




Abstract
We have directly assessed the ability of interferon regulatory factor-1 (IRF-1) to act as a tumor suppressor gene in human breast cancer cells and explored whether this suppressor function is mechanistically conferred by affecting cell cycle transition, apoptosis and/or caspase activation. We have used a dual approach, measuring whether overexpression of wild-type IRF-1 or a dominant negative IRF-1 (dnIRF-1) produce opposing effects on breast cancer cell proliferation in vitro or tumorigenicity in athymic nude mice. Mechanistic studies determined the effects of blocking endogenous IRF-1 expression on cell cycle transition by flow cytometry, on apoptosis by Annexin V staining, and on caspase activation by fluorescent substrate cleavage. IRF-1 mRNA ( P ≤ 0.001) and protein ( P ≤ 0.001) are highly expressed in non-tumorigenic, normal, mammary epithelial cells, with intermediate expression in tumorigenic, but non-metastatic, cells and very low expression in metastatic cell lines. In MCF-7 cells transfected with a wild-type IRF-1 (MCF-7/IRF-1), IRF-1 mRNA expression inversely correlates with the rate of cell proliferation ( r = −0.91; P = 0.002). Conversely, expression of dnIRF-1 in both MCF-7 (MCF-7/dnIRF-1; p53 wild-type) and T47D cells (T47D/dnIRF-1; p53 mutant) increases cell proliferation ( P ≤ 0.001). In athymic nude mice, the incidence of MCF-7/IRF-1 xenografts is reduced ( P = 0.045), whereas MCF-7/dnIRF-1 xenografts exhibit a significantly higher tumor incidence ( P ≤ 0.001). Effects of IRF-1/dnIRF-1 are mediated through changes in the rates of apoptosis and not through cell cycle regulation. MCF-7/dnIRF-1 cells exhibit a 50% decrease in basal apoptosis ( P = 0.007) and a significant reduction in caspase 8 activity ( P = 0.03); similar effects occur in T47D/dnIRF-1 cells, where the effects on apoptosis appear to be mediated through inhibition of caspases 3/7 ( P < 0.001) and caspase 8 ( P = 0.03). These data establish a functional role for IRF-1 in the growth suppression of breast cancer cells and strongly implicate IRF-1 as a tumor suppressor gene in breast cancer that acts, independent of p53, to control apoptosis.
Introduction
Among women living to age 80, ∼11% develop breast cancer and over 40,000 women die of this disease each year in the USA ( 1 ). The precise molecular events responsible for affecting breast cancer risk and disease progression remain to be established. While an increasing number of oncogenes have been identified, relatively few tumor suppressor genes have been directly implicated in the development and/or progression of this disease. Altered expression or function of established tumor suppressor genes such as BRCA1 , BRCA2 and p53 do not fully account for the high prevalence of spontaneous breast cancers. For example, BRCA1 is lost or mutated in <10% of all breast cancers ( 2 ). While p53 is mutated in up to 30% of breast cancers ( 3 , 4 ), the functional relevance of the loss of p53 activity remains to be fully established. Clearly, the search for new tumor suppressor genes is important to improving our understanding of the etiology and natural progression of breast cancer.
The transcription factor interferon regulatory factor-1 (IRF-1) is lost, mutated, or rearranged in several cancers including some hematopoietic ( 5 ) and gastric cancers ( 6 ). IRF-1 can reverse the oncogenic transformation of cells induced by the overexpression of oncogenes including both ras and myc in mouse models ( 7 , 8 ). Since functional roles for ras and myc are established in human breast cancer ( 9 – 11 ), a loss of IRF-1 function also might be important in this disease.
The activities of IRF-1 may be related to its ability to regulate apoptosis ( 12 ), which can occur in a p53-dependent or p53-independent manner ( 13 , 14 ), with or without IRF-1-mediated induction of p21 waf1/cip1 ( 13 ), p27 kip1 ( 15 ), protein kinase R ( 16 ), or 2′,5′-oligoadenylate synthase (2,5-OAS) ( 17 ). This regulation of apoptosis probably involves an IRF-1-mediated caspase cascade, since IRF-1 is known to activate caspase 1 ( 14 ), caspase 7 ( 18 ), caspase 8 ( 19 ) and Fas ligand ( 20 ). Many of these genes are reported to have functionally relevant activities in breast cancer ( 14 , 21 – 25 ).
IRF-1 mRNA is induced by IL-6 treatment in the human breast cancer cell line T47D, but the functional consequences of this induction are not known ( 26 ). Consistent with its regulation by interferons, IRF-1 is also implicated in the growth inhibitory effects of interferon-γ in MCF-7 and MDA-MB-231 breast cancer cells ( 27 – 29 ). We have previously reported a role for IRF-1 in mediating sensitivity to antiestrogens in breast cancer cells ( 30 ), and the implication of IRF-1 as a key player in a broader gene network involved in mediating the effects of antiestrogens ( 31 ) and retinoids ( 32 ). More recently, we have shown that IRF-1 is repressed by estrogen and induced by the antiestrogen ICI 182,780 in antiestrogen-sensitive human breast cancer cells, and blocking the activity of IRF-1 decreased the sensitivity of human breast cancer cells to ICI 182,780 through alterations in apoptosis ( 33 ). Similar effects may also occur in normal human mammary cells ( 34 ).
While most studies have been done in experimental models, IRF-1 also may be relevant in clinical breast cancer. For example, Doherty et al . ( 35 ) measured IRF-1 expression by immunohistochemistry and found IRF-1 to be expressed less frequently in the tumor tissue of high grade ductal carcinoma in situ or node-positive invasive ductal cancer compared with normal breast epithelium. However, IRF-1 expression was not assessed in association with established prognostic markers or clinical outcome. In a recent study from our laboratory, both cytosolic and nuclear IRF-1 staining is detected in breast cancer specimens, with a significant association between estrogen receptor-alpha (ERα) expression and cytosolic (and thus potentially inactive) IRF-1. We found no association with other prognostic markers including lymph node status, tumor grade, tumor size, DNA index and S-phase fraction (Y. Zhu, B. Singh, S. Hewitt, A. Liu, B. Gomez, A. Wang and R. Clarke, manuscript submitted).
We now show that overexpression of IRF-1 is associated with growth suppression of human breast cancer cells in vitro and decreases the tumorigenicity of cells inoculated into athymic nude mice. In addition, we show that loss of IRF-1 activity, through the use of a dominant negative IRF-1, enhances both the tumorigenicity and tumor growth rate of human breast cancer xenografts by affecting IRF-1 dependent apoptosis and caspase activation. Taken together these data support a functional role for IRF-1 in the growth suppression of human breast cancer cells and strongly implicate IRF-1 as a tumor suppressor gene active in breast cancer.
Materials and methods
Cell culture and reagents
The ER + cell lines MCF-7, T47D, and ZR-75–1 and ER- cell lines MDA-MB-231, MDA-MB-435, and HBL100 were routinely grown in improved minimal essential medium (IMEM, Biofluids, Rockville, MD) with phenol red and supplemented with 5% fetal bovine serum (Gibco Life Technologies/Invitrogen, Carlsbad, CA). A1N4 normal mammary epithelial cells (ER-) were grown in IMEM supplemented with 0.5% fetal calf serum, 0.5 μg/ml hydrocortisone, 5 μg/ml insulin, and 10 ng/ml epidermal growth factor. Functional studies were done in both MCF-7 (wild-type p53) and T47D cells (mutant p53) ( 36 ). All cells were maintained in a humidified incubator at 37°C in an atmosphere containing 95% air: 5% CO 2 . MCF-7 cells were originally obtained from Dr Marvin Rich (Michigan Cancer Foundation, Detroit, MI). T47D, ZR-75–1, MDA-MB-231, MDA-MB-435, HBL100 and A1N4 cells were obtained from the Lombardi Comprehensive Cancer Center's Tissue Culture Shared Resource and the American Type Culture Collection (ATCC, Manassas, VA).
RNA extraction
Cells were plated in T-75cm 2 plastic tissue culture flasks at a density of 1 × 10 6 cells/flask and grown for 24 h prior to RNA isolation. Total RNA was extracted from proliferating subconfluent cells using the TRIazol reagent (Life Technologies, Gaithersburg, MD). Briefly, cells were rinsed with 1× PBS to remove serum and lysed by the addition of the TRIazol reagent. RNA was isolated by chloroform extraction and precipitated using isopropanol. Total RNA was quantified by comparing the optical density ratios (OD 260 /OD 280 ) obtained spectrophotometrically using a Beckman DU640 Spectrophotometer (Beckman, Fullerton, CA).
Generation of IRF-1 riboprobe and RNase protection analysis
The IRF-1 riboprobe was generated by RT-PCR amplification of a portion of the IRF-1 sequence from MCF-7 cell mRNA, as previously reported ( 33 ). 36B4, an estrogen-independent mRNA that encodes for the human acidic ribosomal protein P0, was used as a loading control for the RNase protection assays ( 37 ). Riboprobes were linearized following digestion with EcoR1 and transcribed with either SP6 or T7 polymerase (IRF-1 and 36B4, respectively). The IRF-1 and 36B4 riboprobes protect 360 and 220 bp fragments, respectively. The IRF-1 riboprobe also detects the dominant negative IRF-1 construct, which produces a 115 bp protected fragment ( 33 ).
RNase protection assays were conducted as previously described ( 30 , 38 ). Briefly, 30 μg of total RNA was hybridized to 1.5 × 10 5 dpm of probe for 12–16 h at 50°C and digested with 40 μg/ml of RNase A for 30 min at 25°C. Digestion was terminated by the addition of proteinase K (0.25 mg/ml) and 0.5% (w/v) SDS. Samples were then phenol–chloroform/isoamyl alcohol (25:24:1) extracted and ethanol precipitated. Pellets were boiled in loading buffer, fractionated in 6% TBE-urea polyacrylamide gels, and radioactivity detected by autoradiography and quantified using phosphorimage analysis (Molecular Dynamics 445SI, Sunnyvale, CA).
Cell lysis and immunoblotting
For the determination of basal IRF-1 expression, cells were grown in T-75 cm 2 plastic tissue culture flasks. Subconfluent monolayers were lysed in modified radioimmunoprecipitation assay (RIPA) buffer (150 mM NaCl, 50 mM Tris pH 7.5, 1% Igepal CA-630, and 0.5% deoxycholate) supplemented with Complete Mini protease inhibitor cocktail tablets (Roche, Mannheim, Germany). Lysates were clarified by centrifugation, and quantified using the bicinchoninic acid assay (Pierce, Rockford, IL). Forty μg of total protein were combined with 4X Laemmli sample buffer prior to boiling and loading onto pre-cast 10% Bis–Tris acrylamide gels (NuPAGE Electrophoresis System, Invitrogen). Proteins were transferred to nitrocellulose membrane and incubated with primary antibody (IRF-1 C-20 at 1:200; Santa Cruz Biotechnology, Santa Cruz, CA) in TBST (10 mM Tris–HCl (pH 8.0), 150 mM NaCl, and 0.05% Tween-20) containing 5% non-fat dry milk overnight at 4°C. Membranes were then incubated with horseradish peroxidase (HRP)-conjugated secondary antibody for 1 h at room temperature followed by enhanced chemiluminescence (Amersham Biosciences, Piscataway, NJ) and exposure to film. To confirm equal loading, membranes were reprobed as described above using a monoclonal β-actin antibody (1:5000, Sigma, St Louis, MO). Images were quantified by densitometry on a FluorChem 800 digital imaging system (Alpha Innotec, San Leandro, CA).
Generation of the dnIRF-1
The dnIRF-1 construct was generated as described previously ( 33 ), from the wild-type IRF-1 cDNA. dnIRF-1 has a deletion of bp 647–1173 and lacks the transcription-activating domain and the region implicated in targeting the molecule for degradation through the ubiquitin proteasome pathway ( 39 ). The construct retains both the 3′ and 5′ untranslated regions, the DNA binding domain, repression domain, homodimerization domain and the nuclear localization sequences of IRF-1. dnIRF-1 was subcloned into the XhoI site of both the pcDNA3 mammalian expression vector (Invitrogen) and a vector expressing green fluorescent protein (EGFP) under the control of a tetracycline-responsive promoter (pBI-EGFP-tet, Clontech, Palo Alto, CA).
Stable transfection of dominant negative IRF-1
For stable transfections, cells were plated in T-75cm 2 plastic tissue culture flasks at a density of 0.5 × 10 6 cells/flask and grown for 24 h prior to transfection. Then, 8 μg of plasmid DNA was transfected into the breast cancer cells using the FuGENE6 transfection reagent (Roche Diagnostics, Indianapolis, IN). Cells were co-transfected with a pBABE plasmid encoding for puromycin resistance (kindly provided by Dr Matthew Ellis, Washington University, St Louis, MO) and either an empty pBI-EGFP plasmid (Clontech) or one containing the dnIRF-1 cDNA. Stably transfected cells were selected for growth in the presence of 1 μg/ml puromycin. Since the pBI-EGFP plasmid contains a green fluorescent protein selectable marker, fluorescence activated cell sorting was used as a secondary method for selection. Colonies that were both puromycin resistant and expressed EGFP were expanded and screened for expression of the dnIRF-1 by RNase protection assay, as described above. All assays were conducted with pooled transfectants. Smaller scale stable transfections, using 1 μg total DNA, also were done in these cells in 6-well tissue culture plates according to the above protocol.
Cell proliferation
Cells were plated in 96-well plastic tissue culture plates at a density of 1 × 10 2 cells/well. Twenty-four hours after plating, one plate was stained with 100 μl/well of a solution containing 0.5% crystal violet and 25% methanol, rinsed with deionized water, dried overnight and resuspended in 100 μl citrate buffer (0.1 M sodium citrate in 50% ethanol) to assess plating efficiency. Remaining cultures were allowed to grow for an additional 1–7 days before staining. Intensity of crystal violet staining, assessed at 570 nm and quantified using a Vmax Kinetic Microplate Reader and Softmax software (Molecular Devices Corp., Menlo Park, CA), is directly proportional to cell number ( 40 ). For the study comparing IRF-1 mRNA expression with rate of proliferation (Figure 2), cells from four empty vector control clones and four IRF-1 transfectants were plated in 96-well plates (10 3 cells/well). Cells were grown for 5 days and stained with crystal violet as described above.
In vivo tumorigenicity assays
Ovariectomized, NCr nu/nu nude mice were supplemented with 17β-estradiol pellets (60-day release pellets; 0.72 mg estradiol; Innovative Research of America, Sarasota, FL) and used to determine the tumorigenicity of MCF-7 pooled stable transfectants. We used an established approach where 2 × 10 6 control (MCF-7 or T47D transfected with an empty vector control), MCF-7/IRF-1, MCF-7/dnIRF-1, or T47D/dnIRF-1 cells were inoculated into opposite thoracic mammary glands (orthotopic sites) of the same animal ( 41 ). Inoculum size was selected to produce <100% tumor incidence under optimal conditions, based on our experience with these cells, so that we could detect either an increase or decrease in tumor incidence. The in vivo studies were independently performed. For the MCF-7 studies, the dnIRF-1 and control study used n = 36 inoculations per group; the IRF-1 and control study used n = 17 inoculations per group. The primary endpoint was the incidence of proliferating tumors as previously defined ( 42 ); secondary endpoints were tumor area and tumor doubling times. Tumor areas were estimated from the product of the two longest perpendicular measurements ( 41 , 42 ). All in vivo studies were conducted in accordance with Georgetown University Animal Care and Use Committee approved protocols.
Cell cycle analyses
Cells stably transfected with the dnIRF-1 or empty control plasmids were plated in T-75 cm 2 plastic tissue culture flasks at a concentration of 0.5 × 10 6 and allowed to grow for 3 days. Cells were then analyzed for alterations in cell cycle via fluorescence activated cell sorting (FACS). FACS analysis was conducted by the Lombardi Comprehensive Cancer Center Flow Cytometry Shared Resource following the method of Vindelov et al . ( 43 ).
Apoptosis
Annexin V assays were performed according to the manufacturer's instructions (Annexin V-PE Apoptosis Detection Kit I; BD Biosciences, Palo Alto, CA). This method is often considered an optimal assay for detecting apoptosis in MCF-7 cells ( 44 , 45 ). For each dish, 5 × 10 5 cells were seeded in 100 mm diameter plastic tissue culture dishes and allowed to grow for 3 days, at which time cells were harvested by trypsinization and pelleted by centrifugation. Then, 10 5 cells in 100 μl of 1× binding buffer were stained with Annexin V-PE and 7-aminoactinomycin D as directed. Flow cytometric analysis (FACStar Plus flow cytometer, Becton–Dickinson, Mountain View, CA) was performed to determine the proportion of EGFP-positive apoptotic cells in each sample.
Caspase activity assays
Caspases 3/7 (Promega, Madison, WI) and Caspase 8 (BD Biosciences) assays were performed according to the manufacturers' recommendations. For caspase 8, cells were seeded in 100 mm diameter plastic tissue culture dishes and allowed to grow for 2 days, at which time cells were harvested by trypsinization and pelleted by centrifugation. The following caspase activity reactions were run for each cell line in black 96-well plates: (i) IETD-AFC (fluorescent caspase 8 substrate), (ii) IETD-AFC plus the caspase 8 inhibitor IETD-fmk and (iii) no substrate. All samples and a standard curve of fluorescent, free AFC (7-amino-4-trifluoromethyl coumarin) were incubated at 37°C for 2–3 h. Fluorescence was quantified using an XFluor4 Ultra 384 fluorescence plate reader (Tecan USA, Research Triangle Park, NC) at 405 nm (excitation)/508 nm (emission). Caspase activity was determined by the formula (ΔFU/h)/standard curve slope, where ΔFU/h is the difference in Fluorescence Units between the reactions with and without caspase 8 inhibitor. For caspases 3/7, 7500 cells per well were plated in black 96-well dishes. Forty-eight hours later the fluorescent caspases 3/7 substrate Z-DEVD-R110 was added to each well, contents were mixed by gentle shaking at 300 r.p.m. for 5 min, and the plates incubated at room temperature in the dark for 1 h. Samples were read on the Xfluor4 Ultra 384 fluorescence plate reader at 485 nm (excitation)/535 nm (emission).
Statistical analyses
Where appropriate, mRNA and protein expression, cell growth, apoptosis, and caspase activity data from in vitro studies were compared using either Student's t -test or ANOVA. Correlation between IRF-1 mRNA and proliferation was estimated by Pearson's correlation coefficient; the linear regression shown for these data was estimated by the least squares method. Data from in vivo studies were analyzed as previously discussed ( 41 , 46 ). For tumor growth rate, doubling times were estimated following transformation of the data by a Gompertz function and experimental groups compared for statistical significance by one-way ANOVA. Cumulative incidences of proliferating tumors in each experimental group were visualized by the Kaplan–Meier method and compared by the log rank test. Tumor areas between groups and across time were compared using repeated-measures ANOVA to account for the effects of collecting consecutive measurements of each tumor over time.
Results
IRF-1 mRNA and protein are differentially expressed in breast cell lines
Using an RNase protection assay, we compared the expression levels of IRF-1 mRNA in a series of cell models. Among the breast cancer cell lines, the level of IRF-1 mRNA expression is inversely correlated with metastatic or invasive potential ( Figure 1A and B ). IRF-1 is most highly expressed in the non-tumorigenic mammary epithelial cell line A1N4 derived from normal human mammary epithelium (8-fold higher than MCF-7 cells) ( 3 ). IRF-1 mRNA expression in both of the estrogen receptor negative (ER-), estrogen-unresponsive, and invasive MDA-MB-231 and MDA-MB-435 cell lines is, respectively, 3-fold and 3.4-fold lower than in the ER+, estrogen-dependent, non-invasive, MCF-7 cells ( Figure 1B ; P ≤ 0.001, one-way ANOVA). We found a similar association with an invasive phenotype and IRF-1 protein expression ( Figure 1C–D ). Similarly, IRF-1 protein levels in the invasive cell lines are lowest, those of the three tumorigenic but non-invasive cells lines (MCF-7, ZR-75–1, HBL-100) are intermediate and expression of IRF-1 is highest in the A1N4 cells ( Figure 1D ; P ≤ 0.001, one-way ANOVA). We determined that the HBL-100 cells we used form non-metastatic tumors in ovariectomized, NCr thymic nude mice (data not shown). Therefore, IRF-1 mRNA and protein levels are closely correlated with each other and with IRF-1 transcriptional activity as measured previously in standard promoter-reporter assays ( 33 , 47 ).
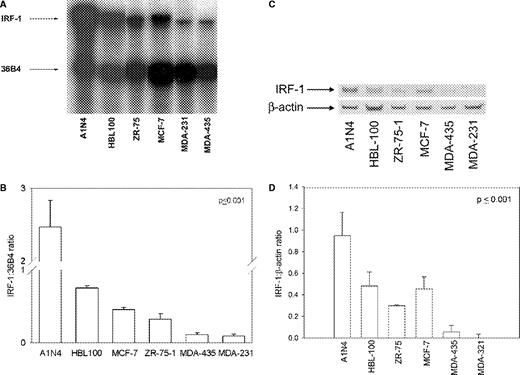
IRF-1 expression in five human breast cancer and one normal breast cell lines. (A) Representative RNase protection assay. (B) RNase protection analyses of IRF-1 mRNA; data represent mean ± SE of three independent determinations, where absorbance is expressed as a ratio of IRF-1:36 B4; P ≤ 0.001 (one-way ANOVA). ( C ) Representative immunoblot of IRF-1 protein; β-actin = loading control. ( D ) Immunoblot analyses of IRF-1 protein; data represent mean ± SE of three independent determinations, where densitometry values are expressed as a ratio of IRF-1:β-actin; P ≤ 0.001 (one-way ANOVA).
IRF-1 mRNA expression correlates inversely with MCF-7 proliferation in vitro
To explore whether IRF-1 expression may be growth inhibitory, MCF-7 cells were stably transfected with an IRF-1 cDNA expression vector. The rate of anchorage-dependent cell proliferation in transfectants (MCF-7/IRF-1) was compared with IRF-1 mRNA levels as measured by RNase protection; overexpression of IRF-1 in the transfectants is apparent in the x -axis of Figure 2 . A significant inverse correlation is evident between IRF-1 mRNA expression and cell proliferation ( Figure 2 ; P = 0.002, r = −0.92, Pearson's correlation coefficient), consistent with a growth suppressive effect of IRF-1 in these cells. During the selection of the MCF7/IRF-1 cells, we observed ∼40% fewer anchorage dependent colonies in the IRF-1 transfected plates than in the vector control transfected plates, which is consistent with the growth suppressive effects of IRF-1 in these cells (not shown).
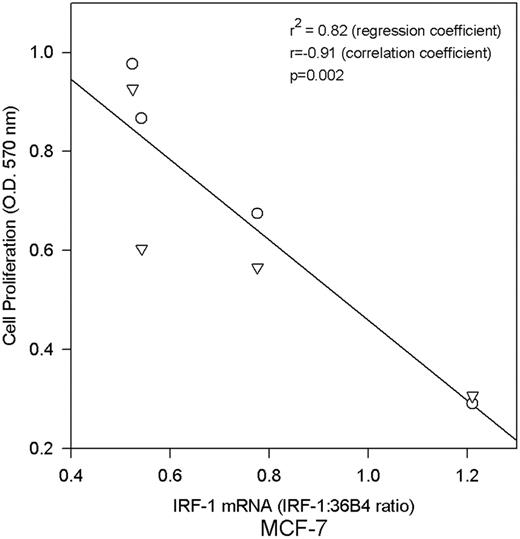
IRF-1 mRNA expression inversely correlates with anchorage dependent growth in MCF-7 cells. Growth rates of MCF-7 cells stably transfected with wild-type IRF-1 were compared with IRF-1 mRNA expression measured by RNase protection assay; P = 0.002 (Pearson's correlation coefficient r = −0.91); r2 = 0.82 (linear regression coefficient).
Overexpression of IRF-1 reduces tumorigenicity in athymic nude mice
Reduced cell proliferation, as seen in MCF-7 cells stably transfected with wild-type IRF-1, suggests a potential tumor suppressive activity of IRF-1. To determine directly whether IRF-1 expression can act as a tumor suppressor in vivo , MCF7/IRF-1 and MCF-7/control cells were inoculated into ovariectomized NCr nu/nu mice supplemented with estradiol. The cumulative incidence of proliferating tumors is significantly lower for the MCF-7/IRF-1 cells compared with the MCF-7/control cells ( Figure 3 ; P = 0.045, log rank test).
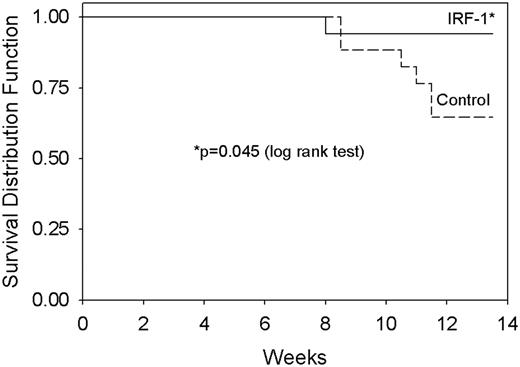
IRF-1 expression reduces tumor incidence in MCF-7 xenografts in athymic nude mice. MCF-7/control or MCF-7/IRF-1 transfected cells were inoculated into ovariectomized NCr nu/nu mice supplemented with 17β-estradiol. Data are expressed as the proportion of mice without a proliferating tumor at the inoculation site. No tumors grew at sites other than where cells were inoculated; P = 0.045 for cumulative tumor incidence (log rank test).
dnIRF-1 increases the proliferation of MCF-7 and T47D cells in vitro
Data obtained with the direct overexpression of IRF-1 could be confounded by the selection of cell populations that include some cells now resistant to the growth inhibitory effects of constitutive IRF-1 expression. We used a dominant negative that inhibits the activity of endogenous IRF-1 to address this concern, applying an alternative but fully complementary approach to further establish a functional role for IRF-1 in affecting breast cancer cell proliferation. We studied two different cell lines (MCF-7 and T47D) in which we had previously measured basal IRF-1 mRNA expression ( Figure 1 and ( 33 )).
A dnIRF-1 was previously constructed by PCR amplification of a portion of the wild-type IRF-1 cDNA and shown to inhibit the transcriptional activation of IRF-1 in an interferon-stimulated response element promoter-reporter assay in both MCF-7 and T47D cells ( 33 ). The dnIRF-1 construct was then constitutively expressed in both MCF-7 (MCF-7/dnIRF-1) and T47D (T47D/dnIRF-1) human breast cancer cells. MCF-7/dnIRF-1 cells proliferate significantly more rapidly when compared with control transfectants ( Figure 4A ; P ≤ 0.001, Student's t -test). Similarly, significant differences are seen in the growth of T47D/dnIRF-1 and T47D/control transfectants ( Figure 4B ; P ≤ 0.001, Student's t -test). We also observed a 25–30% increase in anchorage-dependent colony formation in both of these cell lines (not shown), consistent with the inhibitory effects of wild-type IRF-1 overexpression seen in MCF-7 cells.
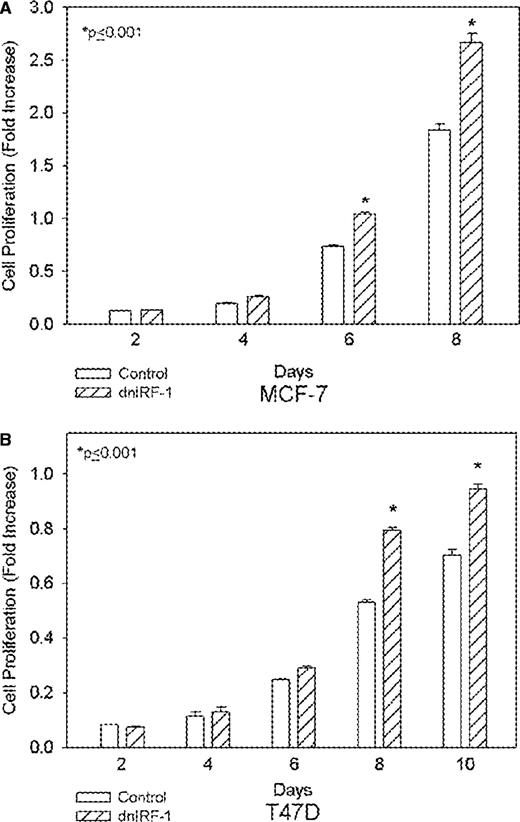
dnIRF-1 enhances the growth of MCF-7 and T47D cells. ( A ) MCF-7 cells stably transfected with the dnIRF-1 or empty vector control were assessed for growth at various time points by crystal violet growth assays. ( B ) Growth rates of T47D cells stably transfected with dnIRF-1 or empty vector control were determined at various time points by crystal violet growth assays. Data are expressed as spectrophotometric absorbance at 570 nm and represent mean ± SE of three independent determinations; P ≤ 0.001 (Student's t -test).
dnIRF-1 increases tumorigenicity in athymic nude mice
As with the study of the constitutive expression of the wild-type IRF-1 cDNA, we assessed the effects of constitutively expressing dnIRF-1 on tumorigenicity in athymic nude mice. Tumors are formed in estradiol-supplemented mice by both the control (MCF-7/control) and MCF-7/dnIRF-1 cells. A significant increase (26% average difference in tumor number over all weeks) is seen in the cumulative incidence of proliferating tumors formed in the MCF-7/dnIRF-1 compared with the MCF-7/control transfectants ( Figure 5 ; P < 0.001, log rank test). There is also a statistically significant difference in tumor growth rates: MCF-7/dnIRF-1 transfectants grow 2.5-fold faster than the MCF-7/control transfectants (not shown; P = 0.042, one-way ANOVA) as assessed following Gompertzian transformation of the growth data for all proliferating tumors ( 41 ). We observed a statistically significant difference in tumor volume; MCF-7/dnIRF-1 tumors exhibit an overall increase of ∼38% compared with MCF-7/control tumors ( P < 0.001, repeated-measures ANOVA). Preliminary data also show an increase in tumor incidence in the T47D/dnIRF-1 cells compared with their respective controls (not shown). The increased tumorigenicity in both cell models is consistent with the effects on cell proliferation in vitro and shows that dnIRF-1 exhibits opposite effects from the constitutive expression of wild-type IRF-1. When considered together, these observations provide compelling evidence of putative tumor suppressor action for IRF-1 in human breast cancer cells.
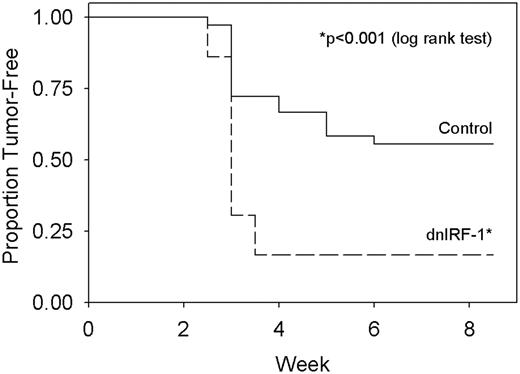
dnIRF-1 enhances the tumorigenicity of MCF-7 cells. MCF-7/control or MCF-7/dnIRF-1 transfected cells were inoculated into ovariectomized NCr nu/nu mice supplemented with 17β-estradiol. Data are expressed as the proportion of mice without a proliferating tumor at an inoculation site. No tumors grew at sites other than where cells were inoculated. P ≤ 0.001 for cumulative tumor incidence (log rank test).
dnIRF-1 alters the rate of basal apoptosis but not the cell cycle profiles of MCF-7 cells
IRF-1 can induce several antiproliferative and proapoptotic genes in non-breast cancer cells. To determine the mechanism by which dnIRF-1 increases the tumorigenicity of breast cancer cells, we compared the cell cycle profiles and rates of basal apoptosis in MCF-7/dnIRF-1 and MCF-7/control cells. While fewer MCF-7/dnIRF-1 transfectants are in S-phase compared with MCF-7/control transfectants, no significant differences are seen between the cell cycle profiles as measured by flow cytometry ( Table I ). Basal apoptosis was then measured by Annexin V staining. MCF-7/dnIRF-1 cells exhibit a ∼50% decrease in apoptosis compared with MCF-7/control cells ( P = 0.007, Student's t -test). Results similar to those seen with MCF-7/dnIRF-1 and MCF-7/control cells are also apparent in our studies with T47D/dnIRF-1 cells and T47D/control cells (not shown). Thus, the increased growth and tumorigenicity of cells constitutively expressing the dnIRF-1 construct is probably due primarily to differences in the basal rate of apoptosis.
Cell cycle distribution in MCF-7/control and MCF7/dnIRF-1 cells growing in vitro
| Cells | G 0 /G 1 | S | G 2 /M |
|---|---|---|---|
| Control | 60.6 ± 4.6 | 26.8 ± 3.9 | 12.6 ± 0.8 |
| dnIRF-1 | 67.5 ± 3.1 | 20.7 ± 3.0 | 11.7 ± 0.3 |
| P -value | NS | NS | NS |
| Cells | G 0 /G 1 | S | G 2 /M |
|---|---|---|---|
| Control | 60.6 ± 4.6 | 26.8 ± 3.9 | 12.6 ± 0.8 |
| dnIRF-1 | 67.5 ± 3.1 | 20.7 ± 3.0 | 11.7 ± 0.3 |
| P -value | NS | NS | NS |
NS = not significant.
Cell cycle distribution in MCF-7/control and MCF7/dnIRF-1 cells growing in vitro
| Cells | G 0 /G 1 | S | G 2 /M |
|---|---|---|---|
| Control | 60.6 ± 4.6 | 26.8 ± 3.9 | 12.6 ± 0.8 |
| dnIRF-1 | 67.5 ± 3.1 | 20.7 ± 3.0 | 11.7 ± 0.3 |
| P -value | NS | NS | NS |
| Cells | G 0 /G 1 | S | G 2 /M |
|---|---|---|---|
| Control | 60.6 ± 4.6 | 26.8 ± 3.9 | 12.6 ± 0.8 |
| dnIRF-1 | 67.5 ± 3.1 | 20.7 ± 3.0 | 11.7 ± 0.3 |
| P -value | NS | NS | NS |
NS = not significant.
dnIRF-1 differentially inhibits caspases 3/7 and 8 activities in MCF-7 and T47D cells
To discern the mechanism by which dnIRF-1 inhibits the basal rate of breast cancer cell apoptosis, we measured the activation of several caspases. Caspases 3 and 7 are characterized as effector caspases; their activation by the upstream apical or initator caspases (like caspase 8) is one of the final events that occur prior to cell death ( 48 ). First, the basal caspases 3/7 activity of MCF-7/dnIRF-1 and T47D/dnIRF-1 cells was compared with control-transfected cells by assaying the cleavage of the fluorescence-conjugated caspases 3/7 substrate Z-DEVD-R110. While dnIRF-1 expression had no significant effect on caspases 3/7 substrate cleavage in MCF-7 cells ( Figure 6 ; P = 0.85, Student's t test), there was a modest (yet significant) 30% inhibition of caspase activity observed in T47D cells ( P < 0.001, Student's t test). Using a different fluorescent substrate specific for caspase 8 (IETD-AFC), we next determined whether activity of this upstream initiator caspase was altered by the expression of dnIRF-1 and found that in both cell lines there was strong inhibition of caspase 8 activity in the presence of dnIRF-1 ( Figure 6 ; P = 0.03, Student's t test). These data imply that the decreased basal rate of apoptosis in cells constitutively expressing the dnIRF-1 construct is due to differential inhibition of caspase enzymatic activity.
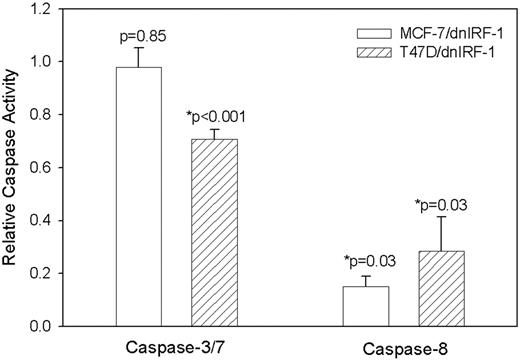
dnIRF-1 differentially inhibits basal caspases 3/7 and caspase 8 activities in MCF-7 and T47D cells. Activity was measured in MCF-7 and T47D transfectants by cleavage of the fluorescent caspases 3/7 substrate Z-DEVD-R110, or the caspase 8 substrate IETD-AFC. For caspases 3/7, data from a representative experiment performed in sextuplicate is presented as relative caspase activity (normalized to control-transfected cells); P = 0.85 and P < 0.001, respectively, Student's t test. For caspase 8, data from four independent determinations are presented as relative caspase activity (normalized to control-transfected cells); P = 0.03, Student's t test.
Discussion
Our prior observations and the data published by other investigators suggesting tumor suppressor effects of IRF-1 in other cancers led to the hypothesis that IRF-1 may have tumor suppressor activities in human breast cancer. We have applied several criteria to assessing the potential tumor suppressor activities of IRF-1 including patterns of IRF-1 mRNA and protein expression in normal, non-metastatic and metastatic cell lines, the ability of IRF-1 to affect cell proliferation in vitro , and tumorigenesis of human breast cancer cell xenografts in athymic nude mice.
Existing tumor suppressor genes have multiple functions, with one widely observed characteristic being a loss or reduction in gene expression that can occur via transcriptional repression, gene deletion or loss of heterozygosity. The inverse pattern of IRF-1 mRNA and protein expression in breast cancer cell lines is consistent with tumor suppressor activity. For example, the highest expression levels are seen in the normal A1N4 cells, intermediate expression is evident in the tumorigenic—but non-metastatic—MCF-7 and ZR-75–1 cells lines, and the lowest expression levels are detected in the highly invasive and metastatic MDA-MB-231 and MDA-MB-435 cells. This pattern of mRNA and protein expression is also consistent with data from immunohistochemistry studies showing lower IRF-1 protein expression in breast cancer tissue compared with normal breast epithelium ( 35 ) and that the expression pattern of IRF-1 in breast cancers is predominantly cytosolic and, therefore, potentially inactive (Y. Zhu, B. Singh, S. Hewitt, A. Liu, B. Gomez, A. Wang and R. Clarke, manuscript submitted).
Reduced IRF-1 mRNA expression in breast cancer cell lines appears primarily transcriptional, since it remains inducible by the steroidal ER antagonist ICI 182,780 in endocrine-responsive breast cancer cells and by the cytotoxic drug doxorubicin in both ER+ and ER− breast cancer cells ( 33 ). However, we cannot exclude the possibility that other mechanisms contribute to the low expression of IRF-1 protein reported in breast tumors ( 35 ). The IRF-1 locus is 5q31.1; in 11% of sporadic breast cancers 5q12–31 is deleted ( 49 ), and 5q is deleted in 86% of tumors with mutated BRCA1 ( 50 ). These chromosomal regions were found by comparative genomic hybridization and are not mapped in detail. While 5q31.1 is likely to be consistently deleted, these deleted regions probably contain many other genes and we are actively looking for specific deletion or loss of heterozygosity at the IRF-1 locus in breast cancer tissues. Effects of the absence of IRF-1 expression in humans are not known. However, loss of IRF-1 in mice that are either p53 null or overexpress the ras oncogene significantly increases the rate of tumor formation ( 7 )—evidence supporting a tumor susceptibility function for IRF-1 independent of p53 (see below).
Reduced IRF-1 activity could also result from overexpression of an endogenous inhibitor. One such inhibitor is the nucleolar phosphoprotein nucleophosmin (NPM), which blocks the transcriptional activity of IRF-1 in leukemic cells ( 51 ). Preliminary data from our laboratory show that this interaction also occurs in breast cancer cells. We have previously shown NPM to be an estrogen-regulated protein in breast cancer cells and that the level of NPM expression increases with acquisition of a more progressed phenotype ( 52 ) and antiestrogen resistance ( 30 ). Autoantibodies to NPM are present in serum from breast cancer patients, and the levels of these autoantibodies increase 6 months prior to recurrence ( 53 ). Thus, IRF-1 activity may be affected by transcriptional regulation, altered expression of an endogenous inhibitor, and perhaps also by mutational events. Such redundancy in disrupting IRF-1 expression or activity is a characteristic of key functional genes, including tumor suppressors, and recent evidence supports the hypothesis that IRF-1 represents a key node in a broader signaling network associated with endocrine responsiveness in some breast cancers ( 33 ).
To assess the functional relevance of differential IRF-1 expression, we studied whether reduced IRF-1 activity affects the phenotype of breast cancer cells. Stable transfection with dnIRF-1 should eliminate the potential problems associated with the use of transient short-term siRNA or antisense approaches, which can induce marked interferon and stress responses that could confound data interpretation ( 54 – 56 ). We show that expression of an exogenous wild-type IRF-1 significantly decreases the rate of cell proliferation in vitro , whereas expression of dnIRF-1 significantly increases both MCF-7 (MCF-7/dnIRF-1) and T47D (T47D/dnIRF-1) cell proliferation. While the data show functional relevance for IRF-1 in affecting human breast cancer cell proliferation in vitro , consistent with its role in mediating growth inhibitory effects of interferon-γ ( 27 , 28 ), this evidence alone is not sufficient to establish tumor suppressor activity. Therefore, we explored the ability of both wild-type IRF-1 expression and dnIRF-1 expression to directly affect tumor incidence and growth in breast cancer xenografts in athymic nude mice. Fully consistent with the data in vitro , constitutive IRF-1 expression significantly reduces, and dnIRF-1 expression significantly increases, tumor incidence. These data clearly show a direct tumor suppressor activity of IRF-1 in human breast cancer cells in vivo .
The growth inhibitory effects of IRF-1 as measured in vitro and in vivo do not distinguish between effects on cell cycle arrest or apoptosis as the primary mechanism for these activities. For example, an increased rate of apoptosis can decrease the measured rate of proliferation by increasing cell loss and reducing the number of viable cells produced over time. Hence, we measured directly the effects of IRF-1 activity on both cell cycle distribution and apoptosis. We found no difference between the cell cycle profiles of control and dnIRF-1 transfected MCF-7 and T47D cells. These data suggest that the primary effect of IRF-1 activity is most likely through signaling to apoptosis. In support of this, we found significant differences in the rates of basal apoptosis between these cells, reflecting the known activity of IRF-1 to induce apoptosis through inducing a caspase cascade. These data are also consistent with our recent observations in both MCF-7 and T47D cells, where IRF-1 activity is strongly implicated in affecting responsiveness to the antiestrogen ICI 182,780. IRF-1 does not contribute to the G 0 /G 1 cell cycle arrest activities of ICI 182,780 but accounts for the induction of apoptosis by this antiestrogen ( 33 ).
Since IRF-1 can affect the expression and/or activity of several caspases, including caspase 1, 7 and 8 ( 14 , 18 , 19 ), we sought to determine which caspases may be downregulated during dnIRF-1-mediated cell proliferation. We observed that dnIRF-1 has differential effects on caspase activity in MCF-7 and T47D cells. Caspase 8 is usually considered to be an apical or initiator caspase that is activated in response to death receptor signaling. Caspases 3 and 7 are characterized as effector caspases whose activation by the upstream initator caspases is one of the final events that occurs prior to cell death ( 48 ). Due to a deletion in the CASP-3 gene ( 57 ), MCF-7 cells do not express caspase 3 protein but can induce apoptosis through sequential activation of other caspases including caspases 9, 7 and 6 ( 24 ). In MCF-7 cells, dnIRF-1 inhibits the basal activity of caspase 8 but not caspase 7. In T47D, which express both caspases 3 and 7, caspases 3/7 and caspase 8 are inhibited by dnIRF-1. Thus, caspase 3 is not an absolute requirement for IRF-1-mediated apoptosis.
It is not immediately clear why basal caspase 7 activity is not inhibited by dnIRF-1 in MCF-7 cells when inhibition of caspase 3 and/or 7 activity is seen in T47D cells. The fluorescent molecule used here (Z-DEVD-R110) is a substrate for both caspases 3 and 7. It may be that there is sufficient expression of both caspases in the T47D cells, whereas the low levels of caspase 7 expressed in our MCF-7 cells do not allow the assay to detect any further suppression by dnIRF-1. Perhaps more likely, caspase 8 may act independent of caspase 3 (and/or 7), since this has been clearly demonstrated in the context of TGF-β-induced apoptosis in other cell systems ( 58 ). Substrates for caspase 8 other than the effector caspases have also been identified, including the receptor tyrosine kinase HER2 ( 59 ), the F-actin-associated protein plectin ( 60 ), and the cyclin-dependent kinase inhibitor p27(KIP1) ( 1 ); cleavage of these molecules could also play a role in apoptosis induction independent of caspases 3/7. Finally, it is also possible that suppression of basal apoptosis by introduction of the dnIRF-1 is due to dysregulation of other apoptotic mediators that are differentially expressed in MCF-7 and T47D cells, and in turn these molecules are directly responsible for the observed reduction in caspase activity.
Differential activation of caspases has significant consequences for the induction of cell death. Interferon-γ sensitizes MCF-7 cells to Fas-mediated apoptosis through the expression of caspase 8 but independent of p53 ( 25 , 29 ), and IRF-1 is required for the interferon-γ-mediated increase in caspase 8 expression and activity in MCF-7 cells due to an interferon-responsive element present in the caspase 8 promoter ( 29 ). Thus, induction of caspase 8 can be an important function of IRF-1 and our data suggest that this plays a key role in the tumor suppressor activities of IRF-1 in breast cancer cells. Indeed, caspase 8 is a tumor suppressor in several different neoplasms, and its dysregulation has been implicated in resistance to cytotoxic chemotherapy ( 61 ). The induction of one putative tumor suppressor (caspase 8) by another (IRF-1) is suggestive of an important regulatory network that controls the death of breast cancer cells. This also lends support to our previous studies showing that IRF-1 (and potentially caspase 8) plays a critical role in antiestrogen response and subsequent apoptosis in responsive breast cancer cells ( 33 ).
The ability of IRF-1 to affect signaling to apoptosis with or without functional p53 could allow IRF-1 to act in breast cancers despite the frequent loss of p53 activity in these tumors. We see broadly comparable effects of IRF-1 in MCF-7 (wild-type p53) and T47D cells (p53 mutant) in this study and in our prior study of antiestrogen resistance ( 33 ), supporting the hypothesis that IRF-1 can function independent of p53 in some breast cancer cells. Experiments to identify specific p53-independent IRF-1-mediated apoptotic pathways in breast cells, and how these influence responsiveness to hormonal and cytotoxic stressors, are in progress.
The notable changes in tumor incidence seen in the xenograft studies (MCF-7/IRF-1, MCF-7/dnIRF-1, T47D/dnIRF-1) would have a major impact if extrapolated to the human population, since the activities of IRF-1 are a consequence of its effects on apoptosis. For example, tumor suppression that results primarily from cell cycle arrest leaves viable neoplastic cells alive. Over time, these surviving cells have the opportunity to adapt and progress, eventually recurring as more aggressive lesions. Tumor suppression driven by a sufficient increase in apoptosis could produce a rate of cell loss that exceeds the rate of cell replacement, eventually leading to the elimination of all neoplastic cells. The ability of antiestrogens to reduce the incidence of ER+ breast cancers in the chemoprevention setting ( 62 , 63 ) may well be driven by such an effect. Indeed, we have shown recently that antiestrogen signaling to apoptosis is driven by an ICI 182 780-induced increase in IRF-1 expression/activation; loss of this regulation confers resistance to the proapoptotic effects of antiestrogens. These observations implicate IRF-1 as a key player in hormonal signaling to apoptosis in ER+ breast cancer cells ( 33 ) and are consistent with a recent study reporting a correlation between wild-type IRF-1 overexpression and cell death in the ER- cells ( 64 ). Overall, given the tumor suppressor effects of IRF-1 in other cancers, the lower expression seen in neoplastic versus normal breast, and the data presented here, IRF-1 appears to be a potent tumor suppressor gene in some breast cancers.
These two authors contributed equally to this work.
This work was supported in part by the American Cancer Society award IRG-97-1520-01-IRG (T.S.), the Ella and Charles O. Lathum Charitable Trust (T.S.) Public Health Service awards R01-CA/AG58022–10 and R01-CA096483–01 (R.C.), Department of Defense awards DAMD17–99–9189 (K.B.B.), BC030280 and BC010619 (R.C.) from the United States Army Medical Research and Material Command. Technical services also were provided by the Animal Shared Resource (R.Clarke) and Flow Cytometry and Cell Sorting and Macromolecular Shared Resources funded through Public Health Service award P30-CA51008–14 (Lombardi Comprehensive Cancer Center Support Grant).
Conflict of Interest Statement : None declared.
References
Frost,V., Al Mehairi,S. and Sinclair,A.J. (
de Jong,M.M., Nolte,I.M., te Meerman,G.J., van der Graaf,W.T., Oosterwijk,J.C., Kleibeuker,J.H., Schaapveld,M. and de Vries,E.G. (
Elledge,R.M. and Allred,D.C. (
Willman,C.L., Sever,C.E., Pallavicini,M.G., Harada,H., Tanaka,N., Slovak,M.L., Yamamoto,H., Harada,K., Meeker,T.C., List,A.F. and Taniguchi,T. (
Nozawa,H., Oda,E., Ueda,S., Tamura,G., Maesawa,C., Muto,T., Taniguchi,T. and Tanaka,N. (
Nozawa,H., Oda,E., Nakao,K. et al . (
Tanaka,N., Ishihara,M. and Taniguchi,T. (
Clark,G.J. and Der,C.J. (
de Bono,J.S., Tolcher,A.W. and Rowinsky,E.K. (
Tanaka,N., Ishihara,M., Kitagawa,M., Harada,H., Kimura,T., Matsuyama,T., Lamphier,M.S., Aizawa,S., Mak,T.W. and Taniguchi,T. (
Tanaka,N., Ishihara,M., Lamphier,M.S., Nozawa,H., Matsuyama,T., Mak,T.W., Aizawa,S., Tokino,T., Oren,M. and Taniguchi,T. (
Tamura,T., Ishihara,M., Lamphier,M.S., Tanaka,N., Oishi,I., Alzawa,S., Matsuyama,T., Mak,T.W., Taki,S. and Taniguchi,T. (
Moro,A., Santos,A., Arana,M.J. and Perea,S.E. (
Kumar,A., Yang,Y.-L., Flati,V., Der,S., Kadereit,S., Deb,A., Haque,J., Reis,L., Weissmann,C. and WIlliams,B.R.G. (
Coccia,E.M., Del Russo,N., Stellacci,E., Orsatti,R., Benedetti,E., Marziali,G., Hiscott,J. and Battistini,A. (
Sanceau,J., Hiscott,J., Delattre,O. and Wietzerbin,J. (
Suk,K., Chang,I., Kim,Y.H., Kim,S., Kim,J.Y., Kim,H. and Lee,M.S. (
Chow,W.A., Fang,J.J. and Yee,J.K. (
Savinova,O., Joshi,B. and Jagus,R. (
Boudreau,N., Sympson,C.J., Werb,Z. and Bissell,M.J. (
Keane,M.M., Ettenberg,S.A., Lowrey,G.A., Russell,E.K. and Lipkowitz,S. (
Liang,Y., Yan,C. and Schor,N.F. (
Ruiz-Ruiz,C., Munoz-Pinedo,C. and Lopez-Rivas,A. (
Harroch,S., Revel,M. and Chebath,J. (
Yim,J.H., Ro,S.H., Lowney,J.K., Wu,S.J., Connett,J. and Doherty,G.M. (
Hoshiya,Y., Gupta,V., Kawakubo,H., Brachtel,E., Carey,J.L., Sasur,L., Scott,A., Donahoe,P.K. and Maheswaran,S. (
Ruiz-Ruiz,C., Ruiz,d.A., Rodriguez,A., Ortiz-Ferron,G., Redondo,J.M. and Lopez-Rivas,A. (
Gu,Z., Lee,R.Y., Skaar,T.C., Bouker,K.B., Welch,J.N., Lu,J., Liu,A., Zhu,Y., Davis,N., Leonessa,F., Brunner,N., Wang,Y. and Clarke,R. (
Clarke,R., Liu,M.C., Bouker,K.B., Gu,Z., Lee,R.Y., Zhu,Y., Skaar,T.C., Gomez,B., O'Brien,K., Wang,Y. and Hilakivi-Clarke,L.A. (
Skaar, T. C. and Clarke, R. (
Bouker,K.B., Skaar,T.C., Fernandez,D.R., O'Brien,K.A. and Clarke,R. (
Bowie,M.L., Dietze,E.C., Delrow,J., Bean,G.R., Troch,M.M., Marjoram,R.J. and Seewaldt,V.L. (
Doherty,G.M., Boucher,L., Sorenson,K. and Lowney,J. (
Bartek,J., Iggo,R., Gannon,J. and Lane,D.P. (
LaBorda,J. (
Clarke,R., Brünner,N., Katz,D., Glanz,P., Dickson,R.B., Lippman,M.E. and Kern,F. (
Nakagawa,K. and Yokosawa,H. (
Frandsen,T.L., Boysen,B.E., Jirus,S., Spang-Thomsen,M., Dano,K., Thompson,E.W. and Brunner,N. (
Clarke,R. (
James,M.R., Skaar,T.C., Lee,R.Y., MacPherson,A., Zwiebel,J.A., Ahluwalia,B.S., Ampy,F. and Clarke,R. (
Vindelov,L.L., Christensen,I.J. and Nissen,N.I. (
Lindner,D.J., Ma,X., Hu,J., Karra,S. and Kalvakolanu,D.V. (
Del Bino,G., Darzynkiewicz,Z., Degraef,C., Mosselmans,R., Fokan,D. and Galand,P. (
Hanfelt,J. (
Kano,A., Haruyama,T., Akaike,T. and Watanabe,Y. (
Strasser,A., O'Connor,L. and Dixit,V.M. (
Tirkkonen,M., Tanner,M., Karhu,R., Kallioniemi,A., Isola,J. and Kallioniemi,O.P. (
Tirkkonen,M., Johannsson,O., Agnarsson,B.A., Olsson,H., Ingvarsson,S., Karhu,R., Tanner,M., Isola,J., Barkardottir,R.B., Borg,A. and Kallioniemi,O.P. (
Kondo,T., Minamino,N., Nagamura-Inoue,T., Matsumoto,M., Taniguchi,T. and Tanaka,N. (
Skaar,T.C., Prasad,S.C., Sharaeh,S., Lippman,M.E., Brünner,N. and Clarke,R. (
Brankin,B., Skaar,T.C., Trock,B.J., Berris,M. and Clarke,R. (
Bridge,A.J., Pebernard,S., Ducraux,A., Nicoulaz,A.L. and Iggo,R. (
Fujimoto,T., Onda,M., Nagai,H., Nagahata,T., Ogawa,K. and Emi,M. (
Mirmohammadsadegh,A., Maschke,J., Basner-Tschakarjan,E., Bar,A. and Hengge,U.R. (
Janicke,R.U., Sprengart,M.L., Wati,M.R. and Porter,A.G. (
Inman,G.J. and Allday,M.J. (
Benoit,V., Chariot,A., Delacroix,L., Deregowski,V., Jacobs,N., Merville,M.P. and Bours,V. (
Beil,M., Leser,J., Lutz,M.P., Gukovskaya,A., Seufferlein,T., Lynch,G., Pandol,S.J. and Adler,G. (
Kim,P.K., Mahidhara,R. and Seol,D.W. (
Fisher,B., Costantino,J.P., Wickerham,D.L. et al . (
Cummings,S.R., Eckert,S., Krueger,K.A. et al . (
Pizzoferrato,E., Liu,Y., Gambotto,A., Armstrong,M.J., Stang,M.T., Gooding,W.E., Alber,S.M., Shand,S.H., Watkins,S.C., Storkus,W.J. and Yim,J.H. (
Author notes
1Lombardi Comprehensive Cancer Center and Department of Oncology and 2Department of Biostatistics and Biomathematics, Georgetown University School of Medicine, 3970 Reservoir Road NW, Washington, DC 20057, USA and 3Department of Medicine, Division of Clinical Pharmacology and Indiana University Cancer Center, Indiana University, Indianapolis, IN 46202, USA

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}