






Abstract
Traditionally, many invasive fungal infections were associated with a poor prognosis, because effective therapeutic options were limited. The recent development of new antifungal agents has significantly contributed to the successful treatment of fungal diseases. These drugs offer novel mechanisms of action and expanded spectrums of activity over traditional treatment options. However, with these new agents comes the need for increased awareness of the potential interactions and toxicities associated with these drugs. Therefore, an understanding of the pharmacokinetic and pharmacodynamic properties of the classes of antifungal compounds is vital for the effective management of invasive fungal infections. This review provides a summary of the pharmacologic principles involved in treatment of fungal diseases.
The number of agents available to treat invasive fungal infections has increased by 30% since the turn of the millennium. Although that statistic is impressive, it brings the total number of approved systemic antifungal drugs to only 14 [1], with the potential for 1 more product to possibly emerge this year. These recent additions have provided clinicians with a tool previously lacking in the management of these life-threatening infections: therapeutic alternatives.
Along with new options, however, comes the need to understand the uniqueness of each agent, including its role in therapy, toxicity profile, and interactions with concomitant medications. To attain the maximum effect from these agents, clinicians should also become familiar with strategies to optimize efficacy through an understanding of pharmacokinetic and pharmacodynamic properties. These characteristics are unique for each class of antifungal drug and even for each member within a class. In many cases, this variability is not subtle and merits careful attention.
An additional concern related to the increasing number of antifungal drugs is the rapid increase in expenditures associated with their use. Many institutions throughout the United States are struggling with the increased financial burden related to the prescribing of these antifungal drugs [2]. Optimization of therapies through targeted application of various kinetic and dynamic principles may be one strategy to maximize the cost-effectiveness of treatment of invasive fungal infections. This review focuses on the pharmacologic principles involved in treatment of fungal disease and compares and contrasts the differences among the available agents. Given its role as an agent used primarily to treat superficial infections, terbinafine will not be included in this discussion.
History And Mechanisms of Action
Amphotericin B has been the mainstay of antifungal therapy since its release in the 1950s [3]. This agent emerged as the preferred polyene over the more toxic agent in this class, nystatin. Nystatin has since been relegated to topical and localized therapy because of its unfavorable adverse effect profile. The polyene agents exert their antifungal activity via binding to ergosterol in the fungal cell membrane (figure 1). This disrupts cell permeability and results in rapid cell death [4]. To date, amphotericin B remains the broadest-spectrum antifungal agent available, with activity against many clinically relevant yeasts and moulds (table 1). During the 1990s, newer lipid preparations of amphotericin B, including amphotericin B lipid complex (Abelcet; Enzon), liposomal amphotericin B (AmBisome; Astellas Pharma US), and amphotericin B colloidal dispersion (Amphotec; Three Rivers Pharmaceuticals) were developed to alleviate drug toxicity [27]. These agents possess the same spectrum of activity as does amphotericin B deoxycholate. Each agent has been shown to decrease nephrotoxicity in comparison with the conventional preparation of amphotericin B [28]. However, with the exception of findings for histoplasmosis, data supporting increased efficacy of the lipid products against common opportunistic fungal pathogens are lacking [29].
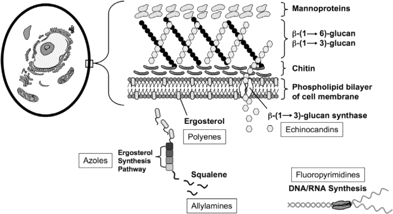
Targets of systemic antifungal agents
Amphotericin B and griseofulvin remained the only systemic therapeutic options for invasive fungal disease until the early 1970s, when flucytosine (Ancobon; Valeant Pharmaceuticals) was released. Flucytosine is a pyrimidine analog that exerts antifungal activity via inhibition of both DNA synthesis and protein synthesis in the fungal cell [30]. It also holds the distinction of being part of the only routinely recommended combination antifungal regimen for treatment of cryptococcal meningitis [31]. Unfortunately, the toxicity of this agent and the rapid development of resistance when used as monotherapy have precluded its routine use for the treatment of other invasive infections [5, 32, 33].
In 1979, the first systemic azole antifungal agent, ketoconazole, was introduced [4]. Azole agents exert their antifungal activity by blocking the demethylation of lanosterol, thereby inhibiting ergosterol synthesis [4]. Ketoconazole was followed chronologically by fluconazole (Diflucan; Pfizer), itraconazole (Sporanox; Janssen Pharmaceuticals), and voriconazole (Vfend; Pfizer) [34]. Four investigational agents remain in development: posaconazole, which has been submitted for US Food and Drug Administration approval, and ravuconazole, BAL8557, and albaconazole, which remain under study. Each agent offers a specific antifungal spectrum. Earlier agents in the class demonstrated potent activity against some, but not all, yeasts, and itraconazole had some activity against moulds, including Aspergillus species (table 1). The newer, expanded-spectrum triazoles have been shown to have cidal activity against a wide spectrum of moulds, as well as enhanced activity against Candida species and other yeasts [6, 7, 35].
The echinocandins represent the newest class of antifungals. Caspofungin (Cancidas; Merck) was released in 2001. This was followed by micafungin (Mycamine; Astellas Pharma US) in 2005 and anidulafungin (Eraxis; Pfizer). The mechanism of activity of the echinocandins is inhibition of the production of (1→3)-β-D-glucan, an essential component in the fungal cell wall [4]. The spectrum of activity is therefore limited to pathogens that rely on these glucan polymers and is less broad than the spectrums of the polyene or azole agents. The echinocandins exhibit fungicidal activity against many Candida species, making this drug class a desirable alternative to the azole agents, which exhibit only static activity against yeasts [35, 36]. Because mammalian cells have no cell wall, the echinocandins have very few toxic adverse effects in humans.
Pharmacokinetics and Pharmacodynamics
The selection of an appropriate antifungal agent depends on multiple factors in addition to the spectrum of activity. As with antibacterial therapy, the routes of administration and elimination are often important considerations in selecting a drug. This is particularly true when the optimal therapy for a patient with a fungal infection is being determined. Alterations in gastrointestinal tract integrity, impaired renal or hepatic function, and limited intravenous access are frequent issues for patients who are at high risk of acquiring fungal disease.
Further complicating the clinical picture is the variability in available formulations among different antifungal agents. Many drugs are available only as intravenous preparations (e.g., amphotericin B preparations and echinocandin agents) or only as oral preparations (e.g., posaconazole and flucytosine) because of differences in solubility and oral bioavailability. For the agents that can be administered by multiple routes (e.g., fluconazole, itraconazole, and voriconazole), there are often difficulties in administration of these preparations because of toxicities, drug interactions, and variability with different product formulations. Therefore, it is important to have an appreciation of the differences among these drugs with regard to their pharmacokinetic properties, including absorption, distribution, metabolism, and excretion.
Absorption. Several of the antifungal agents, including the polyene and echinocandin classes, do not have appreciable oral bioavailability. Until the early 1990s, the lack of oral treatment options left intravenous therapy as the only alternative for the treatment of invasive fungal infections. Today, each member of the azole class can be administered orally; however, the degree of absorption and optimal administration conditions vary for each of these drugs (table 2). Differences can even exist between various formulations of the same agent.
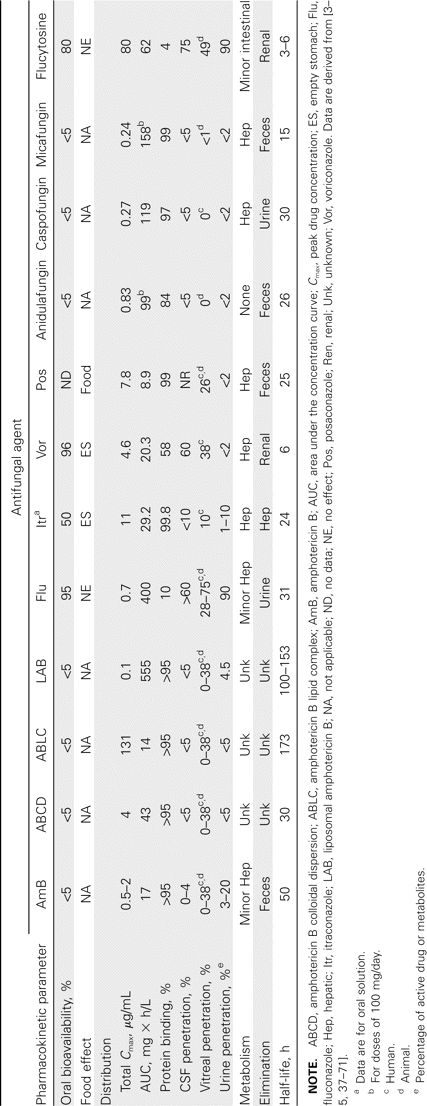
Comparative pharmacokinetics of the antifungal agents.
Fluconazole is readily absorbed, with oral bioavailability easily achieving concentrations equal to 90% of those achieved by intravenous administration [37]. Absorption is not affected by food consumption, gastric pH, or disease state [38, 39]. Variable gastrointestinal absorption does occur with the other members of this class, however, and, for one compound (itraconazole), it varies according to the specific formulation. Oral bioavailability of these agents can be also be affected by food consumption and changes in gastric pH.
Itraconazole capsules demonstrate optimal absorption in the presence of gastric acid and, therefore, cannot be coadministered with agents known to raise gastric pH, such as H2 receptor antagonists or proton pump inhibitors [72, 73]. Furthermore, itraconazole capsules should be administered after a full meal to optimize absorption [74]. In general, the cyclodextrin solution is more efficiently absorbed (i.e., the area under the concentration curve [AUC] is increased by 30%) than is the capsule formulation [40]. In addition, antacid therapy does not have a negative effect on absorption [41, 75]. Food can decrease serum concentrations of itraconazole solution; therefore, this preparation should be administered on an empty stomach [42, 43].
The oral bioavailability of voriconazole is >90% when the stomach is empty, but it decreases when food is present [44, 45]. Thus, this agent should be administered on an empty stomach. In contrast, posaconazole absorption is optimized when administered with a high-fat meal or a similar composition nutritional supplement, such as Boost Plus (Novartis Nutrition) [46].
Distribution. The distribution of antifungal agents in the body is another important factor to consider in the treatment of invasive fungal infections, because these infections may occur at physiologically sequestered sites. As demonstrated by relatively large volumes of distribution, the available antifungal agents are widely distributed throughout the body, with a few significant exceptions discussed below [37, 41, 45, 47, 48]. The main factors affecting drug distribution are molecular size, charge, degree of protein binding, and route of elimination.
Fungal infections of the CNS are associated with high morbidity and mortality and are difficult to treat. Many antifungal agents have large molecular weights that preclude their ability to penetrate the blood-brain barrier and achieve therapeutic CSF concentrations. Currently, flucytosine, fluconazole, and voriconazole have the best CSF penetration, with each resulting in concentrations of at least 50% of those seen in serum [49, 76, 77]. The concept of CSF concentrations predicting the efficacy of antifungal agents for CNS infections is a bit misleading. For example, amphotericin B, a drug that is essentially undetectable in CSF, has been the mainstay of treatment for cryptococcal meningitis, despite the lack of detectable drug concentrations in the CSF [31, 78]. In these instances, it is postulated that tissue concentrations of these agents are adequate to allow efficacy. Recent investigations have suggested that brain parenchymal concentrations of the azole agents and echinocandins may be more meaningful for prediction of therapeutic response.
Ophthalmologic fungal infections are also difficult to treat. Traditionally, topical therapies have been used for these infections, especially when the disease is limited to superficial infection. Many of the available systemic antifungal therapies can achieve intraocular concentrations adequate for treatment of more invasive disease (table 2). For other agents, localized therapy, such as intravitreal injections, is required for reliable concentrations within the vitreous body.
Relatively few available antifungal agents are renally eliminated as unchanged drug or active metabolite and, therefore, do not provide high concentrations of microbiologically active drug in the urine (table 2). Currently, fluconazole and flucytosine are the only drugs that can achieve reliable urine concentrations >50% of serum exposure when given systemically [48, 50]. It is important to note that, because many of these agents produce adequate tissue concentrations, a lack of detectable urine concentrations does not necessarily preclude use when the disease involves renal parenchyma.
The degree of protein binding is another characteristic that alters systemic exposure to drug; a protein-bound drug is not available for microbiologic activity. Thus, this factor plays an important role in determining the amount of active drug present at a given site of infection [79]. Unfortunately, the majority of available pharmacokinetic data for the antifungal agents reflect concentrations of total drug. Therefore, clinicians are left to hypothesize about the amount of measured drug actually available to fight infection (i.e., the portion of free, unbound drug). The polyene agents and many azole antifungals, with the exception of voriconazole and fluconazole, are highly protein bound (>90%). Protein binding with the echinocandin class varies from 85% to 99% for anidulafungin, caspofungin, and micafungin [4, 46, 47, 51–53, 80, 81].
The major protein that binds these drugs is albumin, although other serum proteins may also play a role [82, 83]. Many patients who are at risk for fungal infection are malnourished and, as a result, have low levels of serum albumin. The effect of this on protein binding of drugs may result in higher concentrations of available or active drug; however, this concept has not been sufficiently studied with regard to a potential effect on antifungal drug dosing or efficacy [79, 84–86].
Metabolism and elimination. Many systemic antifungal agents undergo some degree of hepatic metabolism before elimination. One notable exception is flucytosine, which is not known to be metabolized hepatically, because urine excretion of unchanged drug accounts for >90% of its elimination [48]. For the amphotericin B products, the exact routes of metabolism and elimination are largely unknown [4].
All azole antifungals undergo some degree of hepatic metabolism (table 2). For fluconazole, the role of metabolism in drug elimination is minimal, but this is not the case with itraconazole, voriconazole, and posaconazole, which are highly dependent on metabolism for drug elimination. Given that there are few active antifungal metabolites, this results in production of inactive compounds that provide no clinically meaningful activity, with the notable exception of hydroxyitraconazole (a metabolite of itraconazole) [87]. Although oxidative metabolism is the primary process involved in azole metabolism, glucuronide conjugation does occur with some of these drugs, especially posaconazole [88].
Each of the 2 available echinocandins (caspofungin and micafungin) undergoes metabolism to produce 2 distinct inactive metabolites. For caspofungin, these processes are hepatic hydrolysis and N-acetylation [89]. Micafungin undergoes nonoxidative metabolism to produce 2 distinct compounds [90]. Although it is a weak substrate for cytochrome P450 (CYP450), the metabolism of micafungin does not appear to be affected by inhibitors or substrates of this enzyme system. Unlike caspofungin and micafungin, anidulafungin is not hepatically metabolized but undergoes nonenzymatic degradation [91].
Effect of organ dysfunction on drug dosing. The effect of organ dysfunction on the elimination of the antifungal agents is summarized in table 3. Although the various formulations of amphotericin B are known for their ability to cause nephrotoxicity, they do not require dose adjustment for patients with decreased renal function. In fact, of all the available systemic antifungal agents, only fluconazole and flucytosine require dosing modification when given to patients with decreased levels of creatinine clearance [4, 48]. In some instances, such as with amphotericin B, dosing regimens may be altered in attempts to ameliorate toxicity, but this is not done as a result of altered drug clearance. Another example is the cyclodextrins, which are present in the intravenous preparations of itraconazole and voriconazole and can accumulate in renal disease. Therefore, the use of these formulations in patients with creatinine clearance <50 mL/min, in the case of voriconazole, and 30 mL/min, in the case of itraconazole, is cautioned for these formulations [41, 45].
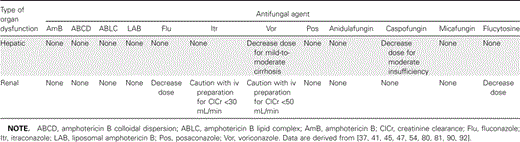
Suggested dose modifications for antifungal agents, by type of organ dysfunction.
Hepatic disease can also affect the elimination of several antifungal agents. For the majority of these agents, however, no dose alteration is recommended. Of the azoles, only voriconazole requires dose reduction for patients with mild-to-moderate cirrhosis [45]. Similarly, caspofungin is the only echinocandin with recommendations for dose modification in severe hepatic disease [47]. The metric used to determine appropriate dosing in hepatic disease is the Child-Pugh scoring system, which is appropriate for patients with chronic liver dysfunction but not for patients who have acute hepatic injury. Currently, information is not available to guide drug dosing in this clinical scenario.
Drug-drug interactions. The effect of antifungal agents on other therapeutic regimens merits serious consideration when therapy is being initiated or discontinued. Antifungal drugs can alter the safety or efficacy of concomitant therapies through several mechanisms. The first of these involves additive toxicities associated with concomitant administration; the most apparent example is nephrotoxicity caused by amphotericin B. This toxicity can enhance the renal effect of many other agents, including cyclosporine and the aminoglycosides [93].
A more complicated issue relates to the inhibition of drug metabolism that occurs as a result of these drug interactions. A complete review of CYP450-mediated drug interactions is beyond the scope of this article, but the importance of this effect should not be minimized [94]. Because of their mechanism of action, all the azole antifungals inhibit CYP450 enzymes to some degree (table 4). As a result, careful consideration must be given when an azole agent is added to a patient's drug regimen. Similarly, when an azole agent is discontinued, the change in metabolism that occurs may have profound clinical implications. For example, organ rejection has been reported after discontinuation of an azole antifungal that was not accompanied by the necessary upward dose adjustments in the affected immunosuppressant agent (e.g., calcineurin inhibitor) [97].
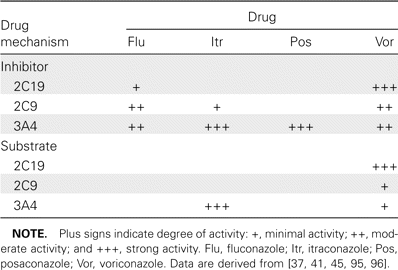
Summary of azole-mediated cytochrome P450 drug-drug interactions.
Although caspofungin and micafungin are not major substrates for the CYP450 enzyme system, they both have interactions that appear to be mediated via this mechanism. Caspofungin concentrations are decreased when administered with CYP450 inducers, such as rifampin and phenytoin [98]. Micafungin does have weak inhibitory properties against CYP3A4 and has been shown to increase serum concentrations of substrates of this enzyme. This phenomenon has been specifically assessed with sirolimus and nifedipine, and, in both cases, the AUC of the target drug was significantly elevated [54]. Anidulafungin does not appear to exhibit these CYP450-mediated interactions [91].
Initial product labeling for caspofungin indicated that a drug-drug interaction occurred when this agent was administered in combination with cyclosporine. This was based on data obtained in studies of healthy volunteers who received these drugs in combination as part of phase 1 development of caspofungin. After coadministration, an unacceptable elevation in hepatic enzymes was seen; therefore, cyclosporine was prohibited in clinical trials of caspofungin. This prohibition was reflected in the warning section of the initial caspofungin package insert [47]. More recent data, however, suggest that this effect is not reproducible in infected patients receiving the drug [99, 100]. In consideration of these data, these warnings have recently been revised in the product labeling.
Another mechanism involved in drug-drug interactions relates to the role of P-glycoprotein (P-gp). P-gp is a transporter protein involved in the absorption and distribution of drugs, as well as drug resistance. In a fashion similar to their interactions via the CYP450 enzyme system, azole antifungals have affinity for P-gp, because of their mechanism of action at the fungal cell membrane. More specifically, itraconazole is both a substrate and an inhibitor of P-gp, whereas fluconazole does not inhibit P-gp but may be a weak substrate. Therefore, interactions among the azole antifungals with P-gp may affect response to therapy or may play a role in interactions with other medications [101].
Pharmacodynamics. Another important consideration in the optimization of antifungal treatment regimens is the interaction between the fungal pathogen, the antifungal agent, and host factors. These pharmacodynamic principles have not been described for antifungal agents with the same level of detail as for the antibacterial agents. However, fairly extensive in vitro and animal model investigations have been undertaken with agents from the triazole, polyene, and echinocandin antifungal classes.
A series of reports has defined the pharmacokinetic exposure of these compounds relative to the MIC of the infecting pathogen as a means of optimizing treatment efficacy. In animal models of disseminated candidiasis, killing of fungal organisms with echinocandins and polyenes is optimized by achieving peak drug concentrations 2–10-fold in excess of the MIC [102, 103]. Treatment outcome with the triazole antifungals has been shown to correlate with the drug exposure over time, which is similar to the concentration needed to inhibit the organism in vitro, or the MIC. The pharmacokinetic index that best accounts for the entire exposure over time is the ratio of the 24-h AUC to the MIC (24-h AUC : MIC). In preclinical infection models, a free drug 24-h AUC : MIC value near 25 : 1 has been shown to reproducibly predict outcome with each of the triazole compounds [104]. Examination of clinical trial data with Candida infections has suggested that this pharmacodynamic relationship is similarly helpful for prediction of treatment efficacy in humans [105, 106]. The clinical relevance of the relationships between a specific drug exposure, the MIC, and outcome is less clear for other fungal pathogens and drug classes.
The relationship between antifungal pharmacokinetics and certain host toxicities has been demonstrated for a few compounds. Several decades ago, the relationship between flucytosine serum concentrations and bone marrow toxicity was elucidated [107]. More recently, the toxicodynamic relationship between higher-than-anticipated voriconazole exposure and hepatoxicity has been suggested [108]. More extensive evaluation of clinical data will be necessary to better understand these important pharmacodynamic relationships.
To aid clinicians in implementing these principles, serum drug concentration monitoring is now available for several of the available antifungal agents. Table 5 outlines the appropriate conditions for monitoring, target concentrations, and the association between this information and clinical outcomes, either therapeutic or toxic.
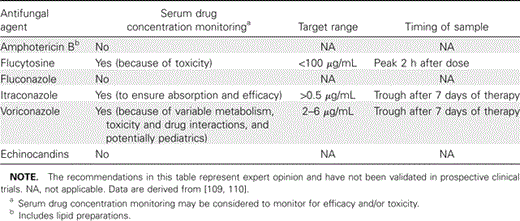
Serum drug concentration monitoring for antifungal agents.
Toxicities
Table 6 reviews the major comparative toxicities of the systemic antifungal agents available for management of invasive fungal disease. In addition to the types of toxicities presented (i.e., hepatic, renal, hematologic, and infusional toxicities and electrolyte abnormalities), each agent is associated with a set of unique adverse events, as described below.
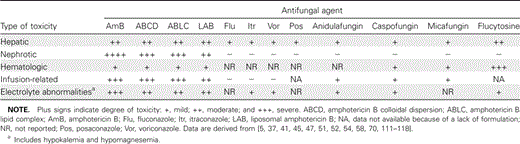
Comparative toxicities of antifungal agents.
Amphotericin B preparations. The toxicity of amphotericin B is well known. In addition to the nephrotoxicity and acute infusion-related reactions associated with the drug, a unique pulmonary reaction can be seen, particularly with certain lipid preparations. With the liposomal preparation of amphotericin B, a triad of infusional toxicity has been characterized. This toxicity can manifest as a combination of the following clinical scenarios: pulmonary toxicity (i.e., chest pain, dyspnea, and hypoxia); abdominal, flank, or leg pain; or flushing and urticaria [119, 120]. Similarly, with amphotericin B colloidal dispersion, severe hypoxia has been reported in patients; in one study, hypoxia occurred more commonly in association with the use of amphotericin B colloidal dispersion than with amphotericin B deoxycholate [121]. Hypoxia has also been reported in association with use of the lipid complex of amphotericin B. In one study, up to 20% of patients experienced this toxicity. Unique characteristics in this case included onset of symptoms beyond the second day of therapy for >70% of patients [111].
Azole antifungal agents. Fluconazole is an extremely well-tolerated agent that lacks significant toxicity, despite having been used for treatment and prophylaxis in many patient populations for more than a decade. However, reversible alopecia is not uncommon with this agent [122].
Oral itraconazole solution is also relatively safe but can be associated with nausea and diarrhea severe enough to force discontinuation. This reaction is caused by the excipient hydroxypropyl-β-cyclodextrin, which is used to increase solubility of the parent drug [123]. Itraconazole has been described as causing a unique triad of hypertension, hypokalemia, and edema, mostly in older adults [124]. A negative inotropic effect resulting in congestive heart failure has also been described and has prompted changes to the package labeling to avoid administration of itraconazole to patients with a history of heart failure [41, 125].
Two unique adverse events have been associated with the use of voriconazole: visual disturbances and cutaneous phototoxicity. The mechanism for visual disturbances is not known but manifests itself as photopsia (i.e., the appearance of bright lights, color changes, or wavy lines) or abnormal vision in up to 45% of patients receiving the treatment [126]. This effect is usually mild and transient, and it abates with continued treatment. In addition, this effect appears to be associated with higher doses of voriconazole [112]. Rash has been reported in association with voriconazole use in up to 8% of subjects; phototoxicity-related rash occurs less frequently but is a significant problem for ambulatory patients [127, 128]. This effect is not prevented through the use of sunscreens but is reversible after discontinuation of therapy.
Posaconazole has been well tolerated in clinical trials to date. The most frequently reported adverse events attributed to the drug have been associated with hepatic toxicities. These toxicities seem to occur less frequently than with other members of the triazole class [113]. Fatal hepatotoxicity has been reported with itraconazole, voriconazole, and posaconazole. Therefore, close monitoring of hepatic function is warranted with all members of the azole class [41, 45].
Echinocandins. The echinocandins are associated with few toxicities, making them safe agents to administer. The most notable, yet uncommon, event reported is a histamine-mediated infusion-related reaction. As with vancomycin, this reaction can be relieved by slowing the rate of infusion or premedicating with an antihistamine, such as diphenhydramine.
Conclusion
Clinicians now have access to an expanded number of antifungal agents; however, the panacea of antifungal therapy remains to be found. Therefore, a keen appreciation of the properties associated with each antifungal agent is imperative in the selection and administration of antifungal therapy. Differences in the pharmacokinetics of each unique drug render effective administration a challenge, particularly given the complex regimens that patients who are at risk for fungal infection receive because of their underlying disease states. Toxicity profiles also play a major role in the treatment of fungal disease, and differences among the antifungal classes, as well as agents within a given class, must be understood. With judicious use of the available agents, we are able to successfully and safely treat a growing number of life-threatening infections.
Acknowledgments
Financial support. This article was supported by an educational grant from Schering-Plough.
Potential conflicts of interest. E.S.D.A. has received grants, research support, consultation fees, and honoraria from Pfizer, Astellas Pharma US, Enzon Pharmaceuticals, Merck, and Schering-Plough and is on the paid speakers' bureaus of Pfizer, Astellas Pharma US, Enzon Pharmaceuticals, and Schering-Plough. R.L. has received grants and research support from Merck, Pfizer, Astellas Pharma US, and Enzon Pharmaceuticals; has received consultation fees from Merck, Schering-Plough, Pfizer, and Enzon Pharmaceuticals; has received honoraria from Merck, Pfizer, and Schering-Plough; and is on the paid speakers' bureaus of Merck and Astellas Pharma US. J.S.L. has received honoraria and consultation fees from Pfizer, Astellas Pharma US, and Schering-Plough and is on the paid speakers' bureaus of Pfizer, Astellas Pharma US, and Schering-Plough. C.M. has received grants and research support from Ortho-McNeil; has received consultation fees from Wyeth Pharmaceuticals, Ortho-McNeil, and Elan Pharmaceuticals; has received honoraria from Pfizer and Ortho-McNeil; and is on the paid speakers' bureaus of Ortho-McNeil, Wyeth Pharmaceuticals, and Elan Pharmaceuticals. D.A. has received grants and research support from Vicuron Pharmaceuticals, Schering-Plough, Pfizer, GlaxoSmithKline, Astellas Pharma US, and Enzon Pharmaceuticals; has received consultation fees from Vicuron Pharmaceuticals, Merck, Schering-Plough, Bristol-Myers Squibb Cytogenetics, Conjugon, Pfizer, and Astellas Pharma US; and has received honoraria from Merck and Pfizer.
References
Figures and Tables
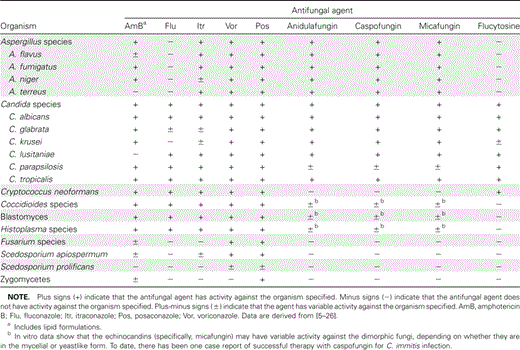
Antifungal spectrum of activity against common fungi.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments