the country9,14. These apparently conflicting results prompted us to investigate in further detail the amino acid composition of HCV subtype 1a clades worldwide. In the present work, we investigated the phylogenetic relationships of worldwide HCV subtype 1a NS3 sequences, and assessed their association with the 80 K polymorphism, as well as other underlying amino acid substitutions that have been reported as related to the former13.
Results
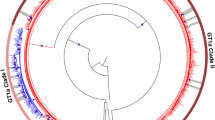
The nucleotide sequences of NS3 protease from worldwide HCV subtype 1a strains split into clades I and II, as expected (Fig. 1A). Interestingly, we found that clade I was further structured into three well-supported sub-clades that were named clades IA, IB and IC. Sequences from the US branched basal to both clades I and II, as well to sub-clades IA, IB and IC (Fig. 1A). Basal to clade I, a few US and European sequences did not form a specific sub-clade, but are clearly suggestive of being ancestral to the three sub-clades currently observed.

(A) Maximum likelihood phylogeny of HCV subtype 1a NS3 gene sequences isolated worldwide showing the geographic distribution of HCV 1a clades and sub-clades. Branches are colored according to the geographic origin of sequences as indicated in the legend (upper right). Arcs indicate the positions of HCV 1a clades and sub-clades. Asterisks point to key nodes with high support (aLRT ≥ 0.90). The tree was rooted at the midpoint. (B) Charts depicting the frequency of HCV 1a clades and sub-clades in the United States of America (US), Europe, Australia/New Zeland (AU/NZ) and Brazil (BR).
The regional distribution of HCV subtype 1a clades was widely heterogeneous with a clear predominance of clade II in Europe (67%), clade I in the US (73%) and in Brazil (96%), and a comparable frequency of both clades in Oceania (Australia / New Zealand) (Fig. 1B). With respect to clade I sub-clades, the geographic discrepancy was even higher, with clade IA being prevalent in the US (65%) and clade IC in Brazil (96%). Interestingly, clade IB was only found in the US, the only country were all three clade I sub-clades coexist.
The newly found HCV 1a clade I sub-clades also displayed disparate occurrence of natural NS3 polymorphisms related to simeprevir resistance. The proportion of the major resistance mutation 80 K was 64% in clade IA, 0% in clade IB and 3% in clade IC (Table 1). In addition, the presence of this mutation in clade II was uncommon (<1%). The compensatory mutation 91S/T shared similar proportions: the highest in sub-clade IA (59%) and lower in the remaining clade I sub-clades and in clade II (0–8%) (Table 1). However, the compensatory mutation 174N was more prevalent in all three clade I sub-clades I (75–92%) than in clade II (14%) (Table 1).
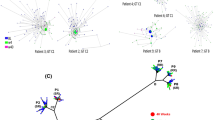
When analyzing the appearance of 80 K in the clade I phylogeny, we observed that some basal sequences to this clade harbored that mutation, as well as its occasional emergence in sub-clade IC (Fig. 2). The emergence of 80 K was correlated with basal sequences of sub-clade IA, with some reversions to Q across the phylogeny. The same pattern was observed for the emergence of 91S/T (Fig. 2). The 174N mutation appeared early in all three clade I sub-clades, with some rare amino acid changes to serine or others, while it was rarely found in clade II sequences (Fig. 2).

Arcs indicate the positions of HCV 1a clades and sub-clades. The trees were rooted at the midpoint.
Discussion
The subdivision of subtype 1a into two distinct genetic clades has been previously shown8. In the present study, we show for the first time the subdivision of clade 1a-I into three well-structured sub-clades (A, B and C). While previous studies have used similar phylogenetic approaches to study the relationships between HCV 1a sequences 8,9,10, they have failed to evidence a substructure within clade subtype 1a clade I, likely due to a limited number10 or representativeness8,9 of sequences from worldwide locations. That was also likely the reason by which a more recent study11 has not pinpointed such structure. A previous study with Brazilian HCV NS5A sequences from treatment-naïve subjects evidenced a monophyletic cluster within subtype 1a clade I15. Upon the analyses conducted herein, we infer those sequences as belonging to subtype 1a sub-clade IC, corresponding to 96% of all Brazilian subjects infected with subtype 1a from whom sequence information is available.
The majority of HCV subtype 1a sequences from the US and Brazil fell within clade I, while sequences from Oceania belonged to both clades I and II at roughly similar proportion, as described previously11,12. Clade II represented 67% of European subtype 1a sequences while clade I enclosed 27% of US sequences, frequencies also similar to those previously reported11. Some sequences from US and Europe were basal even to the split of the three observed clade I sub-clades, representing the likely ancestors to the whole clade I and corroborating the hypothesis postulated by De Luca et al.11. However, in our study, US and European sequences were also placed basal to clade II, indicating again a mixed origin of clade II, in contrast to the previous work that showed an unique European origin for that clade11.
Several studies suggested previously that the HCV NS3 polymorphism 80 K, which confers resistance to the NS3 inhibitor simeprevir16, is heterogeneously distributed within HCV genotype 1, present in 1–43% of HCV subtype 1a and 0–6% in subtype 1b depending of the geographic region analyzed14,17,18. It was been recently proposed that 80 K emerged in the US around 1940–1955 and that sequences harboring that polymorphism grouped together in a monophyletic clade13. Herein we classified most of HCV 1a sequences harboring the 80 K mutation within clade IA, while in other clades its prevalence is very low (3% in sub-clade IC, 0% in sub-clade IB and <1% in clade II). In our analyses, 64% of sub-clade IA sequences harbor 80 K, including some sequences branching at the root of that sub-clade. Therefore, our data revealed that the 80 K mutation emerged multiple times during the radiation of HCV subtype 1a clades and sub-clades, including one correlated with the genesis of sub-clade IA. The 80 K mutation was probably fixed at the base of sub-clade IA with further genetic reversion to 80Q in a number of viral isolates.
The NS3 A91S/T and S174N polymorphisms were strongly associated with 80 K13. In our study, A91S/T appeared in a similar proportion to that of 80 K in sub-clade IA (59% and 64%, respectively), while it was rare in other clade I sub-clades and in clade II (0–8%). The co-occurrence of A91S/T and 80 K did not differ from a random association (data not shown). On the other hand, 174N was found in high proportion in all clade I sub-clades (75–92%), but less frequently in clade II (14%). In this case, an association between 174N and 80 K would be obvious in clade IA, where the majority of the sequences harbor both polymorphisms, but not in clades IB and IC, with high frequency of 174N but no 80 K. Our data suggest that these mutations are not necessarily biologically associated with 80 K, but further association studies are necessary to establish the co-evolutionary patterns between these two polymorphisms and 80 K in HCV NS3.
Patients infected with HCV subtype 1a carrying 80 K present a reduced sustained virological response compared to those not carrying the polymorphism when treated with simeprevir-containing regimens7. Because of that, American and European guidelines for treatment of HCV infection currently indicate genotyping of patients infected with subtype 1a for 80 K detection prior to simeprevir usage19,20. Recently, 80 K was shown to correlate with HCV subtype 1a clade I strains11. Now we show that the majority of 80 K-harboring strains are classified into sub-clade IA, an observation that is congruent with the heterogeneity in the prevalence of 80 K in different parts of the world (1.3% in Brazil; 43% in the US and approximately 14% in Europe)14. Despite all these areas are dominated by clade I strains, the prevalence of sub-clade IA is null in Brazil, while it accounts for 65% and 24% in the US and Europe, respectively.
In conclusion, we showed that HCV subtype 1a clade I is further structured into three definite sub-clades (IA, IB and IC) with sequences geographically segregated, suggesting a founder effect in some cases, like sub-clade IC in Brazil and Oceania. We also show that the presence of the NS3 80 K variation is biased in specific sub-clades, being found majorly in sub-clade IA sequences. These data may improve induced treatment access with simeprevir in areas where subtype 1a sub-clades IB and IC prevail without the need for previous virus genotyping assessment.
Methods
Sequences
A total of 2,185 NS3 protease sequences from treatment-naïve patients infected with HCV subtype 1a of different countries has been described previously14 and used in the present study. To obtain reliable phylogenetic relationships among sequences, only those with the complete HCV NS3 protease sequence were selected and used in the herein analyses, totaling 1,140 sequences: 363 from Europe, 573 from North America (US), 110 from South America (Brazil) and 94 from Oceania. Sequences from Africa and Asia were limited in number and were excluded from the analyses. Sequence alignment was performed with ClustalX v.2.0 and the BioEdit platform v.7.0.5.3 was used for editing and sequence analysis21. References sequences of HCV subtype 1a of clades I and II were extracted from Pickett et al.8.
Phylogenetic analysis
To investigate the relative prevalence of different HCV subtype 1a clades and protease mutations across different geographic regions we performed a Maximum Likelihood (ML) phylogenetic analysis of HCV 1a NS3 sequences. The ML phylogenetic tree was inferred using PhyML program22,23, employing the GTR+I+G nucleotide substitution model selected by the Akaike information criterion implemented in jModelTest24, the subtree pruning and regrafting (SPR) branch swapping algorithm of tree search, and the approximate likelihood ratio test (aLRT) of statistical support for individual nodes.
Additional Information
How to cite this article: Santos, A. F. et al. In-depth phylogenetic analysis of hepatitis C virus subtype 1a and occurrence of 80K and associated polymorphisms in the NS3 protease. Sci. Rep. 6, 31780; doi: 10.1038/srep31780 (2016).
References
Gower, E., Estes, C., Blach, S., Razavi-Shearer, K. & Razavi, H. Global epidemiology and genotype distribution of the hepatitis C virus infection. J. Hepatol. 61, S45–S57 (2014).
Smith, D. B. et al. Expanded classification of hepatitis C virus into 7 genotypes and 67 subtypes: updated criteria and genotype assignment web resource. Hepatology 59, 318–327 (2014).
Papastergiou, V. & Karatapanis, S. Current status and emerging challenges in the treatment of hepatitis C virus genotypes 4 to 6. World J Clin Cases 3, 210–220 (2015).
Alexopoulou, A. & Karayiannis, P. Interferon-based combination treatment for chronic hepatitis C in the era of direct acting antivirals. Ann. Gastroenterol. 28, 55–65 (2015).
De Luca, A., Bianco, C. & Rossetti, B. Treatment of HCV infection with the novel NS3/4A protease inhibitors. Curr. Opin. Pharmacol. 8, 9–17 (2014).
Wyles, D. L. & Gutierrez, J. A. Importance of HCV genotype 1 subtypes for drug resistance and response to therapy. J. Viral Hepat. 21, 229–240 (2014).
Forns, X. et al. Simeprevir with peginterferon and ribavirin leads to high rates of SVR in patients with HCV genotype 1 who relapsed after previous therapy: a phase 3 trial. Gastroenterology 146, 1669–79 e3 (2014).
Pickett, B. E., Striker, R. & Lefkowitz, E. J. Evidence for separation of HCV subtype 1a into two distinct clades. J. Viral Hepat. 18, 608–618 (2011).
Peres-da-Silva, A., Almeida, A. J. & Lampe, E. Genetic diversity of NS3 protease from Brazilian HCV isolates and possible implications for therapy with direct-acting antiviral drugs. Mem. Inst. Oswaldo Cruz 107, 254–261 (2012).
Vicenti, I. et al. Naturally occurring hepatitis C virus (HCV) NS3/4A protease inhibitor resistance-related mutations in HCV genotype 1-infected subjects in Italy. J. Antimicrob. Chemother. 67, 984–987 (2012).
De Luca, A. et al. Two Distinct Hepatitis C Virus Genotype 1a Clades Have Different Geographical Distribution and Association With Natural Resistance to NS3 Protease Inhibitors. Open Forum Infect. Dis. 2, ofv043 (2015).
Lampe, E. et al. Genetic diversity of HCV in Brazil. Antivir. Ther. 18, 435–444 (2013).
McCloskey, R. M. et al. Global origin and transmission of hepatitis C virus nonstructural protein 3 Q80 K polymorphism. J. Infect. Dis. 211, 1288–1295 (2015).
Vidal, L. L., Santos, A. F. & Soares, M. A. Worldwide distribution of the NS3 gene 80 K polymorphism among circulating hepatitis C genotype 1 viruses: implication for simeprevir usage. J. Antimicrob. Chemother. 70, 2024–2027 (2015).
Peres-da-Silva, A., de Almeida, A. J. & Lampe, E. NS5A inhibitor resistance-associated polymorphisms in Brazilian treatment-naive patients infected with genotype 1 hepatitis C virus. J. Antimicrob. Chemother. 70, 726–730 (2015).
Lenz, O. et al. In vitro resistance profile of the hepatitis C virus NS3/4A protease inhibitor TMC435. Antimicrob. Agents Chemother. 54, 1878–1887 (2010).
Lisboa-Neto, G. et al. Resistance mutations are rare among protease inhibitor treatment-naive hepatitis C genotype-1 patients with or without HIV coinfection. Antivir. Ther. 20, 281–287 (2015).
Morel, V., Duverlie, G. & Brochot, E. Patients eligible for treatment with simeprevir in a French center. J. Clin. Virol. 61, 149–151 (2014).
United States Food and Drug Administration. Olysio (Simeprevir) for the Treatment of Chronic Hepatitis C in Combination Antiviral Treatment. (2014), Available at: http://www.fda.gov/forpatients/illness/hepatitisbc/ucm377234.htm. (Accessed: 4th January 2016).
European Medical Association. Olysio—Simeprevir. (2014) Available at: http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/human/medicines/002777/human_med_001766.jsp&mid=WC0b01ac058001d124. (Accessed: 4th January 2016).
Larkin, M. A. et al. Clustal W and Clustal X version 2.0. Bioinformatics 23, 2947–2948 (2007).
Guindon, S. & Gascuel, O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst. Biol. 52, 696–704 (2003).
Guindon, S., Lethiec, F., Duroux, P. & Gascuel, O. PHYML Online–a web server for fast maximum likelihood-based phylogenetic inference. Nucleic Acids Res. 33, W557–W579 (2005).
Posada, D. jModelTest: phylogenetic model averaging. Mol. Biol. Evol. 25, 1253–1256 (2008).
Acknowledgements
This work has been funded by the Brazilian Ministry of Heath and the United Nations Organization for Drugs and Crime (CA 114/2014 to M.A.S.) and by Rio de Janeiro State Science Foundations – FAPERJ (E-26/112.647/2012 to M.A.S.). L.L.V. and D.M. are recipients of PhD fellowships from the Brazilian Ministry of Education – CAPES. D.M. is also funded by a PhD fellowship from “Agencia Nacional de Investigación e Innovación – ANII”, Uruguay.
Author information
Authors and Affiliations
Contributions
A.F.S., G.B. and M.A.S. conceived the study. L.L.V., S.L.S. and D.M. retrieved sequences and performed the analyses. A.F.S. and G.B. oversaw and compiled the analyses. A.F.S., G.B. and M.A.S. wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Santos, A., Bello, G., Vidal, L. et al. In-depth phylogenetic analysis of hepatitis C virus subtype 1a and occurrence of 80K and associated polymorphisms in the NS3 protease. Sci Rep 6, 31780 (2016). https://doi.org/10.1038/srep31780
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep31780
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
