
Abstract
Bipolar disorder (BD) and alcohol dependence (AD) frequently co-occur, and co-occurring BD and AD are associated with devastating public health costs. Minimal neurobiological research exists to guide the development of effective treatments for this treatment-resistant population. We believe the present study represents the first investigation of prefrontal gamma-aminobutyric acid (GABA) and glutamate levels in co-occurring BD and current AD. The participants were 78 individuals who met DSM-IV criteria for BD I/II and current AD (n=20), BD I/II alone (n=19), current AD alone (n=20) or no diagnosis (n=19). The participants completed a baseline diagnostic visit, then returned approximately 4 days later for a two-dimensional J-resolved proton magnetic resonance spectroscopy (1H-MRS) acquisition in dorsal anterior cingulate cortex (dACC). All participants were required to demonstrate ⩾1 week of abstinence from alcohol/drugs via serial biomarker testing before 1H-MRS. A 2 × 2 factorial analysis of variance of cerebrospinal fluid (CSF)-corrected GABA/water concentrations demonstrated a significant BD × AD interaction (F=2.91, P<0.05), signifying uniquely low levels of GABA in BD+AD; this effect doubled when the sample was restricted to individuals who consumed alcohol within 2 weeks of 1H-MRS. There were no overall effects of BD/AD on CSF-corrected glutamate/water levels. However, the BD × AD interaction, signifying uniquely low levels of glutamate in BD+AD, approached statistical significance (F=3.83, P=0.06) in individuals who consumed alcohol within 2 weeks of 1H-MRS. The dACC GABA levels were significantly, negatively associated with Barratt Impulsiveness Scale (r=−0.28, P=0.02) and Obsessive Compulsive Drinking Scale (r=−0.35, P<0.01) scores. If replicated, these results may suggest that future treatment studies should preferentially evaluate therapeutics in BD+AD known to increase prefrontal GABA and glutamate levels.
Similar content being viewed by others
Introduction
Individuals with bipolar disorders (BD) have disproportionately high rates of alcohol dependence,1, 2 and the combination of BD and AD (BD+AD) is associated with substantially worse outcomes than either disorder alone, including poorer clinical course,3 treatment resistance,4 suicidality,5 violent crime,6 disability7 and hospitalization.8 Unfortunately, treatment research for co-occurring BD and AD is remarkably limited. Only seven double-blind, placebo-controlled studies of medications for BD+AD have been conducted to date,9, 10, 11, 12, 13, 14, 15 and only one of these studies demonstrated a medication (that is, valproate) effect on drinking.12 Better understanding the mechanisms that give rise to co-occurring BD and AD could help to identify differences in genetic risk, prevention strategies and novel therapeutic targets.16 The BD and AD research literatures support the hypothesis that dysregulated glutamate and γ-aminobutyric acid (GABA) transmission may partially explain the high observed diagnostic co-occurrence between BD and AD, both directly and through their impact on salient neurobehavioral constructs (impulsivity, drug craving) central to BD and AD.
Glutamate and GABA, the main excitatory and inhibitory neurotransmitters in mammals, respectively, are amino acids that are principally involved in the coordination of cortical activity, synaptic plasticity and modulation of most other neurotransmitter systems.17, 18 Data from animal studies suggest that acute alcohol exposure augments the effects of GABA on the GABAA receptor and antagonizes the effects of glutamate on the N-methyl-d-aspartate (NMDA) receptor; prolonged ethanol exposure results in compensatory downregulation of GABAA receptors and upregulation of NMDA receptors, culminating in a hypoGABAergic/hyperglutamatergic state in alcohol withdrawal.19, 20 Proton magnetic resonance spectroscopy (1H-MRS) studies have demonstrated fluctuating brain glutamate levels in individuals with AD alone, depending on time of drinking; low prefrontal glutamate levels have been observed during active drinking21 and 2–50 days since last drink,22, 23, 24 whereas abnormally high glutamate levels observed during acute withdrawal.25 A recent investigation demonstrated that alcohol craving, measured by the Obsessive Compulsive Drinking Scale (OCDS26), was positively correlated with glutamate+glutamine levels in the nucleus accumbens and anterior cingulate cortex (ACC) in AD participants.27 Conversely, 1H-MRS investigations of GABA in AD have been rare and have produced inconsistent findings.23, 28, 29
Studies of adults with BD have demonstrated links between genes responsible for coding ionotropic glutamate receptor subunits and risk of BD,30 as well as response to lithium,31 the gold standard maintenance treatment for BD. Several lines of evidence suggest altered GABA neurotransmission in BD as well, including reduced levels of cerebrospinal fluid and plasma GABA,32, 33 and differences in GABA receptor genes, GABRA1,34 GABRA3 (ref. 35) and GABRA5.36 In animals, mood-stabilizing medications used to treat bipolar disorder, such as lithium and valproate, have been shown to decrease intrasynaptic glutamate via a number of mechanisms (for example, inhibition of NMDA receptor-mediated Ca2+ influx37). 1H-MRS studies in BD have demonstrated that individuals with BD alone have elevated prefrontal glutamate across mood states.38, 39, 40, 41, 42, 43, 44 In contrast, 1H-MRS investigations of GABA in BD have produced mixed,45, 46, 47 and largely null,48, 49, 50 findings.
Only one 1H-MRS study of co-occurring BD+AD has been published to date,51 despite the clinical importance of this population. Male participants with BD alone (that is, no history of AD) were found to have higher glutamate concentrations in the left dorsolateral prefrontal cortex relative to female participants with BD alone, participants with co-occurring BD and AD, and healthy controls. This study was limited to the BD+AD participants in remission (that is, 6 months to 22 years of abstinence) only, and did not include a comparator group of non-bipolar AD participants, making it difficult to distinguish the extent of contribution of alcohol to the finding.
Impulsivity, often defined as the tendency to respond without forethought,52 has been hypothesized as a critical 'link' between BD and SUD.53 Impulsivity has at least two dimensions (impaired response inhibition, impulsive choice), both are elevated in individuals with AD54, 55 and BD53, 56 regardless of mood state.57 Preclinical studies have shown that injections of NMDA receptor antagonists are associated with impulsive behavior;58 they appear to increase extracellular levels of glutamate due to their disinhibition of GABAergic inputs.59 Similarly, a recent study demonstrated increased impulsive behavior with infusion of the GABAA receptor agonist muscimol into rat ventromedial prefrontal cortex.60 Finally, a number of 1H-MRS studies have demonstrated a positive association between ACC glutamate and impulsivity61, 62, 63 and a negative association between dorsolateral prefrontal cortex64 and ACC61, 65 GABA and impulsivity across clinical populations. Elevated impulsivity might provide some basis for substance use and relapse potential in both BD and AD.
The present study was designed to provide a first 1H-MRS study of prefrontal GABA and glutamate levels in individuals with co-occurring BD and current AD, BD alone, current AD alone and healthy control (HC) subjects. We hypothesized a priori that individuals with co-occurring BD and AD would have significantly different prefrontal glutamate and GABA levels relative to individuals with BD or AD alone or healthy controls. We also hypothesized a priori that individuals with co-occurring BD and AD would have significantly higher scores on self-report measures of trait impulsivity and alcohol craving relative to individuals with BD or AD alone or healthy controls, and that impulsivity and alcohol craving would be associated with prefrontal GABA and glutamate levels, across participants.
Materials and methods
Participants
Seventy-eight individuals who met DSM-IV criteria for BD I or II and 'current' AD (that is, full criteria met within the past 6 months) (BD+AD, n=20), BD I or II alone (BD, n=19), current AD alone (AD, n=20), or no psychiatric diagnosis (HC, n=19) were recruited from inpatient and outpatient clinical settings and community advertisements. The sample size was chosen to provide approximately 80% power to detect large (that is, d=0.8–1.0) effect sizes. Written informed consent was obtained from every participant. To avoid confounding with the effects of acute alcohol or drug intoxication or withdrawal, all the participants were required to demonstrate ⩾1 week of abstinence from alcohol and drugs via serial self-report (Timeline Followback, TLFB66), the sensitive urine marker of recent alcohol use, ethyl glucuronide (EtG67) and urine drug screen (UDS) testing. Participants who reported any alcohol or drug use or provided positive EtG or UDS tests over the 1-week abstinence period were given two additional chances to achieve 1 week of abstinence before exclusion. In the case of conflicting TLFB and cannabinoid-UDS data, we used a validated algorithm to distinguish new cannabis use from residual cannabinoid excretion over the course of the 1-week monitored abstinence period.68 Participants whose positive cannabinoid-UDS was determined to be due to residual cannabinoid excretion (n=4) were retained. General exclusions included serious medical illness or history of head injury, psychotic disorder, recurrent major depressive disorder, past month posttraumatic stress disorder, obsessive-compulsive disorder or eating disorder (assessed with the Structured Clinical Interview for DSM-IV Axis I Disorders, SCID-IV69), current use of benzodiazepines or antidipsotropics (that is, naltrexone, acamprosate, disulfiram), past month electroconvulsive therapy, history of delirium tremens or >1 lifetime alcohol withdrawal seizure, acute alcohol withdrawal (that is, Clinical Institute Withdrawal of Alcohol Scale, CIWA-Ar >10) and daily drug use in the past month. Co-occurring drug use disorder was exclusionary for BD and HC, but not BD+AD and AD, participants. BD+AD and AD participants were required to exceed healthy weekly drinking levels (that is, >7 drinks per week for females, >14 drinks per week for males70) in the past month (or, alternatively, the month preceding abstinence), whereas the BD and HC participants exceeding healthy weekly drinking levels in the past month were excluded. Finally, the BD+AD or BD participants with substantial (that is, >20%) medication dose changes ⩽1 week before magnetic resonance imaging (MRI) were excluded.
Procedure
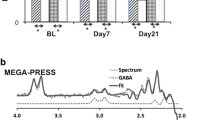
The study was conducted at the Medical University of South Carolina (MUSC) from 17 April 2013 to 24 August 2015. The study protocol was approved by the MUSC institutional review board. The participants completed a baseline diagnostic visit including the SCID-IV,69 Montgomery-Asberg Depression Rating Scale,71 Young Mania Rating Scale,72 TLFB, EtG, UDS, Barratt Impulsiveness Scale (BIS73), OCDS,26 demographic interview and brief abstinence counseling with a licensed PhD/MD clinician. The participants then returned approximately 4 days later (M=3.60, s.d.=0.83) for an MRI visit. Before scanning, the participants repeated the Montgomery-Asberg Depression Rating Scale, Young Mania Rating Scale, TLFB, EtG and UDS. Following these procedures, and with evidence of abstinence from alcohol and drugs, the participants completed the 1H-MRS scan in a Siemens 3.0 T Trio MR scanner with actively shielded magnet, high-performance gradients (45 mT m−1, 200 T m−1 s−1), and a 32-channel head coil. A structural scan was taken for 1H-MRS voxel placement and tissue segmentation (TR/TE(repetition time/echo time) =1900/2.26 ms; field of view =256 mm2; flip angle =9°; spatial resolution =1 × 1 × 1 mm). The 1H-MRS voxel was placed in the dorsal ACC (dACC) on midsagittal T1-weighted images, posterior and superior to the genu of the corpus callosum, with the ventral edge of the voxel aligned with the dorsal edge of the callosum.25 A dACC voxel was chosen because most 1H-MRS studies in AD or BD have focused on this region, and because dACC has been associated with impulsivity61 and alcohol craving27 in 1H-MRS studies. A voxel size of 2.5x2.5x3 cm was selected to ensure adequate signal to noise properties.74 Following placement of six outer-volume suppression bands approximately 1 cm away from the voxel faces and auto-shimming, single-voxel, water-suppressed 1H-MRS spectra were acquired using a two-dimensional J-resolved Spectroscopy sequence: TR/TE=2400/31–229 ms; ΔTE=2 ms; four signal averages per TE step with online averaging; two-dimensional (2D) spectral width =2000−500 Hz; 2D matrix size =2048−100; total acquisition time =13:28 min (ref. 74) water unsuppressed 2D 1H-MRS data were also acquired from the dACC voxel with two signal averages recorded for each TE step (total acquisition time =3:28 min). See Figure 1 for a representative voxel placement and sample spectrum.

Sample dorsal anterior cingulate cortex voxel location (right) and raw (top) and fitted (bottom) two-dimensional J-resolved 1H-MRS spectra analyzed using Prior Knowledge Fitting (ProFit). Colored horizontal bars represent approximate primary spectral locations of Glu, GABA and Gln. Cho, choline; Cre, creatine; GABA, gamma-aminobutyric acid; Gln, glutamine; Glu, glutamate; NAA, n-acetyl aspartate.
Analysis plan
Skull stripping and whole-brain tissue-type segmentation were performed on MP-RAGE images using the FSL BET and FAST tools.75 In-house MATLAB functions were used to extract the three-dimensional volume corresponding to the positioned MRS voxel to obtain within-voxel gray matter, white matter and cerebrospinal fluid tissue content for each subject. Eddy currents and residual water were removed using in-house MATLAB functions. Subsequently, the ProFit algorithm was applied using software-supplied 2D basis sets (which contain 19 metabolites total76). Before Fourier transformation, the raw 2D matrix was zero-filled. CRLB (Cramer-Rao Lower Bound) values, which reflect the uncertainty of estimated model parameters, were provided by the ProFit software. Estimated GABA and glutamate peak areas were normalized to the unsuppressed water signal. Finally, GABA/water and glutamate/water ratios were corrected for (that is, multiplied by) within-voxel cerebrospinal fluid fraction (that is, cerebrospinal fluid proportion+1).74
Participant characteristics were compared between groups using one-way analysis of variance for continuous variables (followed by pairwise independent samples t-tests in the case of significant omnibus tests) and chi-square for categorical variables (followed by pairwise chi-square tests in the case of significant omnibus tests). The associations between select participant characteristics and GABA and glutamate levels were tested using independent samples t-tests. The primary dependent variables (GABA and glutamate) were evaluated for potential violations of statistical test assumptions. The analysis of GABA and glutamate levels was accomplished via 2 (BD) × 2 (AD) factorial analyses of variance. These models were run both in the full sample, as well as in the restricted sample of participants who consumed alcohol within 2 weeks of MRI, due to our previous finding of associations between recent drinking and dACC glutamate in AD.77 The levels of additional estimated metabolites (for example, creatine, N-acetyl aspartate) were not examined as part of planned analyses to control alpha inflation due to multiple comparisons. However, results from exploratory 2 × 2 factorial analyses of variance for other estimated metabolites are provided in Supplementary Table 1. Concerning alcohol craving (OCDS) and impulsivity (BIS), we first compared participant groups on these measures using 2 × 2 factorial analyses of variance. We then estimated Pearson correlations between OCDS and BIS total scores and dACC GABA and glutamate levels across participants. All the analyses were conducted using two-tailed statistical tests.
Results
Ten participants (BD+AD n=3, BD n=1, AD n=5, HC n=1) required one to two additional visits between baseline and MRI to demonstrate ⩾1 week of abstinence from alcohol/drugs. Across groups, 40% of the participants were female, 30% were smokers and 40% reported consuming alcohol ⩽2 weeks before the MRI; mean (s.d.) age =38.5 (11.8).
Preliminary evaluation of potential covariates
See Table 1 for a detailed comparison of patient characteristics by participant group. There were marginal group differences in smoking (P=0.08), but smoking was not independently associated with dACC glutamate (P=0.70) or GABA (P=0.19) levels across the sample. Recency of alcohol consumption (that is, number of days of abstinence preceding the MRI visit) did not differ between BD+AD (M=32.4, s.d.=33.3) and AD (29.5, s.d.=23.0) participants (P=0.75). There were marginal differences in anticonvulsant use (P=0.06; lamotrigine n=11, divalproex sodium n=2, gabapentin n=6, topiramate n=1, oxcarbazepine n=1), with more BD+AD (65%) than BD (35%) participants taking anticonvulsants, but anticonvulsant use was not significantly independently associated with glutamate (BD+AD P=0.77, BD P=0.52) or GABA (BD+AD P=0.92, BD P=0.69) levels. Co-occurring past year drug dependence consisted primarily of cannabis dependence (76.9% of total drug dependence diagnoses; 15.4% stimulant and 7.7% cocaine dependence), and co-occurring drug dependence was not independently associated with glutamate (past year: BD+AD P=0.88, AD P=0.96) or GABA (past year: BD+AD P=0.83, AD P=0.28). Very few participants met criteria for drug dependence within the month preceding the study (that is, BD+AD n=4, AD n=2) and few participants reported any illicit drug use within 2 weeks of MRI (that is, BD+AD n=1, AD n=4, BD n=3). Co-occurring anxiety disorder diagnoses consisted primarily of social phobia (53.8% of total anxiety disorder diagnoses) and GAD (19.2%; 15.4% specific phobia, 7.7% NOS, 3.8% panic disorder), and co-occurring anxiety disorder was not independently associated with glutamate (P=0.24) or GABA (P=0.56) levels.
Primary analysis
The mean water line-width was 6.57 Hz (full width at half maximum; s.d.=0.79 Hz, maximum =9.07 Hz). Average gray matter to brain matter (brain matter=gray matter+white matter) ratio was 61.12% (s.d.=3.24%) and did not differ between the participant groups (F=0.27, P=0.85). Mean glutamate CRLB % was 2.22 (s.d.=1.39, maximum=6.29). Seven participants (n=4 HC, n=2 BD, n=1 AD) were excluded from analyses involving GABA, due to GABA CRLB >20%. Mean GABA CRLB % for the remaining participants was 8.20 (s.d.=3.88, maximum=19.99). A 2 × 2 factorial, general linear modeling analysis of dACC GABA/water concentrations demonstrated a significant main effect of AD (F=5.36, P=0.02) that was qualified by a significant BD × AD interaction (F=2.91, P<0.05), signifying uniquely low levels of dACC GABA/water in BD+AD relative to BD, AD and HC (Cohen’s d for BD+AD vs HC=0.78). This effect nearly doubled when the sample was restricted to individuals who reported consuming alcohol within the 2 weeks before MRI (n=28, BD × AD interaction F=2.15, P=0.03, Cohen’s d for BD+AD vs HC=1.53); see Figure 2. In the overall sample, there were no significant effects of BD and/or AD on dACC glutamate/water levels (BD × AD interaction F=1.46, P=0.231, Cohen’s d for BD+AD vs HC=0.33). However, when the sample was restricted to individuals who reported consuming alcohol within the 2 weeks preceding their scan (n=31), the BD × AD interaction, signifying uniquely low levels of dACC glutamate/water in BD+AD (Cohen’s d for BD+AD vs HC=1.01), approached significance (F=3.83, P=0.06) and reached significance (F=4.21, P=0.05) once within-voxel gray matter/brain matter composition was statistically controlled; see Figure 2. To further explore this finding, we estimated Pearson correlations between glutamate/water levels and number of days of abstinence from alcohol. We restricted this analysis to individuals who reported drinking within 30 days of their scan (n=52) to reduce the influence of outliers on results while retaining enough cases to estimate stable within-group correlations. Glutamate/water levels and days of abstinence were not significantly correlated in the full sample (r=0.18, P=0.19). However, examination of within-group associations revealed that glutamate/water levels and days of abstinence were significantly correlated in the BD+AD group (r=0.63, P=0.02) but not in any of the other groups (AD r=0.19, P=0.58; BD r=−0.02, P=0.96; HC r=−0.14, P=0.63).

Mean (±1 s.e.) GABA/water (left) and glutamate/water (right) concentrations, in institutional units, for AD+BD, AD, BD and HC who reported drinking within 2 weeks of their MRI. *P<0.05, †P<0.10. AD, alcohol dependence; BD, bipolar disorder; GABA, gamma-aminobutyric acid; HC, healthy control.
As hypothesized, scores on the BIS were highest in BD+AD relative to BD, AD or HC individuals, with both BD and AD demonstrating significant main effects on BIS (BD F=24. 25, P<.01; AD F=9.13, P<0.01; BD × AD F=1.07, P=0.30); see Figure 3. BD+AD also scored highest on the OCDS followed by AD, despite similar recent alcohol consumption between BD+AD and AD participants (for example, drinks per drinking day, P=0.44); BD and HC had similarly low OCDS scores (BD F=4.25, P=0.04; AD F=49.86, P<0.01; BD × AD F=3.36, P=0.07); see Figure 3. The dACC GABA/water levels were significantly negatively correlated with BIS (r=−0.28, P=0.02) and OCDS (r=−0.35, P<0.01) scores across groups; see Figure 4. Although glutamate/water concentrations were not significantly associated with BIS (r=−0.03, P=0.83) or OCDS (r=−0.18, P=0.11) scores in the full sample, glutamate/water levels were significantly negatively associated with OCDS (r=−0.35, P=0.05) scores in individuals who reported consuming alcohol within 2 weeks of MRI; association between glutamate/water and BIS in this subsample was similar in magnitude but not significant (r=−0.22, P=0.25). Although BIS was normally distributed, OCDS was not, because HC and BD without AD had predictably low OCDS scores (that is, all under '5'). However, correlations involving OCDS were very similar when HC and BD were removed from the sample (GABA r=−0.36, glutamate r=−0.20; past 2 week drinkers: GABA r=−0.30, glutamate r=−0.48). In sum, these results demonstrate that BD+AD have abnormally low levels of dACC GABA and glutamate, and that low levels of GABA and glutamate are associated with assessments of salient neurobehavioral constructs (that is, impulsivity and craving) in BD+AD.

Mean (±1 s.e.) Barratt Impulsiveness Scale version 11, total score (BIS-11; left) and Obsessive Compulsive Drinking Scale, total score (OCDS; right) for AD+BD, AD, BD and HC. **P<0.01, ***P<0.001. AD, alcohol dependence; BD, bipolar disorder; HC, healthy control.

Associations between GABA/water and Barratt Impulsiveness Scale version 11, total score (BIS-11; left) and GABA/water, in Institutional Units, and Obsessive Compulsive Drinking Scale, total score (OCDS; right). AD, alcohol dependence; BD, bipolar disorder; GABA, gamma-aminobutyric acid; HC, healthy control.
Discussion
The present study demonstrated that individuals with co-occurring BD and current AD had uniquely low levels of prefrontal GABA and glutamate, and that low levels of GABA and glutamate were associated with both impulsivity and craving. These effects were particularly strong in individuals who drank within 2 weeks of the 1H-MRS scan. Despite different 1H-MRS methods, brain areas, and populations, extant studies have consistently demonstrated abnormally low levels of prefrontal glutamate in AD alone individuals21, 22, 23, 24 not experiencing acute alcohol withdrawal. Although studies of BD alone have reported elevated prefrontal glutamate levels,38, 39, 40, 41, 42, 43, 44 1H-MRS research on co-occurring BD and AD (that is ref. 51 and the present study) suggests that when BD and AD are combined within the same individual, the effects of AD may modify the effects of BD on prefrontal glutamate levels leading to a net decrease in glutamate to levels even less than in AD alone. Though evidence for prefrontal GABA abnormalities in 1H-MRS studies of AD23, 28, 29 or BD45, 46, 47, 48, 49, 50 alone has been inconsistent, our data suggest that individuals with co-occurring BD and AD have uniquely low levels of prefrontal GABA.
Although there are several points of convergence between our findings and the extant literature, there are also notable points of divergence. For example, although the extant literature has demonstrated increased prefrontal glutamate in BD alone, and decreased prefrontal glutamate in AD alone not experiencing acute alcohol withdrawal, relative to healthy controls, individuals with BD or AD alone in the present study did not have statistically different prefrontal glutamate levels than controls. One potential explanation for the lack of differences between AD alone and controls could be that prefrontal glutamate levels may have normalized during the ⩾1-week monitored period of abstinence that preceded the 1H-MRS scan;23, 25, 77 only 35–40% of AD alone and co-occurring BD and AD participants reported drinking in the 2 weeks preceding 1H-MRS (that is, in the week preceding the monitored 1-week abstinence period). Another potential explanation is that we carefully excluded AD with potentially confounding clinical characteristics, including history of recurrent depression or complicated alcohol withdrawal including seizures, current posttraumatic stress disorder or obsessive-compulsive disorder or prescription of psychotropic medications. Similar potential explanations could be invoked concerning the lack of observed difference in prefrontal glutamate between BD alone and controls. For example, unlike most prior BD 1H-MRS studies, we carefully excluded individuals with any history of SUD as well as any alcohol/drug consumption in the week preceding MRI. Furthermore, our BD, and co-occurring BD and AD, participants were not severely depressed or manic, and most BD and co-occurring BD and AD participants were taking mood-stabilizing medications (that is, lithium, anticonvulsant medications, atypical antipsychotic medications), which may have reduced potential effect sizes of BD on glutamate levels.78 The present investigation represents a foundation for future studies to follow-up on these potential explanations by, for example, scanning participants without a preceding period of abstinence or focusing on unmedicated BD individuals.
If replicated, the findings from the present study may support preferential evaluation of therapeutics known to increase prefrontal GABA and glutamate in co-occurring BD and AD. Traditionally, treatment trials for co-occurring BD and AD have investigated medications FDA-approved to treat either BD or AD in hopes that such medications would prove efficacious in individuals with both disorders. These hopes have largely not been realized.12 A different approach to selecting and developing medications for clinical trials would be to target neurobiological dysfunctions characteristic of individuals with both BD and AD.16 Our findings not only suggest that individuals with co-occurring BD and AD have unique deficits in prefrontal GABA and glutamate, but that these deficits may be associated with salient neurobehavioral constructs including impulsivity and alcohol craving. Future studies can expand this research further by evaluating more objective/behavioral measures of impulsivity and craving and by including 1H-MRS imaging in clinical trials with specific glutamate/GABA-acting medications.
Though our findings may provide potential avenues for treatment development in individuals with co-occurring BD and AD, a mechanistic understanding of these findings is, at present, limited. The glutamate–GABA cycle is complex, involving several important enzymes (glutamic acid decarboxylase, glutamine synthetase, phosphate-activated glutaminase) as well as transporters for glutamate, glutamine and GABA selectively localized in astrocytes and glutamatergic/GABAergic neurons, respectively.79 Further, overall homeostasis of glutamate–GABA biosynthetic coupling is regulated by more than one mechanism simultaneously; for instance, upon uptake of synaptically released glutamate into astrocytes, glutamate may be converted to glutamine either by glutamine synthetase or by oxidative metabolism, and the extent to which these pathways are engaged appears to depend in part upon glutamatergic tone.79 Although not the main focus of this investigation, we found no significant differences in glutamine levels between study groups. How the myriad enzymes and transporters involved in the glutamate–GABA cycle are regulated by chronic alcohol use or duration of abstinence is poorly understood in humans, much less in those with both BD and AD. Last, 1H-MRS cannot at present distinguish neuronal vs glial glutamate neurotransmitter pools even under normal physiological conditions in healthy subjects. Nonetheless, each of these macromolecular sites in the tripartite synapse37 represent testable potential therapeutic targets that may be better characterized in the future using selective pharmacologic agents and improving MRS technology (for example, carbon-13 MRS80).
The results of the present study should be interpreted in light of several limitations. First, a number of potentially confounding clinical characteristics (for example, concomitant medications, co-occurring drug dependence) were not exclusionary. Allowing these potential sources of variability into the study was by design, to balance feasibility of recruitment/retention and scientific concerns. Striking this balance is particularly important in research involving co-occurring BD and AD, a notoriously difficult to recruit/retain population for research. Though imperfect, we used a statistical approach to evaluating and controlling for the potential impact of these characteristics on glutamate and GABA levels and we found no evidence that such factors were independently responsible for our findings; sample sizes were too small to evaluate potential interactive effects. Second, our findings demonstrated that prefrontal GABA and glutamate differences between co-occurring BD and AD and all other participant groups were particularly strong in participants who drank within 2 weeks of MRI. Given that only 40% of participants reported drinking within 2 weeks of MRI, these findings were based on small samples (that is, n=28–31), and should be considered tentative until replicated. This concern is particularly salient for findings involving glutamate, as findings involving GABA were significant in both the full and restricted (recent drinking) samples. Third, because we excluded participants with a history of delirium tremens or >1 lifetime alcohol withdrawal seizure, it is unclear whether our findings generalize to individuals with very severe AD. Fourth, the ProFit algorithm used to analyze our 2D J-PRESS 1H-MRS data did not allow for separation of co-edited macromolecules (for example, proteins) from the observed GABA signal. Although it is very unlikely that between-group differences in macromolecules represent a viable alternative explanation for our GABA findings, future studies should evaluate macromolecule-suppressed GABA acquisitions81 in co-occurring BD and AD. Finally, given that we did not acquire data from brain regions other than dACC, we cannot comment on the regional specificity of our findings.
These limitations notwithstanding, the present study extends the literature by demonstrating uniquely low levels of prefrontal GABA and glutamate in individuals with co-occurring BD and current AD, along with associations between prefrontal GABA and glutamate levels and impulsivity and alcohol craving. If replicated, these findings support preferential evaluation of therapeutics known to 'normalize' prefrontal GABA and glutamate levels in co-occurring BD and AD.
References
Grant BF, Stinson FS, Dawson DA, Chou SP, Dufour MC, Compton W et al. Prevalence and co-occurrence of substance use disorders and independent mood and anxiety disorders: results from the National Epidemiologic Survey on Alcohol and Related Conditions. Arch Gen Psychiatry 2004; 61: 807–816.
Regier DA, Farmer ME, Rae DS, Locke BZ, Keith SJ, Judd LL et al. Comorbidity of mental disorders with alcohol and other drug abuse. Results from the Epidemiologic Catchment Area (ECA) Study. JAMA 1990; 264: 2511–2518.
Post RM, Denicoff KD, Leverich GS, Altshuler LL, Frye MA, Suppes TM et al. Morbidity in 258 bipolar outpatients followed for 1 year with daily prospective ratings on the NIMH life chart method. J Clin Psychiatry 2003; 64: 680–690, quiz 738-9.
Goldberg JF, Garno JL, Leon AC, Kocsis JH, Portera L . A history of substance abuse complicates remission from acute mania in bipolar disorder. J Clin Psychiatry 1999; 60: 733–740.
Oquendo MA, Currier D, Liu SM, Hasin DS, Grant BF, Blanco C . Increased risk for suicidal behavior in comorbid bipolar disorder and alcohol use disorders: results from the National Epidemiologic Survey on Alcohol and Related Conditions (NESARC). J Clin Psychiatry 2010; 71: 902–909.
Fazel S, Lichtenstein P, Grann M, Goodwin GM, Langstrom N . Bipolar disorder and violent crime: new evidence from population-based longitudinal studies and systematic review. Arch Gen Psychiatry 2010; 67: 931–938.
Wilk J, West JC, Rae DS, Regier DA . Relationship of comorbid substance and alcohol use disorders to disability among patients in routine psychiatric practice. Am J Addict 2006; 15: 180–185.
Hoblyn JC, Balt SL, Woodard SA, Brooks JO 3rd . Substance use disorders as risk factors for psychiatric hospitalization in bipolar disorder. Psychiatr Serv 2009; 60: 50–55.
Brown ES, Carmody TJ, Schmitz JM, Caetano R, Adinoff B, Swann AC et al. A randomized, double-blind, placebo-controlled pilot study of naltrexone in outpatients with bipolar disorder and alcohol dependence. Alcohol Clin Exp Res 2009; 33: 1863–1869.
Gao K, Wu R, Kemp DE, Chen J, Karberg E, Conroy C et al. Efficacy and safety of quetiapine-XR as monotherapy or adjunctive therapy to a mood stabilizer in acute bipolar depression with generalized anxiety disorder and other comorbidities: a randomized, placebo-controlled trial. J Clin Psychiatry 2014; 75: 1062–1068.
Petrakis IL, Poling J, Levinson C, Nich C, Carroll K, Rounsaville B . Naltrexone and disulfiram in patients with alcohol dependence and comorbid psychiatric disorders. Biol Psychiatry 2005; 57: 1128–1137.
Salloum IM, Cornelius JR, Daley DC, Kirisci L, Himmelhoch JM, Thase ME . Efficacy of valproate maintenance in patients with bipolar disorder and alcoholism: a double-blind placebo-controlled study. Arch Gen Psychiatry 2005; 62: 37–45.
Stedman M, Pettinati HM, Brown ES, Kotz M, Calabrese JR, Raines S . A double-blind, placebo-controlled study with quetiapine as adjunct therapy with lithium or divalproex in bipolar I patients with coexisting alcohol dependence. Alcohol Clin Exp Res 2010; 34: 1822–1831.
Sylvia LG, Gold AK, Stange JP, Peckham AD, Deckersbach T, Calabrese JR et al. A randomized, placebo-controlled proof-of-concept trial of adjunctive topiramate for alcohol use disorders in bipolar disorder. Am J Addict 2016; 25: 94–98.
Tolliver BK, Desantis SM, Brown DG, Prisciandaro JJ, Brady KT . A randomized, double-blind, placebo-controlled clinical trial of acamprosate in alcohol-dependent individuals with bipolar disorder: a preliminary report. Bipolar Disord 2012; 14: 54–63.
Swann AC . The strong relationship between bipolar and substance-use disorder. Ann N Y Acad Sci 2010; 1187: 276–293.
Hassel B, Dingledine R . Glutamate. In: Siegel GJ, Albers RW, Brady ST, Price DL (eds). Basic Neurochemistry: Molecular, Cellular, and Medical Aspects. Elsevier Academic Press: London, UK, 2006, pp 267–290.
Olsen RW, Betz H . GABA and glycine. In: Siegel GJ, Albers RW, Brady ST, Price DL (eds). Basic Neurochemistry: Molecular, Cellular, and Medical Aspects. Elsevier Academic Press: London, UK, 2006, pp 291–301.
Gass JT, Olive MF . Glutamatergic substrates of drug addiction and alcoholism. Biochem Pharmacol 2008; 75: 218–265.
Krystal JH, Staley J, Mason G, Petrakis IL, Kaufman J, Harris RA et al. Gamma-aminobutyric acid type A receptors and alcoholism: intoxication, dependence, vulnerability, and treatment. Arch Gen Psychiatry 2006; 63: 957–968.
Ende G, Hermann D, Demirakca T, Hoerst M, Tunc-Skarka N, Weber-Fahr W et al. Loss of control of alcohol use and severity of alcohol dependence in non-treatment-seeking heavy drinkers are related to lower glutamate in frontal white matter. Alcohol Clin Exp Res 2013; 37: 1643–1649.
Bagga D, Khushu S, Modi S, Kaur P, Bhattacharya D, Garg ML et al. Impaired visual information processing in alcohol-dependent subjects: a proton magnetic resonance spectroscopy study of the primary visual cortex. J Stud Alcohol Drugs 2014; 75: 817–826.
Mon A, Durazzo TC, Meyerhoff DJ . Glutamate, GABA, and other cortical metabolite concentrations during early abstinence from alcohol and their associations with neurocognitive changes. Drug Alcohol Depend 2012; 125: 27–36.
Thoma R, Mullins P, Ruhl D, Monnig M, Yeo RA, Caprihan A et al. Perturbation of the glutamate-glutamine system in alcohol dependence and remission. Neuropsychopharmacology 2011; 36: 1359–1365.
Hermann D, Weber-Fahr W, Sartorius A, Hoerst M, Frischknecht U, Tunc-Skarka N et al. Translational magnetic resonance spectroscopy reveals excessive central glutamate levels during alcohol withdrawal in humans and rats. Biol Psychiatry 2012; 71: 1015–1021.
Anton RF, Moak DH, Latham P . The Obsessive Compulsive Drinking Scale: a self-rated instrument for the quantification of thoughts about alcohol and drinking behavior. Alcohol Clin Exp Res 1995; 19: 92–99.
Bauer J, Pedersen A, Scherbaum N, Bening J, Patschke J, Kugel H et al. Craving in alcohol-dependent patients after detoxification is related to glutamatergic dysfunction in the nucleus accumbens and the anterior cingulate cortex. Neuropsychopharmacology 2013; 38: 1401–1408.
Behar KL, Rothman DL, Petersen KF, Hooten M, Delaney R, Petroff OA et al. Preliminary evidence of low cortical GABA levels in localized 1H-MR spectra of alcohol-dependent and hepatic encephalopathy patients. Am J Psychiatry 1999; 156: 952–954.
Mason GF, Petrakis IL, de Graaf RA, Gueorguieva R, Guidone E, Coric V et al. Cortical gamma-aminobutyric acid levels and the recovery from ethanol dependence: preliminary evidence of modification by cigarette smoking. Biol Psychiatry 2006; 59: 85–93.
Le-Niculescu H, Patel SD, Bhat M, Kuczenski R, Faraone SV, Tsuang MT et al. Convergent functional genomics of genome-wide association data for bipolar disorder: comprehensive identification of candidate genes, pathways and mechanisms. Am J Med Genet B Neuropsychiatr Genet 2009; 150b: 155–181.
Perlis RH, Dennehy EB, Miklowitz DJ, Delbello MP, Ostacher M, Calabrese JR et al. Retrospective age at onset of bipolar disorder and outcome during two-year follow-up: results from the STEP-BD study. Bipolar Disord 2009; 11: 391–400.
Gerner RH, Fairbanks L, Anderson GM, Young JG, Scheinin M, Linnoila M et al. CSF neurochemistry in depressed, manic, and schizophrenic patients compared with that of normal controls. Am J Psychiatry 1984; 141: 1533–1540.
Petty F, Kramer GL, Dunnam D, Rush AJ . Plasma GABA in mood disorders. Psychopharmacol Bull 1990; 26: 157–161.
Horiuchi Y, Nakayama J, Ishiguro H, Ohtsuki T, Detera-Wadleigh SD, Toyota T et al. Possible association between a haplotype of the GABA-A receptor alpha 1 subunit gene (GABRA1) and mood disorders. Biol Psychiatry 2004; 55: 40–45.
Massat I, Souery D, Del-Favero J, Oruc L, Noethen MM, Blackwood D et al. Excess of allele1 for alpha3 subunit GABA receptor gene (GABRA3) in bipolar patients: a multicentric association study. Mol Psychiatry 2002; 7: 201–207.
Otani K, Ujike H, Tanaka Y, Morita Y, Katsu T, Nomura A et al. The GABA type A receptor alpha5 subunit gene is associated with bipolar I disorder. Neurosci Lett 2005; 381: 108–113.
Sanacora G, Zarate CA, Krystal JH, Manji HK . Targeting the glutamatergic system to develop novel, improved therapeutics for mood disorders. Nat Rev Drug Discov 2008; 7: 426–437.
Dager SR, Friedman SD, Parow A, Demopulos C, Stoll AL, Lyoo IK et al. Brain metabolic alterations in medication-free patients with bipolar disorder. Arch Gen Psychiatry 2004; 61: 450–458.
Ehrlich A, Schubert F, Pehrs C, Gallinat J . Alterations of cerebral glutamate in the euthymic state of patients with bipolar disorder. Psychiatry Res 2015; 233: 73–80.
Frye MA, Watzl J, Banakar S, O'Neill J, Mintz J, Davanzo P et al. Increased anterior cingulate/medial prefrontal cortical glutamate and creatine in bipolar depression. Neuropsychopharmacology 2007; 32: 2490–2499.
Michael N, Erfurth A, Ohrmann P, Gossling M, Arolt V, Heindel W et al. Acute mania is accompanied by elevated glutamate/glutamine levels within the left dorsolateral prefrontal cortex. Psychopharmacology 2003; 168: 344–346.
Michael N, Erfurth A, Pfleiderer B . Elevated metabolites within dorsolateral prefrontal cortex in rapid cycling bipolar disorder. Psychiatry Res 2009; 172: 78–81.
Ongur D, Jensen JE, Prescot AP, Stork C, Lundy M, Cohen BM et al. Abnormal glutamatergic neurotransmission and neuronal-glial interactions in acute mania. Biol Psychiatry 2008; 64: 718–726.
Soeiro-de-Souza MG, Salvadore G, Moreno RA, Otaduy MC, Chaim KT, Gattaz WF et al. Bcl-2 rs956572 polymorphism is associated with increased anterior cingulate cortical glutamate in euthymic bipolar I disorder. Neuropsychopharmacology 2013; 38: 468–475.
Bhagwagar Z, Wylezinska M, Jezzard P, Evans J, Ashworth F, Sule A et al. Reduction in occipital cortex gamma-aminobutyric acid concentrations in medication-free recovered unipolar depressed and bipolar subjects. Biol Psychiatry 2007; 61: 806–812.
Brady RO Jr, McCarthy JM, Prescot AP, Jensen JE, Cooper AJ, Cohen BM et al. Brain gamma-aminobutyric acid (GABA) abnormalities in bipolar disorder. Bipolar Disord 2013; 15: 434–439.
Wang PW, Sailasuta N, Chandler RA, Ketter TA . Magnetic resonance spectroscopic measurement of cerebral gamma-aminobutyric acid concentrations in patients with bipolar disorders. Acta Neuropsychiatr 2006; 18: 120–126.
Godlewska BR, Yip SW, Near J, Goodwin GM, Cowen PJ . Cortical glutathione levels in young people with bipolar disorder: a pilot study using magnetic resonance spectroscopy. Psychopharmacology 2014; 231: 327–332.
Kaufman RE, Ostacher MJ, Marks EH, Simon NM, Sachs GS, Jensen JE et al. Brain GABA levels in patients with bipolar disorder. Prog Neuropsychopharmacol Biol Psychiatry 2009; 33: 427–434.
Soeiro-de-Souza MG, Henning A, Machado-Vieira R, Moreno RA, Pastorello BF, da Costa Leite C et al. Anterior cingulate Glutamate-Glutamine cycle metabolites are altered in euthymic bipolar I disorder. European neuropsychopharmacology 2015; 25: 2221–2229.
Nery FG, Stanley JA, Chen HH, Hatch JP, Nicoletti MA, Monkul ES et al. Bipolar disorder comorbid with alcoholism: a 1H magnetic resonance spectroscopy study. J Psychiatr Res 2010; 44: 278–285.
Robinson ES, Eagle DM, Economidou D, Theobald DE, Mar AC, Murphy ER et al. Behavioural characterisation of high impulsivity on the 5-choice serial reaction time task: specific deficits in 'waiting' versus 'stopping'. Behav Brain Res 2009; 196: 310–316.
Swann AC, Dougherty DM, Pazzaglia PJ, Pham M, Moeller FG . Impulsivity: a link between bipolar disorder and substance abuse. Bipolar Disord 2004; 6: 204–212.
Mitchell JM, Fields HL, D'Esposito M, Boettiger CA . Impulsive responding in alcoholics. Alcohol Clin Exp Res 2005; 29: 2158–2169.
Rubio G, Jimenez M, Rodriguez-Jimenez R, Martinez I, Avila C, Ferre F et al. The role of behavioral impulsivity in the development of alcohol dependence: a 4-year follow-up study. Alcohol Clin Exp Res 2008; 32: 1681–1687.
Swann AC, Lijffijt M, Lane SD, Steinberg JL, Moeller FG . Increased trait-like impulsivity and course of illness in bipolar disorder. Bipolar Disord 2009; 11: 280–288.
Swann AC, Pazzaglia P, Nicholls A, Dougherty DM, Moeller FG . Impulsivity and phase of illness in bipolar disorder. J Affect Disord 2003; 73: 105–111.
Pattij T, Vanderschuren LJ . The neuropharmacology of impulsive behaviour. Trends Pharmacol Sci 2008; 29: 192–199.
Maeng S, Zarate CA Jr, Du J, Schloesser RJ, McCammon J, Chen G et al. Cellular mechanisms underlying the antidepressant effects of ketamine: role of alpha-amino-3-hydroxy-5-methylisoxazole-4-propionic acid receptors. Biol Psychiatry 2008; 63: 349–352.
Murphy ER, Fernando AB, Urcelay GP, Robinson ES, Mar AC, Theobald DE et al. Impulsive behaviour induced by both NMDA receptor antagonism and GABAA receptor activation in rat ventromedial prefrontal cortex. Psychopharmacology 2012; 219: 401–410.
Ende G, Cackowski S, Van Eijk J, Sack M, Demirakca T, Kleindienst N et al. Impulsivity and aggression in female BPD and ADHD patients: association with ACC glutamate and GABA concentrations. Neuropsychopharmacology 2016; 41: 410–418.
Hoerst M, Weber-Fahr W, Tunc-Skarka N, Ruf M, Bohus M, Schmahl C et al. Correlation of glutamate levels in the anterior cingulate cortex with self-reported impulsivity in patients with borderline personality disorder and healthy controls. Arch Gen Psychiatry 2010; 67: 946–954.
Schmaal L, Veltman DJ, Nederveen A, van den Brink W, Goudriaan AE . N-acetylcysteine normalizes glutamate levels in cocaine-dependent patients: a randomized crossover magnetic resonance spectroscopy study. Neuropsychopharmacology 2012; 37: 2143–2152.
Boy F, Evans CJ, Edden RA, Lawrence AD, Singh KD, Husain M et al. Dorsolateral prefrontal gamma-aminobutyric acid in men predicts individual differences in rash impulsivity. Biol Psychiatry 2011; 70: 866–872.
Silveri MM, Sneider JT, Crowley DJ, Covell MJ, Acharya D, Rosso IM et al. Frontal lobe gamma-aminobutyric acid levels during adolescence: associations with impulsivity and response inhibition. Biol Psychiatry 2013; 74: 296–304.
Sobell LC, Sobell MB . Alcohol Timeline Followback Users' Manual. Addiction Research Foundation: Toronto, ONT, Canada, 1995.
Jatlow P, O'Malley SS . Clinical (nonforensic) application of ethyl glucuronide measurement: are we ready? Alcohol Clin Exp Res 2010; 34: 968–975.
Schwilke EW, Gullberg RG, Darwin WD, Chiang CN, Cadet JL, Gorelick DA et al. Differentiating new cannabis use from residual urinary cannabinoid excretion in chronic, daily cannabis users. Addiction 2011; 106: 499–506.
First MB . Structured Clinical Interview for DSM-IV Axis I Disorders: Patient Edition (February 1996 Final), SCID-I/P. Biometrics Research Department, New York State Psychiatric Institute: New York, NY, USA, 1998.
NIAAA Rethinking Drinking: Alcohol and Your health. NIAAA: Bethesda, MD, USA, 2016.
Montgomery SA, Asberg M . A new depression scale designed to be sensitive to change. Br J Psychiatry 1979; 134: 382–389.
Young RC, Biggs JT, Ziegler VE, Meyer DA . A rating scale for mania: reliability, validity and sensitivity. Br J Psychiatry 1978; 133: 429–435.
Patton JH, Stanford MS, Barratt ES . Factor structure of the Barratt impulsiveness scale. J Clin Psychol 1995; 51: 768–774.
Prescot AP, Renshaw PF . Two-dimensional J-resolved proton MR spectroscopy and prior knowledge fitting (ProFit) in the frontal and parietal lobes of healthy volunteers: assessment of metabolite discrimination and general reproducibility. J Magn Reson Imaging 2013; 37: 642–651.
Smith SM, Jenkinson M, Woolrich MW, Beckmann CF, Behrens TE, Johansen-Berg H et al. Advances in functional and structural MR image analysis and implementation as FSL. Neuroimage 2004; 23 (Suppl 1): S208–S219.
Schulte RF, Boesiger P . ProFit: two-dimensional prior-knowledge fitting of J-resolved spectra. NMR Biomed 2006; 19: 255–263.
Prisciandaro JJ, Schacht JP, Prescot AP, Renshaw PF, Brown TR, Anton RF . Associations between recent heavy drinking and dorsal anterior cingulate N-acetylaspartate and glutamate concentrations in non-treatment seeking individuals with alcohol dependence. Alcohol Clin Exp Res 2016; 40: 491–496.
Hafeman DM, Chang KD, Garrett AS, Sanders EM, Phillips ML . Effects of medication on neuroimaging findings in bipolar disorder: an updated review. Bipolar Disord 2012; 14: 375–410.
Schousboe A, Bak L, Waagepetersen H . Astrocytic control of biosynthesis and turnover of the neurotransmitters glutamate and GABA. Front Endocrinol 2013; 4: 102.
de Graaf RA, Rothman DL, Behar KL . State of the art direct 13C and indirect 1H-[13C] NMR spectroscopy in vivo. A practical guide. NMR Biomed 2011; 24: 958–972.
Aufhaus E, Weber-Fahr W, Sack M, Tunc-Skarka N, Oberthuer G, Hoerst M et al. Absence of changes in GABA concentrations with age and gender in the human anterior cingulate cortex: a MEGA-PRESS study with symmetric editing pulse frequencies for macromolecule suppression. Magn Reson Med 2013; 69: 317–320.
Acknowledgements
The present study was funded by NIAAA K23 AA020842 (PI: JJP). RFA is supported by NIAAA K05 AA017435. JJP thanks his additional K23 mentors and consultants who provided valuable feedback on study design including Drs Kathleen Brady, Mary Phillips, Alan Swann, Jane Joseph, Hugh Myrick and Peter Kalivas. We also thank a number of individuals who assisted us with participant recruitment from clinical and research referral sources, especially Li Hunninghake, James Fox, Chris Kindt, Kristen Williams, Sarah Book, Patricia Latham, Konstantin Voronin, Geoffrey Cheek, Viktoriya Magid, Wood Marchant, Kathryn Bottonari, Emily Bristol, Mark Ghent and Melissa Michel.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
PFR consults for Kyowa Hallo and Tal Medical. RFA has been a consultant in the recent past for Lilly, Lundbeck, Novartis, Indivior and Alkermes, and on Advisory Boards for Alkermes, Indivior and Lundbeck. He has received grant funds from Lilly. He is a member of the Alcohol Clinical Trials Initiative (ACTIVE) sponsored by the American Society of Clinical. Psychopharmacology (ASCP) which has been supported in the last 3 years by AbbVie, Alkermes, Ethypharm, Indivior, Lilly, Lundbeck, Otsuka, Pfizer and XenoPort. The remaining authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on the Translational Psychiatry website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Prisciandaro, J., Tolliver, B., Prescot, A. et al. Unique prefrontal GABA and glutamate disturbances in co-occurring bipolar disorder and alcohol dependence. Transl Psychiatry 7, e1163 (2017). https://doi.org/10.1038/tp.2017.141
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/tp.2017.141
This article is cited by
-
Brain metabolite alterations related to alcohol use: a meta-analysis of proton magnetic resonance spectroscopy studies
Molecular Psychiatry (2022)
-
Molecular landscape of long noncoding RNAs in brain disorders
Molecular Psychiatry (2021)
-
ACC Glu/GABA ratio is decreased in euthymic bipolar disorder I patients: possible in vivo neurometabolite explanation for mood stabilization
European Archives of Psychiatry and Clinical Neuroscience (2021)
-
Anterior cingulate cortex metabolites and white matter microstructure: a multimodal study of emergent alcohol use disorder
Brain Imaging and Behavior (2021)
-
Proton magnetic resonance spectroscopy in substance use disorder: recent advances and future clinical applications
Science China Information Sciences (2020)
