




Abstract
The pathophysiology of spontaneous upbeat (UBN) and downbeat (DBN) nystagmus is reviewed in the light of several instructive clinical findings and experimental data. UBN due to pontine lesions could result from damage to the ventral tegmental tract (VTT), originating in the superior vestibular nucleus (SVN), coursing through the ventral pons and transmitting excitatory upward vestibular signals to the third nerve nucleus. A VTT lesion probably leads to relative hypoactivity of the drive to the motoneurons of the elevator muscles with, consequently, an imbalance between the downward and upward systems, resulting in a downward slow phase. The results observed in internuclear ophthalmoplegia suggest that the medial longitudinal fasciculus (MLF) is involved in the transmission of both upward and downward vestibular signals. Since no clinical cases of DBN due to focal brainstem damage have been reported, it may be assumed that the transmission of downward vestibular signals depends only upon the MLF, whereas that of upward vestibular signals involves both the MLF and the VTT. The main focal lesions resulting in DBN affect the cerebellar flocculus and/or paraflocculus. Apparently, this structure tonically inhibits the SVN and its excitatory efferent tract (i.e. the VTT) but not the downward vestibular system. Therefore, a floccular lesion could result in a disinhibition of the SVN–VTT pathway with, consequently, relative hyperactivity of the drive to the motoneurons of the elevator muscles, resulting in an upward slow phase. UBN also results from lesions affecting the caudal medulla. An area in this region could form part of a feedback loop involved in upward gaze-holding, originating in a collateral branch of the VTT and comprising the caudal medulla, the flocculus and the SVN, successively. Therefore, it is suggested that the main types of spontaneous vertical nystagmus due to focal central lesions result from a primary dysfunction of the SVN–VTT pathway, which becomes hypoactive after pontine or caudal medullary lesions, thereby eliciting UBN, and hyperactive after floccular lesions, thereby eliciting DBN. Lastly, since gravity influences UBN and DBN and may facilitate the downward vestibular system and restrain the upward vestibular system, it is hypothesized that the excitatory SVN–VTT pathway, along with its specific floccular inhibition, has developed to counteract the gravity pull. This anatomical hyperdevelopment is apparently associated with a physiological upward velocity bias, since the gain of all upward slow eye movements is greater than that of downward slow eye movements in normal human subjects and in monkeys.
Vertical nystagmus may be either upbeating or downbeating. When present in the straight-ahead position of gaze (i.e. the primary position) it is referred to as ‘upbeat nystagmus’ (UBN) or ‘downbeat nystagmus’ (DBN) (Leigh and Zee, 1999). DBN is usually greater on looking laterally or in downgaze, whereas UBN often increases on upgaze. Both forms of nystagmus may be affected by head position and by convergence. These types of spontaneous vertical nystagmus should be distinguished from the more common vertical gaze-evoked nystagmus observed only in upgaze or only in downgaze. In spite of many reports of DBN and UBN and multiple hypotheses about possible mechanisms, the pathophysiology is still not understood (Leigh et al., 2002; Halmagyi and Leigh, 2004; Marti et al., 2005b). A general concept is that asymmetries in the cerebello-brainstem network that normally stabilizes vertical gaze could lead to an imbalance in structures such as (i) the vertical cerebello-vestibular ‘neural integrator’, making it possible to hold the eyes steady in upgaze or downgaze against the mechanical pull on the eyes, and/or (ii) the central connections of the vertical vestibulo-ocular reflexes (VOR), including both the semicircular canal and the otolithic responses, or even (iii) the vertical smooth pursuit system. The discussion here will be based on six major clinical facts concerning UBN or DBN, which will be interpreted in the light of experimental data and some other clinical findings. We will successively (i) review UBN due to pontine lesions, (ii) examine the changes in the vertical VOR observed in internuclear ophthalmoplegia (INO), (iii) note the absence of DBN due to clinical focal brainstem lesions, (iv) interpret the mechanism of DBN due to focal cerebellar floccular lesions, (v) consider the mechanism of UBN due to focal caudal medullary lesions, (vi) discuss the influence of head position with respect to gravity on DBN and UBN, and (vii) propose that these two types of nystagmus result from a primary dysfunction of the same upward vestibular pathway.
UBN due to pontine lesions
UBN may be due to focal brainstem lesions. Two main groups of patients predominate, the first with pontine lesions and the second with medullary lesions (see below, UBN due to caudal medullary lesions). In cases of UBN due to pontine damage, the lesions are located in the ventral tegmentum and/or the posterior basis pontis, at the upper pons level, and are usually large and bilateral (Troost et al., 1980; Fisher et al., 1983; Hirose et al., 1991), though a patient with a small lesion has also been reported (C. Pierrot-Deseilligny, D. Milea, J. Sirmai, C. Papeix and S. Rivaud-Péchoux, submitted for publication). These lesions could damage an ascending vestibular tract (Ranalli and Sharpe, 1988a), called the ‘ventral tegmental tract’ (VTT), described in the cat (Carpenter and Cowie, 1985; Sato and Kawasaki, 1987; Uchino et al., 1994) and probably also existing in the monkey (Sato and Kawasaki, 1991). The VTT appears to connect the superior vestibular nucleus (SVN), receiving the excitatory anterior canal inputs generating the upward slow phases, to the superior rectus and inferior oblique motoneurons in the third nerve (IIIrd) nucleus (Sato and Kawasaki, 1991). The course of the VTT in the brainstem (Uchino et al., 1994) initially appears to be slightly ventral and lateral to the brachium conjunctivum (BC) in the lower pons, i.e. in the lateral tegmentum, arching medially and joining the ventral tegmentum in the midpons. The tract decussates slightly above the level of the midpons, close to the upper pole of the nucleus reticularis tegmenti pontis (NRTP), this decussation being located in humans perhaps in the posterior part of the basis pontis (C. Pierrot-Deseilligny, D. Milea, J. Sirmai, C. Papeix and S. Rivaud-Péchoux, submitted for publication). The tract then runs rostrally on the opposite side in the ventral tegmentum of the upper pons, near the medial part of the medial lemnicus, and, in the midbrain, arches medially, near the caudal pole of the red nucleus, before reaching the IIIrd nucleus on both sides. This course could explain how a relatively small unilateral paramedian lesion at the upper (NRTP) level, probably involving the VTT decussation system (i.e. both VTTs), may result in marked UBN (C. Pierrot-Deseilligny, D. Milea, J. Sirmai, C. Papeix and S. Rivaud-Péchoux, submitted for publication). The upward VOR gain is often decreased in cases of UBN (Baloh and Yee, 1989; C. Pierrot-Deseilligny, D. Milea, J. Sirmai, C. Papeix and S. Rivaud-Péchoux, submitted for publication). Therefore, it may be assumed that after a VTT lesion there is relative hypoactivity in the final part of this upward vestibular pathway, eliciting imbalance with the downward vestibular system, which is not directly affected (Fig. 1A). The consequence is a downward slow phase, regularly interrupted by upward quick phases generated by the saccadic system. UBN due to pontine lesions is usually reported to have a large amplitude (between 10° and 15°) (Hirose et al., 1991; C. Pierrot-Deseilligny, D. Milea, J. Sirmai, C. Papeix and S. Rivaud-Péchoux, submitted for publication), and may disappear after 2 or 3 months (Fisher et al., 1983; C. Pierrot-Deseilligny, D. Milea, J. Sirmai, C. Papeix and S. Rivaud-Péchoux, submitted for publication). The eventual disappearance of UBN suggests that an adaptive mechanism can ultimately nullify this type of spontaneous vertical nystagmus. Accordingly, since after such pontine lesions there is a decrease in the upward VOR gain, it may be concluded that the excitatory SVN–VTT pathway is important for mediating both upward eye velocity vestibular signals and upward eye position signals.
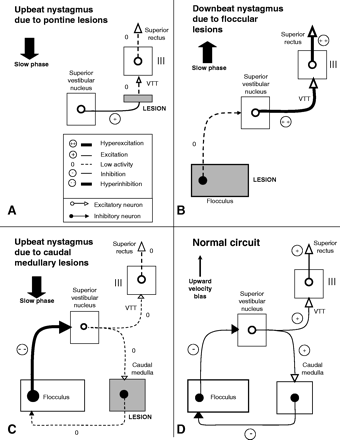
Hypothetical pathophysiology of vertical nystagmus. Only the cerebello-brainstem pathway (on one side), assumed to be mainly involved in primary position upbeat nystagmus (UBN) or downbeat nystagmus (DBN), is shown. (A) UBN due to pontine lesions: the ventral tegmental tract (VTT), originating in the superior vestibular nucleus (SVN), is probably impaired (bilaterally; see text), with consequently relative hypoactivity of the elevator muscle motoneurons, with respect to the unchanged downward system, eliciting a downward slow eye deviation. (B) DBN due to floccular lesions. Since the flocculus normally inhibits the SVN, the lesion results in disinhibition of the downstream pathway, with consequently relative hyperexcitation of the elevator muscle motoneurons, compared with the unchanged downward system, eliciting an upward slow eye deviation. (C) UBN due to caudal medullary lesions. The caudal medulla (nucleus of Roller and/or a cell group of the paramedian tracts), which could receive a collateral branch from the SVN and project to the flocculus via a probably inhibitory pathway, is impaired. The result is disinhibition of the inhibitory flocculovestibular neurons, which are then overactivated, eliciting overinhibition of the downstream pathway (VTT), i.e. low activity with respect to the downward system, with consequently (as in A) a slow downward deviation of the eye. (D) Normal circuit, derived from the clinical results observed in A, B and C and anatomical experimental data known for the cat and the monkey. This circuit could specifically be involved in the upward vestibular system, and does not appear to have an equivalent in the downward system; the result could be a slight upward velocity bias in the normal state.
A few patients with UBN attributed to unilateral BC lesions have also been reported (Nakada and Remler, 1981; Benjamin et al., 1986; Kattah and Dagli, 1990). Such cases are rare and were not fully documented (i.e. without eye movement recordings and/or without modern imaging). Furthermore, they comprised large median tumoral or haemorrhagic lesions, always with associated damage to the cerebellar vermis, which in itself may result in UBN (Baloh and Yee, 1989; Leigh and Zee, 1999). Therefore, these clinical cases are not thoroughly convincing for localization to the BC, especially since this tract and the VTT are near each other in the lower pons. Nevertheless, the role of the BC should be considered since this structure is generally supposed to transmit vertical slow eye movement signals to the IIIrd nucleus, so that theoretically it is possible for UBN to appear after BC damage. However, it seems probable that the transmission of upward vestibular signals is in fact mainly performed by the VTT rather than the BC (Ito, 1982; Sato and Kawasaki, 1991; Uchino et al., 1994). By contrast, the BC could mediate vertical smooth pursuit signals, both upwards and downwards, even though the upward signals could predominate (Chubb and Fuchs, 1982). These signals originate in the cerebellar dentate nucleus and in the y-group region (Zhang et al., 1995a), the latter lying close to the SVN in the brainstem. There are also clinical data supporting the notion that vertical smooth pursuit signals are transmitted mainly through the BC and not through the medial longitudinal fasciculus (MLF): upward and downward smooth pursuit was largely preserved after bilateral MLF lesions (Ranalli and Sharpe, 1988b) and after large bilateral pontine tegmental lesions involving both MLFs and the reticular formations, but sparing the BC (Larmande et al., 1982; Pierrot-Deseilligny et al., 1989) in spite of severe impairment of the vertical VOR in these two types of syndromes. Therefore, the BC appears to be more involved in the transmission of vertical smooth pursuit signals than in that of the vertical (upward) VOR, but this point has no yet been settled.
Results from internuclear ophthalmoplegia
INO is a key syndrome to consider, since the MLF contains vestibular tracts involved in both vertical directions in the cat and the monkey (Carpenter and Cowie, 1985; Graf and Ezure, 1986; McCrea et al., 1987; Highstein and McCrea, 1988; Iwamoto et al., 1990; Sato and Kawasaki, 1990; Büttner-Ennever, 1992). There are excitatory upward and downward vestibular tracts, originating in the medial vestibular nucleus (MVN) and passing through the contralateral MLF, and inhibitory upward and downward vestibular tracts, originating in the SVN and passing through the ipsilateral MLF. Among these tracts, the excitatory upward MVN–MLF tract is of particular interest, since it theoretically plays a role analogous to that of the SVN–VTT tract described above, thus with two different tracts transmitting excitatory upward vestibular signals to the elevator muscle motoneurons. After MLF lesions, the upward and downward VOR gains are severely impaired, reduced to a third of their normal values, and with markedly less impairment of vertical smooth pursuit (Ranalli and Sharpe, 1988b). Analogous results—with a severely impaired vertical VOR (both upwards and downwards)—were observed after experimental lesions in the monkey (Evinger et al., 1977). However, in a more recent report of a single patient with unilateral INO, using head rotations with high acceleration, the upward VOR gain was also impaired but less severely than the downward VOR gain, which is consistent with transmission of the upward vestibular signals through both the MLF and an extra-MLF tract (Cremer et al., 1999). Thus, the MLF transmits vertical eye velocity vestibular signals in both vertical directions but perhaps slightly more so for the downward system. Furthermore, INO is usually associated with a vertical gaze-evoked nystagmus—in downgaze and/or (more frequently) in upgaze—but nystagmus in the straight ahead position of gaze has not been reported after pure bilateral MLF lesions (Evinger et al., 1977; Kirkham and Katsarkas, 1977; Pierrot-Deseilligny and Chain, 1979; Müri and Meienberg, 1985; Ranalli and Sharpe, 1988b; Leigh and Zee, 1999). This confirms that the MLF also transmits both downward and upward eye position signals. Accordingly, after an MLF lesion, no major imbalance exists between the two vertical vestibular systems, at least in the straight-ahead position of gaze, probably because eye velocity and eye position signals are impaired for both vertical directions.
Absence of DBN after focal clinical brainstem lesions
Thus far, our analysis suggests that excitatory upward vestibular signals are transmitted in the brainstem through both the VTT and the MLF, whereas excitatory downward vestibular signals appear to depend only upon the MLF. With such findings, one must ask whether there is in the brainstem an equivalent of the VTT for the downward system. If so, this tract should be anatomically different both from the VTT, involved only in the upward system, and from the MLF, involved in both vertical systems. However, in contrast to reports of about 30 cases of UBN due to focal brainstem (pontine or caudal medullary) lesions, a careful review of the reported cases of DBN leads to the conclusion that there are no cases of DBN due to a focal brainstem lesion, at least located between the vestibular and IIIrd nuclei (Büttner et al., 1995). Since modern imaging methods have existed for many years, it can reasonably be concluded that such cases do not exist. There is, however, a single reported patient with DBN resulting from focal brainstem damage, with small bilateral cavities of syringomyelia located in each lateral part of the medulla (Bertholon et al., 1993). The interpretation, based only on magnetic resonance imaging findings, was that the MVN and/or its efferent tracts, controlling the downward vestibular system, could be directly affected on both sides by these lesions, whereas the SVNs were probably preserved. Thus, the absence of any other focal brainstem lesion resulting in DBN is actually fundamental since it implies that there is no equivalent of the VTT for the downward vestibular system and, therefore, that there is either something missing in the downward vestibular system in terms of central connections or, more likely, that there is something additional (i.e. the VTT) in the upward vestibular system.
Mechanism of DBN
To try to understand how the downward vestibular signals are transmitted to the IIIrd and trochlear nuclei, we must now examine the different causes of DBN. This nystagmus is observed in diverse intoxications and diffuse diseases (encephalitis, etc.), like UBN, but its main causes—cerebellar atrophy and craniocervical anomalies—are relatively focal and involve regions located outside the brainstem (Leigh and Zee, 1999). At the cerebellar level, the flocculus and paraflocculus are involved in slow eye movements and, in the vertical plane, mainly control downward eye movements (for 90% of Purkinje cells) (Leigh and Zee, 1999). It should also be noted that an inhibitory flocculovestibular tract projects to the SVN, exerting permanent tonic inhibition of this nucleus, without known equivalent flocculovestibular inhibition of the parts of the MVN controlling the downward system or the excitatory upward MVN–MLF pathway (Ito et al., 1977; Ito, 1982; Sato and Kawasaki, 1990; Zhang et al., 1995a, b; Leigh et al., 2002). Furthermore, experimental floccular and/or parafloccular lesions in the monkey result in large DBN (Zee et al., 1981). An analogous mechanism could exist in humans, in whom the flocculus and paraflocculus are obviously impaired in the two main, relatively focal causes of DBN. A floccular (or parafloccular) lesion could result in disinhibition of the SVN, with relative hyperexcitation throughout the downstream pathway, including to the superior rectus motoneurons (Marti et al., 2005b) (Fig. 1B). Primary dysfacilitation of the pathways to the depressor muscles is also theoretically possible, but is improbable since the flocculovestibular projections are inhibitory and do not seem to be involved in the downward system. Thus, with floccular lesions, the disinhibited superior rectus motoneurons would be relatively more active than the inferior rectus motoneurons, which remain unchanged. The consequence of this imbalance would be upward slow phases and corrective downward quick phases. In support of a primary hyperactivity in the upward vestibular system in patients with DBN, the upward VOR gain is often increased in these patients (Halmagyi et al., 1983; Gresty et al., 1986; Leigh and Zee, 1999). In patients with DBN, the existence of asymmetry of the VOR in response to rapid pitch rotations (i.e. with downward pitch impulses eliciting a greater response than upward pitch impulses) is a further argument supporting disinhibition of the anterior canal inputs (Walker and Zee, 2005). It should be noted that, in most cases, the upward VOR gain is merely greater than the downward VOR gain (Baloh and Yee, 1989), suggesting that, if the basic disturbance is hyperexcitation of the upward system, the impairment load has probably already been redistributed between both vertical systems by adaptive mechanisms. Of course, in patients who have had DBN for a long time, different types of adaptive mechanisms might change the characteristics of the slow phase of the nystagmus. Therefore, one must be cautious in interpreting the mechanism of DBN at these late stages, in particular in patients with apparently idiopathic DBN. For example, while Glasauer and colleagues suggested, based upon 3D analysis of the slow-phase characteristics in patients with chronic DBN, that there is no evidence of a central vestibular damage but rather an impairment of the cerebello-brainstem integrator (Glasauer et al., 2003), it may be argued that adaptive mechanisms have had time to change the basic pattern of nystagmus. Indeed, any interpretation of a centrally induced nystagmus, especially if the cerebellum or cerebellar pathways are involved, must be tempered with the caveat that adaptive mechanisms, too, may be shaping the response, either as a normal adaptive response or as a maladaptive response associated with the lesions themselves.
The amplitude (eye position) of DBN is variable, ranging between a few degrees and 10–15°. In the case of a small DBN (with an amplitude of a few degrees), it seems surprising that the adaptive mechanisms fail to improve or suppress the abnormal movement. However, where there is progressive (degenerative or malformative) or even diffuse damage, these mechanisms may have already been exhausted, resulting in long-lasting DBN, often increasing with time. Lastly, there is currently no obvious explanation for the increase in DBN amplitude (and UBN amplitude as well) frequently observed in lateral gaze and/or convergence. However, malfunctioning of the translational VOR, which uses otolith cues and normally modulates slow-phase velocity according to the angle of vergence and the positions of the eye in the orbit, could be expected in such damage (Leigh and Zee, 1999), but further specific studies will be needed to resolve this question.
Two particular types of experimental brainstem lesions, without equivalent syndromes in humans, also elicit DBN. First, in the cat, a (primary position) DBN was observed after a muscimol injection made in a subgroup of cells of the paramedian tracts (PMT) (Nakamagoe et al., 2000). The PMT cells are located between the MLFs, both rostrally and caudally to the abducens nuclei (Büttner-Ennever et al., 1989). They receive afferent signals from all premotor structures involved in horizontal and vertical eye movements (i.e. the brainstem reticular formations generating saccades as well as the vestibular nuclei controlling slow eye movements) and they project to the flocculus. The rostral PMT subgroup, where damage resulted in DBN (Nakamagoe et al., 2000), was located just above the level of the abducens nuclei. Electrophysiological recording, performed before the lesion, showed that this area was involved in both upward saccades and VOR. Furthermore, the DBN slow phase induced by the lesion had an exponentially decaying profile, suggesting impaired neural integration (see next section). The interpretation of this DBN was (i) hypoactivity in the (putative excitatory) PMT floccular neurons, which normally receive signals from the SVN, with, therefore, (ii) hypoactivity in the inhibitory flocculo-SVN neurons, and then (iii) disinhibition of the upward vestibular pathway, as after floccular lesions, namely relative hyperexcitation in the drive to the motoneurons of the elevator muscles, and (iv) an upward slow phase. Thus, this rostral subgroup of PMT cells could be involved in the downward gaze-holding system. The absence of an analogous syndrome in humans may be explained by the very particular location of this subgroup of PMT cells, namely between the MLFs, with therefore the impossibility of observing a specific clinical lesion of the PMT cells without associated damage to the MLFs. Since the MLFs are involved in both the upward and the downward vestibular systems, a lesion affecting both these fascicles and the PMT cells probably results in relatively balanced deficits between the two vertical systems, thus eliciting only vertical gaze-evoked nystagmus, not nystagmus in the straight-ahead position of gaze. Secondly, in the monkey, midsagittal pontomedullary sectioning of the posterior tegmentum resulted in bilateral INO and DBN (De Jong et al., 1980). INO was explained by the interruption of abducens nucleus interneurons decussating before ascending in the MLFs. The same lesion could also involve the excitatory upward and downward vestibular neurons passing through the MLFs and decussating at the same caudal brainstem level. The interpretation of the DBN in this experiment is difficult, since it could have resulted (i) from damage to the same PMT cells mentioned above in the cat, and/or (ii) from the interruption of the mainly inhibitory vestibular commissural system (Ito, 1982; Fukushima and Kaneko, 1995), connecting the two SVNs and thus resulting in disinhibition of these nuclei and hyperexcitation of the downstream pathway. However, whatever the mechanism of the DBN in these midsagittal lesions and the actual role of the rostral subgroup of PMT cells, both these experiments probably resulted in hyperactivity of the SVNs, as after floccular lesions, without therefore any further argument for the existence in the downward system of an ascending vestibular tract equivalent to the VTT.
Accordingly, in DBN, both eye velocity and eye position signals appear to be impaired in most cases, suggesting imbalance both in the central vestibular connections and in the vertical gaze-holding system. It may be assumed that the specific inhibitory flocculo-SVN tract involved in the downward VOR normally inhibits the specific excitatory SVN–VTT pathway involved in the upward VOR, as shown by experimental data (Hirai and Uchino, 1984; Sato and Kawasaki, 1990; Uchino et al., 1994). However, such an organization with a specific inhibitory flocculovestibular pathway involved in downward eye movements does not really solve the problem of the apparently missing excitatory downward vestibular tract, compared with the upward vestibular system, in particular for the movements performed between the straight-ahead position of gaze and downgaze, where a simple inhibitory mechanism is usually not sufficient to overcome the orbital viscoelastic forces. Another mechanism, not involving a further downward excitatory vestibular tract, might, however, be involved (see below, Role of gravity in vertical nystagmus and VOR).
Before leaving the mechanisms of DBN, it should be noted that a specific impairment of smooth pursuit has at times been suggested to account for this nystagmus (Leigh and Zee, 1999). In support of the smooth pursuit hypothesis, it has recently been shown that DBN may be transitorily reproduced in healthy subjects after prolonged training using asymmetrical smooth pursuit stimulation (Marti et al., 2005a). However, such a result does not constitute evidence that the cerebellar pathological DBN is also due to specific smooth pursuit impairment. Moreover, the vertical VOR and optokinetic nystagmus (optokinetic nystagmus) were not tested in this study. The results could simply confirm that, at the cerebello-brainstem level, smooth pursuit and all other slow eye movements share similar structures and mechanisms, with an analogous imbalance in favour of the upward system (see Conclusions). Therefore, even though the smooth pursuit system is obviously involved in the vertical slow eye-movement disturbances in DBN (and also in UBN), there is no definite evidence that the smooth pursuit impairment could be the primary cause of spontaneous vertical nystagmus.
UBN due to caudal medullary lesions
We have already noted two different focal clinical causes of vertical nystagmus, the first related to VTT damage in the pons, resulting in UBN, and the second to the impairment of the flocculus, resulting in DBN. However, there is another region involved in the mechanisms of UBN since caudal medullary lesions, usually affecting the paramedian part of the posterior tegmentum bilaterally, result in UBN in humans (Gilman et al., 1977; Keane and Itabashi, 1981; Fisher et al., 1983; Kato et al., 1985; Baloh and Yee, 1989; Munro et al., 1993; Tyler et al., 1994; Janssen et al., 1998; Hirose et al., 1998; Ohkoshi et al., 1998; Minagar et al., 2001; Tilikete et al., 2002). The pathophysiology of this UBN is not yet known. Since most lesions were located inferiorly to the nucleus prepositus hypoglossi (NPH) in the posterior paramedian part of the medulla, it has at times been suggested that the nucleus intercalatus (NI), lying just caudally to the NPH, could be involved. However, if the afferent and efferent tracts of the different lower medullary nuclei (Büttner-Ennever and Büttner, 1988) are carefully examined, no obvious link with UBN can be found if the NPH or NI circuitry is considered. By contrast, the nucleus of Roller (NR), as suggested by Keane and Itabashi (1981), appears to be a better candidate to play a role in upward vestibular eye movements. This small nucleus is located at the same caudal medullary level as the NI, lying slightly anteriorly and medially to the superior part of this nucleus. Therefore, the NR (or one of its adjacent afferent or efferent tracts) was probably also damaged in most, if not all, of the caudal medullary lesions resulting in UBN. This nucleus (i) is particularly well developed in higher primates (chimpanzees and humans) (Büttner-Ennever and Büttner, 1988), (ii) receives a strong projection from the SVN (McCrea et al., 1987), probably via a collateral branch of the vestibulo-oculomotor neurons (the axons of which could pass through the VTT), and (iii) projects strongly to the flocculus (Langer et al., 1985b), via a tract which could be inhibitory (Büttner-Ennever and Büttner, 1988). Such a pathway might explain the mechanism of UBN due to a caudal medullary lesion (Fig. 1C): after a lesion affecting the NR itself or an adjacent region in the rostrocaudal axis (i.e. its immediate afferent tract, originating in the upper medulla, or its efferent tract, running more caudally in the medulla), the activity of the medullofloccular tract would be interrupted, resulting in disinhibition of the inhibitory flocculovestibular neurons and, therefore, in ‘hyperinhibition’ of the whole downstream pathway, namely the vestibulo-oculomotor neurons passing through the VTT. The end result would be similar to that observed after a VTT lesion, in other words relative hypoactivity in the drive to the superior rectus motoneurons with, consequently, downward slow phases and upward quick phases. Another possibility is that the medullary lesions resulting in UBN affect a caudal subgroup of the PMT cells (Büttner et al., 1995). Such PMT cells do exist in the vicinity of the NR and NI, and the general principle of the connectivity of the PMT cells is to receive afferent signals from the premotor (including vestibular) structures and to project to the flocculus (Büttner-Ennever et al., 1989). It has yet to be demonstrated, however, that these putative caudal PMT cells are actually involved in upward gaze-holding. If this is indeed the case, these caudal PMT cells could be the counterpart for the upward ocular motor system of the rostral PMT cells involved in downward gaze-holding in the cat (see above, Mechanism of DBN).
The UBN amplitude (eye position) in patients with caudal medullary lesions was variable, ranging between 1° or 2° and 10°. In those studies where the outcome was reported, the improvement or disappearance of UBN was also variable, ranging between a few weeks (Janssen et al., 1998) and a few months (Tilikete et al., 2002), but with persistence for at least 2 years in one patient (Baloh and Yee, 1989). Therefore, adaptive mechanisms appear to be possible here, too, as with UBN due to pontine lesions. It should be noted that the vertical VOR was never tested in any of these cases of UBN due to medullary lesions. Lastly, no torsional component was described in these patients, which suggests that the lesions were effectively bilateral since a unilateral lesion of the vertical VOR pathways might be expected to cause a mixed vertical torsional nystagmus (Leigh and Zee, 1999), as in one pontine case with UBN (C. Pierrot-Deseilligny, D. Milea, J. Sirmai, C. Papeix and S. Rivaud-Péchoux, submitted for publication).
Since an area in the caudal medulla probably belongs to the vertical gaze-holding network, specific abnormalities in the profile of the UBN slow phase could be expected after a lesion of this area. However, the exponential characteristic of the slow phase, theoretically allowing one to distinguish between damage to the gaze-holding neural integrator and an imbalance in other ocular motor pathways, is probably not a reliable sign: (i) it may be difficult to appreciate even using eye movement recordings, especially if there are frequent quick phases; (ii) the waveform of the slow phase was variable in previously reported patients with caudal medullary lesions; (iii) the exponential waveform of the slow phase was intermittent in a patient with a VTT lesion (C. Pierrot-Deseilligny, D. Milea, J. Sirmai, C. Papeix and S. Rivaud-Péchoux, submitted for publication) and was variable in individual patients with UBN (or DBN) (Leigh and Zee, 1999); and (iv) all the brainstem vestibular pathways are more or less part of the gaze-holding network (Fukushima and Kaneko, 1995). Lastly, the amplitude of UBN (and also of DBN) often depends upon the vertical position of the eyes in the orbits, since, for example, in UBN due to caudal medullary lesions, the nystagmus amplitude increased in upgaze and decreased in downgaze (Alexander's law) in most cases, the reverse being observed, however, in two cases (Ohkoshi et al., 1998; Minagar et al., 2001). Finally, the characteristics of slow phases in UBN due to caudal medullary lesions do not appear to be fundamentally different from those observed in UBN due to pontine lesions.
Accordingly, after this review of the three main clinical focal causes of spontaneous vertical nystagmus—two in the brainstem, causing UBN, one in the flocculus, causing DBN—we suggest that the same pathway is probably involved in these three cases (Fig. 1D). This pathway includes the SVN and the VTT as the excitatory efferent tract. It could also comprise a feedback loop controlled by an area of the caudal medulla, receiving afferent signals from the SVN and projecting to the flocculus through a putative inhibitory tract, and finally, via the well-known inhibitory flocculovestibular tract (Langer et al., 1985a), to the SVN. Organized in this way, this additional pathway could be either excitatory for the upward system when stimulated at the SVN or the caudal medullary levels, or inhibitory for the same upward system when stimulated at the floccular level.
Role of gravity in vertical nystagmus and VOR
There is now accumulating evidence that gravity also plays an important role in vertical vestibular eye movement physiology—maybe largely via the otolithic system (Halmagyi and Leigh, 2004)—since (i) DBN (with upward slow phase) is often increased when the patient's head is upside-down (Baloh and Spooner, 1981; Baloh and Yee, 1989; Leigh and Zee, 1999), i.e. when gravity is reversed for the head and acts in the same direction as the nystagmus slow phase, and DBN may also be increased in an intermediary (prone or supine) position in respect to gravity (Halmagyi et al., 1983; Baloh and Yee, 1989; Marti et al., 2002) even though the results obtained in the supine position sometimes appear to be contradictory; (ii) DBN may occur in healthy subjects with an upside-down position of the head (Leigh et al., 2002) or even when the head is simply no longer in an erect position (Goltz et al., 1997), with similar results in the cat (Rude and Baker, 1996); whereas (iii) UBN (with downward slow phase) appears to be at times improved with a decrease in the gravity effect, namely when the patient's head is upside-down (i.e. with gravity acting in the opposite direction to the slow phase nystagmus), or even when the patient is in the supine or prone position (Fisher et al., 1983; Baloh and Yee, 1989; Hirose et al., 1991; Janssen et al., 1998); (iv) UBN occurs in normal subjects when the gravity load is artificially increased in a centrifuge, i.e. when the downward system is reinforced (Marcus et al., 1989); and (v) the vertical VOR is markedly decreased in microgravity (Vieville et al., 1986). This probable contribution of gravity to the downward system could correspond to a part of the apparently missing excitatory downward vestibular signals that we referred to above, and may explain why the central connections of the downward vestibular system appear to be much simpler than those of the upward system (see Conclusions).
Conclusions
One may first conclude that UBN is due to pontine or medullary lesions directly or indirectly resulting in a primary hypoactivity of the excitatory upward SVN–VTT pathway, whereas DBN probably results from a primary hyperactivity of the same pathway, due to floccular damage, without, apparently, any major primary involvement of the excitatory downward vestibular pathway in either case. Thus, in all these cases of spontaneous vertical nystagmus, a primary vestibular dysfunction appears to affect the SVN–VTT pathway, which could normally supplement the action of the ancillary excitatory upward MVN–MLF pathway. Secondly, as stated above, gravity may influence vertical spontaneous nystagmus and the vertical VOR. Since gravity facilitates the downward vestibular system and restrains the upward vestibular system, it may be hypothesized that the additional excitatory upward SVN–VTT pathway mainly developed in order to counteract gravitational pull. This pathway would provide a supplement of upward eye velocity vestibular signals (via the SVN–VTT) and of upward eye position signals (via the caudal medulla flocculus SVN) to the motoneurons of the elevator muscles. However, the upward VOR, optokinetic nystagmus and smooth pursuit gains are superior to the corresponding downward gains in normal subjects, monkeys and cats (Baloh et al., 1983; Matsuo and Cohen, 1984; Ranalli and Sharpe, 1988b; Baloh and Demer, 1991; Tweed et al., 1994; Maruyama et al., 2004). This upward velocity bias suggests that the upward vestibular system is not only anatomically hyperdeveloped, compared to the downward vestibular system, but also physiologically stronger. This organization might also explain the well-known (but poorly understood) upward eye deviation observed at eyelid closing. However, it has also been proposed, to explain the vertical VOR asymmetry in healthy subjects, that the orientation of the six semicircular canals results in an asymmetry of the spontaneous input from the vestibular periphery and therefore in a constant upward drift (Bohmer and Straumann, 1998).
Accordingly, the hyperactive upward vestibular system could require permanent inhibition, even when the head is erect. The inhibition could be specifically induced by the otoliths and vision, which, via the flocculus, may modulate the circuit gain to adapt it to the various positions of the head. However, since both the additional excitatory upward SVN–VTT pathway and its specific floccular inhibition apparently need to be permanently active to maintain the eyes in the primary position, a lesion affecting the excitatory branches (VTT or caudal medulla) or the inhibitory part (flocculus) is likely to result in UBN or DBN. Compensation is poor in DBN due to progressive floccular lesions because, with the usual degenerative causes and untreated cranio-cervical anomalies, the possibilities of adaptation have probably already been exhausted when the nystagmus occurs: at this stage, the hyperdeveloped upward vestibular system might no longer be inhibited. By contrast, UBN due to acute focal (pontine or medullary) lesions, affecting the additional upward SVN–VTT pathway, may improve after a few weeks or months, probably because adaptive mechanisms could involve the undamaged ancillary upward MVN–MLF pathway.
Many thanks to R. J. Leigh (Cleveland) and D. S. Zee (Baltimore), who read an initial version of the manuscript and made valuable remarks.
References
Baloh RW, Demer JL. Gravity and the vertical vestibulo-ocular reflex.
Baloh RW, Spooner JW. Downbeat nystagmus: a type of central vestibular nystagmus.
Baloh RW, Richman L, Yee RD, Honrubia V. The dynamics of vertical eye movements in normal subjects.
Benjamin EE, Zimmerman CF, Troost BT. Lateropulsion and upbeat nystagmus are manifestations of central vestibular nystagmus.
Bertholon P, Convers P, Barral FG, Duthel R, Michel D. Syringomyélobulbie post-traumatique et nystagmus inférieur.
Bohmer A, Straumann D. Pathomechanism of mammalian downbeat nystagmus due to cerebellar lesion: a simple hypothesis.
Büttner U, Helmchen C, Büttner-Ennever JA. The localizing value of nystagmus in brainstem disorders.
Büttner-Ennever JA. Patterns of connectivity in the vestibular nuclei.
Büttner-Ennever JA, Büttner U. The reticular formation. In: Büttner-Ennever JA, editor. Neuroanatomy of the oculomotor system. Amsterdam: Elsevier;
Büttner-Ennever JA, Horn AKE, Schmidtke K. Cell groups of the medial longitudinal fasciculus and paramedian tracts.
Carpenter MB, Cowie RJ. Connections and oculomotor projections of the superior vestibular nucleus and cell group ‘y’.
Chubb MC, Fuchs AF. Contribution of y-group of vestibular nuclei and dentate nucleus of the cerebellum to generation of vertical smooth eye movements.
Cremer PD, Migliacio AA, Halmagyi GM, Curthoys IS. Vestibulo-ocular reflex pathways in internuclear ophthalmoplegia.
De Jong JMBV, Cohen B, Matsuo V, Uemura T. Midsagittal pontomedullary brainstem section: effects on ocular adduction and nystagmus.
Evinger LC, Fuchs AF, Baker R. Bilateral lesions of the medial longitudinal fasciculus in monkeys: effects on the horizontal and vertical components of voluntary and vestibular induced eye movements.
Fisher A, Gresty M, Chambers B, Rudge P. Primary position upbeating nystagmus. A variety of central positional nystagmus.
Fukushima K, Kaneko CRS. Vestibular integrators in the oculomotor system.
Gilman D, Baloh RW, Tomiyasu U. Primary position nystagmus. A clinicopathologic study.
Glasauer S, Hoshi M, Kempermann U, Eggert T, Büttner U. Three-dimensional eye position and slow phase velocity in humans with downbeat nystagmus.
Goltz HC, Irving EL, Steinbach MJ, Eizeman M. Vertical eye position control in darkness—orbital position and body orientation interact to modulate drift velocity.
Graf W, Ezure K. Morphology of vertical canal related second order vestibular neurons in the cat.
Gresty M, Barratt H, Rudge P, Page N. Analysis of downbeat nystagmus. Otolithic vs semicircular canal influences.
Halmagyi GH, Rudge P, Gresty MA, Sanders MD. Downbeating nystagmus. A review of 62 cases.
Highstein SM, McCrea RA. The anatomy of vestibular nuclei. In: Büttner-Ennever JA, editor. Neuroanatomy of the oculomotor system. Amsterdam; Elsevier;
Hirai N, Uchino Y. Floccular influence on excitatory relay neurons of vestibular reflexes of anterior semicircular canal origin in the cat.
Hirose G, Kawada J, Tsukada K, Yoshioka A, Sharpe JA. Upbeat nystagmus: clinicopathological and pathophysiological considerations.
Hirose G, Ogasawara T, Shirakawa T, Kawada J, Kataoka S. Primary position upbeat nystagmus due to unilateral medial medullary infarction.
Ito M, Nisiramu N, Yamamoto M. Specific patterns of neural connexions involved in the control of the rabbit's vestibulo-ocular reflexes by the cerebellar flocculus.
Iwamoto Y, Kitama T, Yoshida K. Vertical eye movement-related secondary vestibular neurons ascending in the medial longitudinal fasciculus in cat. II. Direct connections with the extraocular motoneurons.
Janssen JC, Larner AJ, Morris H, Bronstein AM, Farmer SF. Upbeat nystagmus: clinicoanatomical correlation.
Kattah JC, Dagli TF. Compensatory head tilt in upbeating nystagmus.
Kato I, Nakamura T, Watanabe Y, Harada K, Aoyagi M, Katagiri T. Primary position upbeat nystagmus. Localizing value.
Keane JR, Itabashi HH. Upbeat nystagmus: clinico-pathological study of two patients.
Kirkham TH, Katsarkas A. An electro-oculographic study of internuclear ophthalmoplegia.
Langer T, Fuchs AF, Chubb MC, Scudder CA, Lisberger SC. Floccular efferents in the rhesus macaque as revealed by autoradiography and horseradish peroxidase.
Langer T, Fuchs AF, Scudder CA, Chubb MC. Afferents to the flocculus of the cerebellum in the rhesus macaque as revealed by retrograde transport of horseradish peroxidase.
Larmande P, Henin D, Jan M, Elie A, Gouaze A. Abnormal vertical eye movements in the locked-in-syndrome.
Leigh RJ, Das VE, Seidmann SH. A neurobiological approach to acquired nystagmus.
Marcus JT, Bles W, Van Holten CR. Influence of gravitoinertial force on vestibular nystagmus in man observed in a centrifuge.
Marti S, Palla A, Straumann D. Gravity dependence of ocular drift in patients with cerebellar downbeat nystagmus.
Marti S, Bockisch CJ, Straumann D. Prolonged asymmetric smooth-pursuit stimulation leads to downbeat nystagmus in healthy human subjects.
Marti S, Straumann D, Glasauer S. The origin of downbeat nystagmus. An asymmetry in the distribution of on-directions of vertical gaze-velocity Purkinje cells.
Maruyama M, Fushiki M, Yasuda K, Watanabe Y. Asymmetric adaptive gain changes of the vertical vestibulo-ocular reflex in cats.
Matsuo V, Cohen B. Vertical optokinetic and vestibular nystagmus in the monkey: up-down asymmetry and effects of gravity.
McCrea RA, Strassman A, May E, Highstein SM. Anatomical and physiological characteristics of vestibular neurons mediating the vertical vestibulo-ocular reflexes in the squirrel monkey.
Minagar A, Sheremata WA, Tusa RJ. Perverted head-shaking nystagmus: a possible mechanism.
Munro NA, Gaymard B, Rivaud S, Majdalani A, Pierrot-Deseilligny C. Upbeat nystagmus in a patient with a small medullary infarct.
Müri RM, Meienberg O. The clinical spectrum of internuclear ophthalmoplegia in multiple sclerosis.
Nakada T, Remler MP. Primary position upbeat nystagmus: another central vestibular nystagmus.
Nakamagoe K, Iwamoto Y, Yoshida K. Evidence for brainstem structures participating in oculomotor integration.
Ohkoshi N, Komatsu Y, Mizusawa H, Kanazawa I. Primary position upbeat nystagmus increased on downward gaze: clinicopathologic study of a patient with multiple sclerosis.
Pierrot-Deseilligny C, Rivaud S, Samson Y, Cambon H. Some instructive cases concerning the circuitry of ocular smooth pursuit in the brainstem.
Ranalli PJ, Sharpe JA. Upbeat nystagmus and the ventral tegmental pathway of the upward vestibulo-ocular reflex.
Ranalli PJ, Sharpe JA. Vertical vestibulo-ocular reflex, smooth pursuit and eye-head tracking dysfunction in internuclear ophthalmoplegia.
Rude SA, Baker JF. Otolith orientation and downbeat nystagmus in the normal cat.
Sato Y, Kawasaki T. Target neurons of floccular caudal zone inhibition in y-group nucleus of vestibular nuclear complex.
Sato Y, Kawasaki T. Operational unit responsible for plane-specific control of eye movement by cerebellar flocculus in cat.
SatoY, Kawasaki T. Identification of Purkinje cell/climbing control by the cerebellar flocculus.
Tilikete C, Hermier M, Pelisson D, Vighetto A. Saccadic lateropulsion and upbeat nystagmus: disorders of caudal medulla.
Troost BT, Martinez J, Abel CA, Heros RC. Upbeat nystagmus and internuclear ophthalmoplegia with brainstem glioma.
Tweed D, Sievering D, Misslisch H, Koenig E. Rotational kinematics of the human vestibulo-ocular reflex. II. Velocity steps.
Tyler KL, Sandberg E, Baum KF. Medial medullary syndrome and meningovascular syphilis: a case report in an HIV-infected man and review of the literature.
Uchino Y, Sasaki M, Isu N, Irai N, Imagawa M, Endo K, Graf W. Second-order vestibular neuron morphology of the extra-MLF anterior canal pathway in the cat.
Vieville T, Clement G, Lestienne F, Berthoz A. Adaptive modifications of optokinetic and vestibulo-ocular reflexes in microgravity. In: Keller EL, Zee DS, editors. Adaptive processes in visual and oculomotor systems. Oxford: Pergamon Press;
Walker MF, Zee DS. Asymmetry of the pitch vestibulo-ocular reflex in patients with cerebellar disease.
Zee DS, Yamazaki A, Butler PH, Gücer G. Effects of ablation of flocculus and paraflocculus in primate.
Zhang Y, Partsalis AM, Highstein SM. Properties of superior vestibular nucleus flocculus target neurons in the squirrel monkey. I. General properties in comparison to flocculus projecting neurons.
Author notes
1INSERM 679 and Service de Neurologie 1 and 2Service d'Ophtalmologie, Hôpital de la Salpêtrière (AP-HP), Paris, France


{kind=link}