




Abstract
Mitomycin C (MMC) induces various types of DNA damages that cause significant cytotoxicity to cells. Accordingly, repair of MMC-induced damages involves multiple repair pathways such as nucleotide excision repair, homologous recombination repair and translesion bypass repair pathways. Nonetheless, repair of the MMC-induced DNA damages in mammals have not been fully delineated. In this study, we investigated potential roles for Xeroderma pigmentosum (XP) proteins in the repair of MMC-induced DNA damages using an assay that detects the ssDNA patches generated following treatment with MMC or 8′-methoxy-psoralen (8-MOP) + UVA (ultraviolet light A). Human wild-type cells formed distinctive ssDNA foci following treatment with MMC or 8-MOP + UVA, but not with those inducing alkylation damage, oxidative damage or strand-break damage, suggesting that the foci represent ssDNA patches formed during the crosslink repair. In contrast to wild-type cells, mutant defective in XPE orXPG did not form the ssDNA foci following MMC treatment, while XPF mutant cells showed a significantly delayed response in forming the foci. A positive role for XPG in the repair of MMC-induced DNA damages was further supported by observations that cells treated with MMC induced a tight association of XPG with chromatin, and a targeted inhibition of XPG abolished MMC-induced ssDNA foci formation, rendering cells hypersensitive to MMC. Together, our results suggest that XPG along with XPE and XPF play unique role(s) in the repair of MMC-induced DNA damages.
Introduction
DNA crosslink is a serious damage to chromosomal DNA that blocks key DNA metabolisms including DNA replication and transcription. Therefore, the ability of cells to repair crosslink damages is not only crucial for cell survival, but also a determining factor for drug sensitivity of cells. Since crosslink damage involves both strands of DNA, its repair process is likely to occur through the coordinated action of multiple DNA repair pathways such as nucleotide excision repair (NER), the double-strand break (DSB)/homologous recombination repair, and/or the translesion bypass repair pathways ( 1 , 2 ). A model of crosslink repair has been proposed, in which mammalian cells use excinuclease complex to uncouple the crosslink followed by homologous recombination or a translesion bypass repair to complete repair of the first strand ( 3 – 5 ). These notions are supported by the fact that Mitomycin C (MMC) or other crosslinking agents cause minimal structural distortion on chromosomal DNA and may not be recognized by known damage recognition factors such as (Xeroderma Pigmentosum, XP) XPC-hHR23B, XPA or RPA ( 6 , 7 ). Instead, the initiation of crosslink repair is possibly coupled to DNA replication or transcription machinery when it stalls at the damaged site ( 8 – 10 ).
A homologous recombination-based crosslink repair assay in vitro identified a number of proteins involved in interstrand cross link (ICL) repair, including ERCC1-XPF, RPA, PCNA, as well as MutS-beta ( 11 ). RPA was found to be essential for both the formation of incision as well as in the subsequent DNA synthesis step of crosslink damage repair, indicating that it is required for lesion recognition and/or for the subsequent endonucleolytic processing. PCNA is required for the DNA synthesis stage and although it is not critical for the incision stage of the reaction it does enhance this step presumably by a stimulation of lesion recognition by MutS-beta ( 11 ). Loss of bloom syndrome (BLM) gene or fanconi anemia (FA) gene(s) also resulted in hypersensitivity to ICL agent(s), suggesting that BLM and FA are either directly or indirectly involved in DNA repair or cell survival in response to crosslink damage ( 10 ). A recent study demonstrated that SNM1/PSO2, a gene encoding a 5′-exonuclease, was also required for incision of ICL repair in yeast ( 12 , 13 ). A siRNA targeting hSNM1B gene rendered cells sensitive to ionizing radiation, suggesting the possibility of hSNM1B involvement in homologous recombination repair of DSB arising as intermediates of ICL repair ( 12 ). Physical interaction between SNM1A, one of the three SNM1 families in higher eukaryotes, and PIAS1 was required for their nuclear localization as well as for the crosslink repair activity ( 14 ).
In an effort to understand the mechanistic details of the ICL repair pathway, we have developed an in vivo assay that detects the ssDNA foci generated in response to ICL agents such as MMC or 8′-methoxy-psoralen (8-MOP). WT cells exhibited distinctive ssDNA foci following ICL damages, while cells lacking XPE or XPG did not. On the other hand, XPF mutant cells showed a much delayed in response to ICL damage in forming the ssDNA foci, suggesting that formation of the ssDNA foci possibly represents the early stage of ICL repair pathway that requires various XP proteins.
Materials and methods
Chemicals and antibodies
MMC, 5′-bromo-2′-deoxyuridine (BrdU), methyl methane sulphonate (MMS), adriamycin (ADR), cis -dichlorodiammineplatinum(II) (CDDP; also known as cisplatin), H 2 O 2 and 8-MOP were purchased from Sigma Chemical (St. Louis, MO). An anti-BrdU monoclonal antibody specific for BrdU on ssDNA not dsDNA was from the BD Biosciences (San Jose, CA). Anti-XPG (monoclonal), anti-XPF (monoclonal), and anti-ERCC1 (monoclonal) antibodies were from the Neo Markers (Fremont, CA). An anti-GFP polyclonal antibody raised against full-length green fluorescent protein (GFP) was from the Santa Cruz Biotechnology (Santa Cruz, CA).
Cell lines and their culture conditions
Human fibroblasts [GM00037F (WT), GM02415B (XPE), GM08437A (XPF) and GM03021B (XPG)] were grown in minimum essential medium (MEM) supplemented with 20% fetal calf serum (FCS). HeLa cells were maintained in Dulbecco's modified Eagle's medium (DMEM) and F12 (1:1) supplemented with 10% FCS at 37°C in a 5% CO 2 incubator.
BrdU tracking and cell cycle progression
HeLa cells were cultured in the presence of 10 µM BrdU for 24 h. After removing BrdU from the media, cells were further incubated for 0, 20 or 40 h. After washing with PBS, cells were fixed in 70% ethanol at −20°C and washed again with PBS containing 0.5% FCS and 0.5% Tween-20. Following 2 M HCl treatment, cells were neutralized in 0.1 M sodium borate and stained with a fluorescein isothiocyanate (FITC)-conjugated anti-BrdU antibody. Following brief washing with PBS, cells were stained with 50 µg/ml propidium iodide (PI) in PBS containing 10 µg/ml RNase A. Samples were analyzed for DNA content (PI) on the x-axis and BrdU (FITC) on the y-axis by a FACScan flow cytometer (Becton Dickinson). Watson Pragmatic analysis was used to quantify cell cycle distribution of BrdU-incorporated cells ( 15 ).
The ssDNA patch assay
Monolayer cells (1–3 × 10 4 ) seeded onto a coverslip (18 mm diameter) were grown for 30 h in culture medium containing 10 µg/ml BrdU. After removing BrdU with fresh media, cells were treated with various DNA damaging agents for 1 h. Cells were briefly washed with fresh media and further incubated for the indicated period. For immunofluorescent analysis, cells were washed with PBS, fixed in ice-cold absolute methanol for 30 min at −20°C, and rinsed in ice-cold acetone for a few seconds. After washing four times with PBS, cells were incubated with an anti-BrdU monoclonal antibody in PBS containing 0.5% BSA for 2 h. After washing with PBS, samples were incubated with FITC-conjugated anti-mouse IgG (Molecular Probes, Eugene, OR) for 1 h. Slides were mounted in 90% glycerol, 0.1 M Tris–HCl (pH 8.0) and 2.3% 1,4-diazobicyclo-2,2,2-octane, 4′,6-diamidino-2-phenylindole (2 mg/ml). Images were collected using a Zeiss LSM-510 confocal microscope.
Transfection of cells with siRNA
The siRNA specific for XPG and XPF, as well as a control-siRNA were prepared from Dharmacon Research (Lafayette, CO). We also synthesized an antisense XPG-morpholino (25mer; 5′-CCAGAGCCCCTGGACCCC CAT GAGG-3′; underlined letters represent translation start site) (Gene Tools, LLC, Philomath, OR). HeLa cells (1–3 × 10 4 ) were plated on 6-well plates and incubated for 12–16 h prior to transfection. Cells were washed once with fresh medium and siRNA (0.2–0.4 µM) diluted in DMEM/F12 to a final volume of 200 µl added. In a separate tube, 6 µl of Oligofectamine transfection reagent (Invitrogen, Carlsbad, CA) was mixed with 15 µl of DMEM/F12 and incubated for 10 min at room temperature. Diluted siRNA samples were combined with the Oligofectamine mixtures and incubated for 20 min at room temperature before adding to cells. After incubation for 4 h, 0.5 ml culture medium containing 30% serum was added without removing the transfection mixture. Following incubation for 24–96 h, cell lysates were prepared and examined for efficacy of siRNA by western blot analysis.
Preparation of cell lysates and immunoblot analysis
Cells (1 × 10 5 ) were collected, washed with PBS, and lysed in a buffer containing 25 mM HEPES (pH 7.5), 0.3 M NaCl, 1.5 mM MgCl 2 , 0.2 mM EDTA, 0.5% Triton X-100, 20 mM β-glycerolphosphate, 1 mM sodium vanadate, 1 mM DTT, protease inhibitor cocktails (Sigma Chemical). Cell lysates (50 µg) were loaded onto a SDS–PAGE and, following gel electrophoresis, proteins were transferred to a PVDF membrane (Millipore, Billerica, MA) and immunoblotted with primary antibody followed by peroxidase-coupled secondary antibody (Amersham, Piscataway, NJ) and an enhanced chemiluminescence (Amersham) reaction prior to visualization on Kodak-o-mat film.
Association of XPG with chromatin following ICL damage
HeLa cells (1 × 10 5 ) were plated in a 100 mm culture dishes and transfected with pEGFP-XPG using a transfection reagent (FuGENE 6). After 24 h, the cells were treated with MMC (20 µM) for 1 h and harvested at various time points for nuclei preparation. Isolated nuclei were washed with a buffer containing 0.3 M NaCl, and chromatin-associated proteins were separated on 8% SDS–PAGE and analyzed for XPG by western blot. Ku80 was used as a loading control. For immunofluorescence analysis of chromatin-associated XPG, HeLa cells were grown on a slide and transfected with pEGFP-XPG using FuGENE 6. After 24 h, cells were treated with DNA damaging agent for 1 h and further incubated for 2 h before cell harvesting. Cells were treated with lysis buffer (25 mM HEPES, pH 7.5, 0.3 M NaCl, 1.5 mM MgCl 2 , 0.2 mM EDTA, 0.5% Triton X-100, 0.5 mM DTT and 1 mM PMSF) for 10 min on ice, washed once with PBS, and fixed in ice-cold acetone–methanol (1:1) for 7 min at −20°C. Slides were mounted in 90% glycerol, 0.1 M Tris–HCl (pH 8.0) and 2.3% 1,4-diazobicyclo-2,2,2-octane, 4′,6-diamidino-2-phenylindole (2 mg/ml). Images were collected using a Zeiss LSM-510 confocal microscope.
Results
Mammalian cells treated with MMC induced formation of unique ssDNA foci
MMC induces guanine-N2 monoadducts or a DNA crosslink in the minor groove between two guanines at their 2-amino groups, which can be repaired through translesion bypass, recombinational repair, or NER pathway ( 16 – 18 ). Regardless of the types of DNA damage, initiation of MMC-induced DNA repair is likely to involve dual incisions of one strand of the damaged DNA site in mammalian cells, which results in the generation of the ssDNA patch ( 2 ). We therefore examined whether MMC treatment induces formation of the ssDNA foci in mammalian cells. To visualize the ssDNA patches, BrdU-labeled cells were treated with MMC and, the ssDNA patches were identified using an anti-BrdU antibody (BD Biosciences) that specifically recognizes BrdU on ssDNA and not dsDNA ( Figure 1A ). DNA histogram analysis showed that intensity of BrdU labeling (y-axis) reduced to half every twenty hours ( Figure 1B ), suggesting that majority of cells were actively involved in cell cycle progression following BrdU incorporation. In the ssDNA foci assay, WT human fibroblast cells formed distinctive foci following MMC treatment ( Figure 2A ). These foci possibly represented the ssDNA patches at the crosslink damage sites since the cells treated with MMS or ADR did not show such foci ( Figure 2A ). Furthermore, WT cells treated with both psoralen and ultraviolet light A (UV-A), but not either alone, also showed ssDNA foci similar to those treated with MMC ( Figure 2B ), suggesting that MMC-induced ssDNA foci represent the ssDNA patches produced during crosslink repair in vivo .
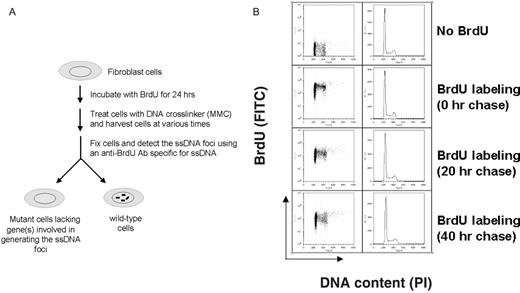
Schematic diagram of the ssDNA foci assay. ( A ) This assay is designed to evaluate the ICL repair activity in mammalian cells. 5′-bromo-2′-deoxyuridine (BrdU)-incorporated fibroblast cells were treated with DNA crosslinker (MMC) for 1 h and further incubated in fresh media for the repair of damaged DNA. An anti-BrdU antibody recognized BrdU on ssDNA patches and exhibited distinctive nuclear foci. An antibody was highly specific for BrdU on ssDNA and did not show any interaction with BrdU on dsDNA (data not shown). ( B ) HeLa cells were monitored for cell cycle progression following labeling with BrdU. After the BrdU labeling (10 µM) for 24 h, cells were harvested at various time points, washed once in PBS, and fixed in 70% ethanol at −20°C. The cells were washed with PBS containing 0.5% FCS and 0.5% Tween-20, denatured in 2 M HCl, neutralized in 0.1 M sodium borate, and stained with an anti BrdU antibody conjugated to FITC. The cells were washed in PBS as above and stained with 50 µg/ml PI in PBS containing 10 µg/ml RnaseA. The samples were analyzed by bivariate FACS analysis, with DNA content (PI) on the x-axis and BrdU (FITC) on the y-axis. The dot plots and the histograms representing the cells are shown.
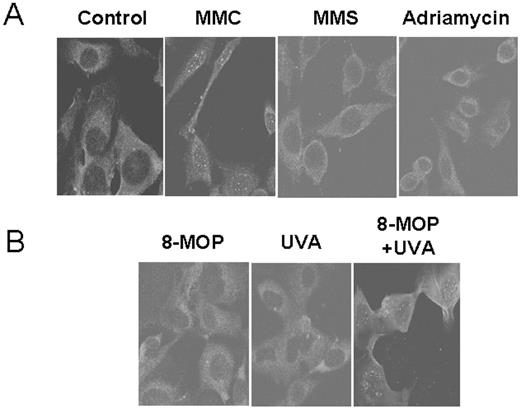
Formation of the ssDNA foci was observed following treatment with MMC, not with ADR or MMS. ( A ) Following BrdU labeling of cells for 30 h, WT human fibroblast cells were treated with various DNA damaging agents (2 µM MMC, 100 µM MMS or 10 µM ADR) for 1 h. Cells were further incubated in fresh media for 24 h prior to cell fixing and immunostaining with an anti-BrdU antibody specific for ssDNA patches. Fluorescence images were analyzed by confocal microscope. ( B ) WT cells were treated with 8-MOP (100 µM), UV-A (500 J/m 2 ), or both 8-MOP + UV-A prior to immunostaining analysis using an anti-BrdU antibody specific for ssDNA patches.
Human mutant cells defective in XPE or XPG failed to form ssDNA foci following MMC treatment
Several XP proteins are involved in either recombination-dependent or recombination-independent crosslink repair in vitro ( 17 , 19 ). We examined WT cells and XP-deficient fibroblast cells for formation of the ssDNA foci following MMC treatment. WT cells and XPF-deficient cells showed distinctive ssDNA-specific patches following MMC treatment, whereas cells defective in XPE or XPG did not ( Figure 3A ), suggesting that the MMC-induced ssDNA patch assay is different from those observed in the NER pathway ( 20 ). Similar results were obtained when cells were treated with both psoralen and UV-A ( Figure 3B ), suggesting that the ssDNA foci represent an essential step in the repair of crosslink damage. It should be pointed out that only 20–30% of WT and XPF-deficient cells were positive in forming the foci following MMC treatment ( Figure 3B ), suggesting that formation of the ssDNA foci may occur when cells are in the S-phase of the cell cycle ( 7 ). This is in keeping with a recent observation that it is crosslink damage induced DSB that forms during DNA replication ( 5 ).
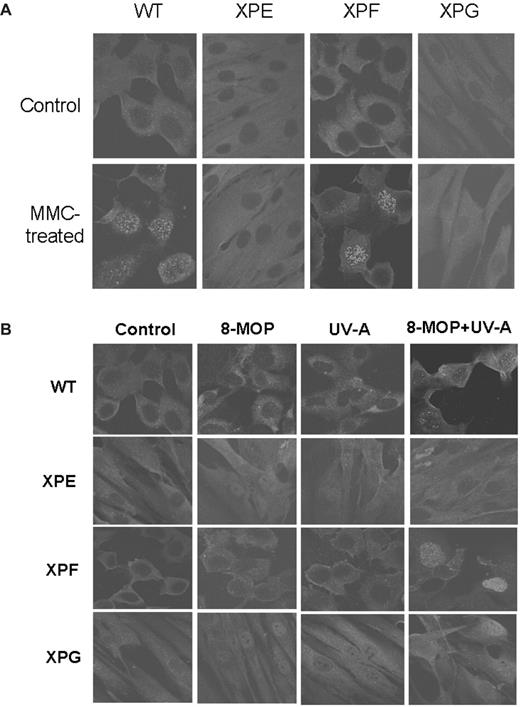
Formation of the ssDNA foci following crosslink damage in WT cells and various XP-mutant cells. ( A ) WT cells or various XP-mutant cells were treated with either 0 (top panels) or 20 µM (bottom panels) MMC for 1 h, and further incubated for 24 h prior to immunofluorescence with an anti-BrdU antibody specific for ssDNA patches. ( B ) WT or XP-mutant cells were mock-treated (first row), 100 µM 8-MOP (second row), 500 J/m 2 UV-A (third row), or 100 µM 8-MOP + 500 J/m 2 UV-A (fourth row), and further incubated for 12 h prior to immunofluorescence as described in A.
Lack of XPF significantly delayed MMC-induced ssDNA foci formation in vivo
Both XPF and XPG are structure-specific endonucleases that play essential role in NER ( 2 , 6 ). A positive role for XPF in the early stage of ICL repair has been reported previously ( 2 , 5 , 11 ). A question arises whether XPG and XPF play a unique role in formation of the ssDNA foci following MMC treatment. We therefore carried out kinetic analysis of the ssDNA foci formation with WT and XP-deficient cells following MMC treatment. The WT cells showed the ssDNA foci within 3 h following MMC treatment ( Figure 4 ). The foci formation reached maximum in 24 h and completely disappeared in 72 h, suggesting that the foci representing ssDNA patches occurred during the early stage of crosslink repair. Repair of MMC-induced damages occurred at a significantly slower rate than NER that usually concludes within 2 h following UV damage ( 21 , 22 ). A mutant defective in XPG or XPE showed no foci formation up to 96 h following MMC treatment, while XPF mutant formed the ssDNA foci at a much later time point (24 h post-treatment) than WT cells (3 h post-treatment) ( Figure 4 ), suggesting that XPF, in addition to XPE and XPG, is also involved in MMC-induced crosslink repair.
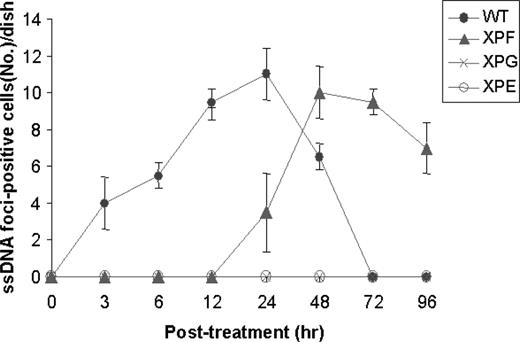
Kinetic analysis of the ssDNA foci formation following MMC treatment. WT and various XP-mutant human fibroblast cells were examined for formation of the ssDNA foci following MMC treatment. Cells were treated with 2 µM of MMC for 1 h prior to cell harvest at various time points and measured for the ssDNA foci-positive cells per coverslip (18 mm diameter) in a dish. Numbers were the mean values from four separate samples.
A targeted inhibition of XPG abolished formation of the ssDNA foci following MMC treatment
Human XPG mutant cells failed to form ssDNA patches following MMC treatment, suggesting that XPG may have a unique role in the crosslink repair. A possibility exists, however, that human XPG-deficient cells may have additional mutation(s) at other gene(s) crucial for inducing ssDNA patches following MMC treatment. To address this concern, WT human fibroblast cells were treated with siRNA specific to XPG or XPF, and examined for MMC-induced ssDNA foci formation. Both XPG- and XPF-siRNA were highly effective since the treatment of HeLa cells with either siRNA significantly lowered protein expression to an undetectable level ( Figure 5A , lanes 3–6) under conditions where a control siRNA showed little or no effect ( Figure 5A , lane 2). A targeted inhibition of XPF also led to a significant decrease in ERCC-1, an XPF-associated protein ( Figure 5B ), suggesting that XPF stabilizes ERCC-1 by forming a complex ( 23 ). Treatment of cells with XPG-siRNA completely abolished formation of the ssDNA foci, whereas XPF-siRNA showed little or no effect ( Figure 5C ), suggesting that XPG has a unique role in the repair of MMC-induced DNA damages that possibly occurred during the early stage of crosslink repair.
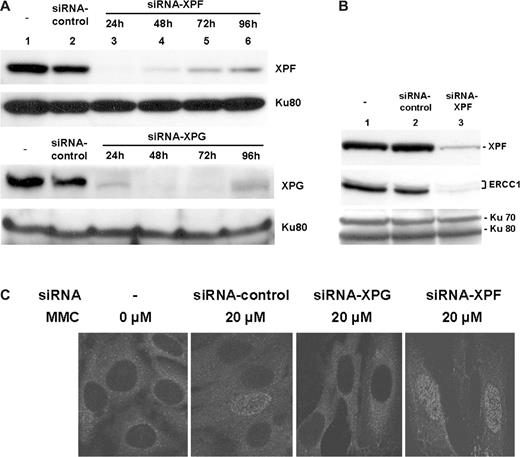
A targeted inhibition of XPG abolished formation of the ssDNA foci following MMC treatment. ( A ) Effect of XPG- or XPF-specific siRNA on protein expression. WT human fibroblast cells were either untreated (lane 1; upper & lower panels), or treated with a control (scrambled) siRNA (lane 2; upper and lower panels), XPF-specific siRNA (lanes 3–6; upper panel) or XPG-specific siRNA (lanes 3–6; lower panel). After harvesting cells at various times, expression of XPG or XPF was analyzed by immunoblot. Expression of Ku80 was included as an internal control for individual lanes. ( B ) A targeted inhibition of XPF also affected expression of its binding partner, ERCC-1. WT human fibroblast cells were either untreated (lane 1), or treated with a control (scrambled) siRNA (lane 2), or XPF-specific siRNA (lane 3) for 48 h prior to harvest. Cell extracts were prepared and analyzed for XPF and ERCC1 by western blot. Expression of Ku70/Ku80 was included as an internal control (bottom panel). ( C ) A siRNA targeting of XPG not XPF abolished formation of the ssDNA foci following MMC treatment. WT human fibroblast cells were treated with indicated siRNA for 24 h prior to treatment with 20 µM MMC for 1 h. Following incubation for 24 h, the ssDNA foci were visualized using immunofluorescence with an anti-BrdU antibody specific for ssDNA patches. Cells were analyzed by confocal microscope.
A targeted inhibition of XPG sensitizes HeLa cells in response to ICL damage
Since siRNA effectively inhibited XPG expression and formation of the ssDNA foci in HeLa cells, we examined whether a targeted inhibition of XPG sensitizes cells following crosslink damage. HeLa cells were treated with either control-antisense RNA or two different XPG-antisense RNAs (Morpholino A or B) for 72 h and plated in a 96-well plate (1 × 10 4 cells/well). Unlike many antisense structural types (e.g. siRNA), Morpholinos do not degrade their target RNA molecules; instead, they sterically block binding to a target sequence within an RNA and simply get in the way of molecules which might otherwise interact with the RNA. Treatment of cells with the target (XPG)-morpholino lowered the XPG expression to undetectable level (lanes 3 and 4, Figure 6 ) under conditions where the control morpholino showed a little or no effect (lane 2, Figure 6 ), suggesting that the antisense morpholino strategy was effective in targeting XPG in cancer cells.
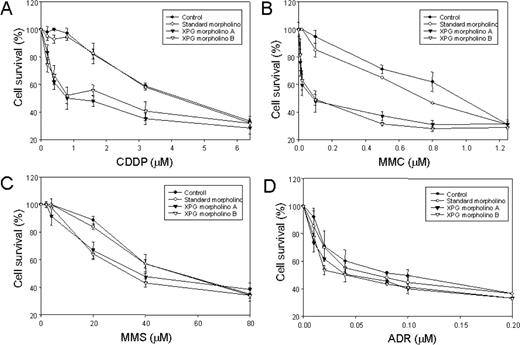
Effect of a targeted inhibition of XPG on cell sensitivity following treatment with various DNA damaging agents. HeLa cells were incubated with either control-specific antisense RNA or two different XPG-specific antisense RNA for 48 hrs prior to treatment with DNA damaging agent. Cells were treated with various concentrations of cisplatin ( A ), MMC ( B ), MMS ( C ) or adriamycin (ADR) ( D ) for 72 h before cell survival assay. Percentage of cell survival (%) was obtained from MTT assay at the indicated concentration and the results were the averages of three assays.
Cells were treated with various DNA damaging agents and incubated further at 37°C for 72 h. Cells treated with mock or a control antisense RNA were resistant up to 1 µM of cisplatin (CDDP) treatment. On the other hand, cells treated with XPG-specific probe were sensitive to cisplatin treatment (IC 50 = 0.5 µM; Figure 6A ). Similar to cisplatin, cells also showed extreme sensitivity to MMC treatment only when treated with the XPG-specific probe ( Figure 6B ). In contrast, cells showed relatively mild sensitivity in response to alkylating agent (MMS) or ADR when pretreated with the XPG-specific probe ( Figure 6C and D ). This result also supports a positive role for XPG in the repair of crosslink damage.
An association of XPG with chromatin was induced upon crosslink damage
To further investigate a potential role for XPG in the repair of crosslink damage, nuclei were isolated from HeLa cells following treatment with MMC or cisplatin, and examined for association of XPG. Western blot analysis indicated that most of XPG was not associated with chromatin under normal conditions ( Figure 7A , lane 1 and Figure 7B , first panel), whereas MMC treatment immediately induced the interaction of XPG with chromatin ( Figure 7A , lanes 2 and 3). An immunofluorescence study with isolated nuclei from HeLa cells also showed an induced association of XPG with chromatin following MMC or cisplatin treatment ( Figure 7B ). These observations support a positive role for XPG in perhaps an early stage of crosslink repair.
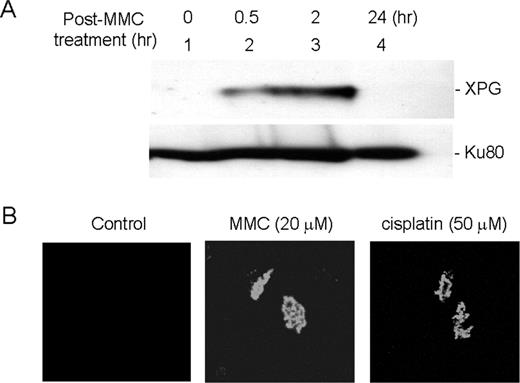
Chromatin association of XPG was induced upon MMC damages. ( A ) Western blot analysis of chromatin-associated XPG following MMC damage. HeLa cells were treated with MMC (20 µM) for 1 h and harvested at various time points. Isolated nuclei were washed with a buffer containing 0.3 M NaCl, and chromatin-associated proteins were separated on 8% SDS–PAGE and analyzed for XPG by western blot. Ku80 was used for western analysis as a loading control. ( B ) Immunofluorescence analysis of chromatin-associated XPG following treatment with either MMC or cisplatin. HeLa cells were grown on slides and treated with nothing (first row), 20 µM MMC (middle row) and 50 µM cisplatin (third row) for 1 h and incubated for 2 h prior to chromatin preparation. Images were collected using a Zeiss LSM-510 confocal microscope.
Discussion
MMC induces both intra- and inter-strand DNA crosslinks that block DNA replication and transcription. It is not known how MMC-induced DNA damages are repaired in mammals; however, the initiation process possibly involves dual incision on the first strand and generates the ssDNA patch on one strand, while the damaged DNA piece hangs on the second strand ( 2 , 11 , 23 ). Filling the gap of the first strand DNA can be accomplished by either translesion bypass or recombinational repair mechanism ( 18 ). In this study, we described an in vivo analysis of MMC damage and its repair using an assay that detects the ssDNA patches produced during the crosslink repair. We found that MMC-induced formation of the ssDNA patches was completely dependent on the presence of XPG and XPE, suggesting that these two proteins may have unique role(s) in the early stage of crosslink repair.
Both XPF and XPG are structure-specific endonucleases essential for the release of an oligonucleotide containing the lesion in the early stage of NER ( 6 ). Following MMC treatment, however, XPG (3′-endonuclease) was crucial for formation of the ssDNA foci, whereas XPF (5′-endonuclease) affects kinetics of the foci formation ( Figure 4 ). Although detailed mechanism of MMC-induced ssDNA foci is yet to be defined, our in vivo finding was somewhat contradictory to the previous in vitro observations that the excinuclease complex involving XPF-ERCC1 was responsible for the recognition of crosslink damage and generation of dual incisions ( 2 , 5 , 17 , 19 ). This discrepancy could be explained by the fact that the in vivo crosslink repair may be complex and involve more than one repair pathway ( 18 ), whereas the previous in vitro studies were reconstituted predominantly by one ICL repair pathway. Alternatively, XPG and XPF-ERCC1, unlike NER, may have separate role(s) in forming the ssDNA patches that represent an early stage of crosslink repair pathway, whilst both participate in the later stage of ICL repair such as incision of the second strand.
It is intriguing that MMC-induced ssDNA foci formation in XPF mutant cells occurred much later than that observed in WT cells ( Figure 4 ). ERCC1-XPF, through its role in NER, is essential for the repair of crosslink DNA damage. ERCC1-XPF is also involved in recombinational repair processes distinct from NER. Earlier genetic data implicate that the XPF-ERCC1 was required for crosslink repair via homologous recombination-mediated DSB repair pathway ( 5 ). However, an increased amount of DSB and the phosphorylation of histone variant H2AX (gamma-H2AX) were observed in both WT and Ercc1(−/−) cells following MMC treatment, although in Ercc1(−/−) cells MMC-induced gamma-H2AX foci persisted at least 48 h longer than in WT cells ( 5 ), suggesting that XPF-ERCC1 is required for the resolution of MMC-induced DSBs, although DSBs occur after crosslink damage via an ERCC1- (or XPF-) independent mechanism ( 5 ). It would be interesting to see whether the MMC-induced ssDNA foci actually represent the early stage of crosslink repair via the homologous recombination-mediated DSB repair pathway.
We do not know what specific role XPG plays in forming the ssDNA foci following MMC treatment in vivo . XPG is not only essential for dual incisions at the crosslink lesion, but is also required in a non-enzymatic capacity for occurrence of the second, 5′-incision by the ERCC1-XPF heterodimer, another structure-specific endonuclease of opposite polarity ( 2 , 24 , 25 ). XPG interacts with a number of repair factors involved in NER/transcription-coupled repair (TCR) [transcription factor II-H (TFII-H), RPA and PCNA] ( 26 – 29 ). XPG is also involved in base excision repair of oxidative DNA damages ( 30 , 31 ). Our findings described here show that XPG was exclusively localized in the nucleus and its interaction with chromatin was induced following MMC treatment ( Figure 7 ). Furthermore, a targeted inhibition of XPG significantly increased cell sensitivity in response to crosslinking agents ( Figure 6 ), suggesting that XPG is also involved in ICL repair pathways. XPG and its endonuclease activity may directly be involved in generation of the ssDNA patches following MMC damages; however, a possibility exists that a unique role for XPG in crosslink repair may be through mediating physical interaction with other repair factors. MMC-induced formation of the ssDNA patches was also dependent on the presence of XPE ( Figure 3 ). XPE was originally identified as a damage-specific DNA-binding protein ( 32 ) and may be involved in recognizing MMC-induced DNA damage, although we do not know what specific role it plays in the repair of MMC-induced DNA damage.
We would like to thank Drs Randy Legerski and Laura S. Haneline for critical reading of the manuscript. This research was supported by grants from NIH (CA92111 to SHL) and the US Army (DAMD17-00-1-0295 to S-HL). S-JP was supported by NIH postdoctoral fellowship (F32 GM20167-01).
Conflict of Interest Statement : None declared.
References
De Silva,I.U., McHugh,P.J., Clingen,P.H. and Hartley,J.A. (
Sancar,A., Lindsey-Boltz,L.A., Unsal-Kacmaz,K. and Linn,S. (
McHugh,P.J., Spanswick,V.J. and Hartley,J.A. (
Reardon,J.T. and Sancar,A. (
Niedernhofer,L.J., Odijk,H., Budzowska,M. et al . (
Lee,S.H. (
Rothfuss,A. and Grompe,M. (
Pichierri,P., Averbeck,D. and Rosselli,F. (
Pichierri,P. and Rosselli,F. (
Pichierri,P., Franchitto,A. and Rosselli,F. (
Zhang,N., Lu,X. and Legerski,R.J. (
Demuth,I., Digweed,M. and Concannon,P. (
Li,X., Hejna,J. and Moses,R.E. (
Ishiai,M., Kimura,M., Namikoshi,K. et al . (
Watson,J.V., Chambers,S.H. and Smith,P.J. (
Warren,A.J., Maccubbin,A.E. and Hamilton,J.W. (
Li,L., Peterson,C.A., Lu,X., Wei,P. and Legerski,R.J. (
Zheng,H., Wang,X., Warren,A.J., Legerski,R.J., Nairn,R.S., Hamilton,J.W. and Li,L. (
Bessho,T., Mu,D. and Sancar,A. (
Rubbi,C.P. and Milner,J. (
McCarthy,M.J., Rosenblatt,J.I. and Lloyd,R.S. (
Mone,M.J., Bernas,T., Dinant,C., Goedvree,F.A., Manders,E.M., Volker,M., Houtsmuller,A.B., Hoeijmakers,J.H., Vermeulen,W. and van Driel,R. (
Bessho,T., Sancar,A., Thompson,L.H. and Thelen,M.P. (
Araujo,S.J., Tirode,F., Coin,F., Pospiech,H., Syvaoja,J.E., Stucki,M., Hubscher,U., Egly,J.M. and Wood,R.D. (
Riedl,T., Hanaoka,F. and Egly,J.M. (
He,Z., Henricksen,L.A., Wold,M.S. and Ingles,C.J. (
Gary,R., Ludwig,D.L., Cornelius,H.L., MacInnes,M.A. and Park,M.S. (
Le Page,F., Kwoh,E.E., Avrutskaya,A., Gentil,A., Leadon,S.A., Sarasin,A. and Cooper,P.K. (
Thorel,F., Constantinou,A., Dunand-Sauthier,I. et al . (
Klungland,A., Hoss,M., Gunz,D., Constantinou,A., Clarkson,S.G., Doetsch,P.W., Bolton,P.H., Wood,R.D. and Lindahl,T. (
Bessho,T. (
Author notes
1Department of Biochemistry and Molecular Biology, 2Microbiology and 3Walther Oncology Center, Indiana University School of Medicine, Indianapolis, IN 46202, USA and 4Inje University, Kimhae, Korea

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}