




Abstract
The dystroglycanopathies are a novel group of human muscular dystrophies due to mutations in known or putative glycosyltransferase enzymes. They share the common pathological feature of a hypoglycosylated form of α-dystroglycan, diminishing its ability to bind extracellular matrix ligands. The LARGE glycosyltransferase is mutated in both the myodystrophy mouse and congenital muscular dystrophy type 1D (MDC1D). We have transfected various cell lines with a variety of LARGE expression constructs in order to characterize their subcellular localization and effect on α-dystroglycan glycosylation. Wild-type LARGE co-localized with the Golgi marker GM130 and stimulated the production of highly glycosylated α-dystroglycan (hyperglycosylation). MDC1D mutants had no effect on α-dystroglycan glycosylation and failed to localize correctly, confirming their pathogenicity. The two predicted catalytic domains of LARGE contain three conserved DxD motifs. Systematically mutating each of these motifs to NNN resulted in the mislocalization of one construct, while all failed to have any effect on α-dystroglycan glycosylation. A construct lacking the transmembrane domain also failed to localize at the Golgi apparatus. These results indicate that LARGE needs to both physically interact with α-dystroglycan and function as a glycosyltransferase in order to stimulate α-dystroglycan hyperglycosylation. We have also cloned and overexpressed a homologue of LARGE, glycosyltransferase-like 1B (GYLTL1B). Like LARGE it localized to the Golgi apparatus and stimulated α-dystroglycan hyperglycosylation. These results suggest that GYLTL1B may be a candidate gene for muscular dystrophy and that its overexpression could compensate for the deficiency of both LARGE and other glycosyltransferases.
INTRODUCTION
The last 3 years have seen an explosion in our understanding of the pathogenic mechanisms underlying a group of human muscular dystrophies. These forms, collectively termed dystroglycanopathies, all have mutations in known or putative glycosyltransferase enzymes and share the common pathological feature of a hypoglycosylated form of α-dystroglycan in skeletal muscle ( 1 – 3 ). They include three severe forms of congenital muscular dystrophy (CMD) with eye abnormalities and neuronal migration defects: muscle-eye-brain disease [MEB (OMIM 253280)] ( 4 ), Walker–Warburgh–syndrome [WWS (OMIM 236670)] ( 5 ) and Fukuyama CMD [(FCMD OMIM 253800)] ( 6 ). In addition both congenital muscular dystrophy type 1C [a form with variable brain involvement; MDC1C (OMIM 606612)] ( 7 ) and the milder allelic variant limb girdle muscular dystrophy 2I [LGMD2I (OMIM 607155)] ( 8 ) are due to mutations within the Fukutin-related protein ( FKRP ) gene.
Along with these human diseases an identical pathomechanism exists in the myodystrophy mouse (Large myd ). These mice are characterized by a relatively severe phenotype with kyphoscoliosis and shortened life span due to a progressive muscular dystrophy with markedly elevated serum creatin kinase (CK). Other features include histological evidence of cardiac involvement and a neuronal migration defect ( 9 , 10 ). The spontaneously arising myodystrophy mutation is an autosomal recessive 100 kb genomic deletion within the Large gene ( 11 ). We recently identified two heterozygous mutations in the human orthologue of this gene in one patient with muscular dystrophy and reduced α-dystroglycan immunolabelling ( 12 ). These were a missense mutation [G1525A (E509K)] and a single nucleotide insertion that is predicted to result in the premature termination of translation (1999insT). The clinical features of this novel form of CMD, termed MDC1D (OMIM 608840), were similar to other α-dystroglycanopathies with congenital onset, mental retardation and evidence of structural brain involvement together with white matter dysmyelination on brain magnetic resonance imaging (MRI).
Dystroglycan is a crucial component of the dystrophin–glycoprotein complex ( 13 ). The DAG1 gene encodes a precursor protein that is post-translationally cleaved into α and β subunits. α-Dystroglycan is a peripheral membrane glycoprotein that associates with transmembrane β-dystroglycan at the sarcolemma, together forming a link between the extracellular matrix and the actin-associated cytoskeleton ( 14 , 15 ). Both α- and β-dystroglycan are glycosylated, but whereas the β subunit is consistently detected with a molecular mass of 43 kDa, the mass of α-dystroglycan is variable. Developmental and tissue-specific glycosylation occurs within the central mucin-like domain of the 74 kDa α-dystroglycan core polypeptide, giving rise to mature proteins with molecular masses ranging from 120 to 200 kDa ( 15 , 16 ). α-Dystroglycan glycosyation appears to consist largely of O -linked oligosaccharides, and these structures form the receptors for many of its extracellular matrix ligands including laminin-1 and 2, agrin, neurexin and perlecan ( 17 , 18 ). In the dystroglycanopathies these carbohydrate moieties are either absent or significantly reduced, resulting in a decrease in high affinity binding of extracellular matrix ligands ( 19 ).
Although the biochemical activity of LARGE has yet to be established, recent research from the group of Kevin Campbell has identified two intriguing activities of this protein. Firstly, the transient overexpression of LARGE in cultured fibroblasts demonstrated a marked increase in α-dystroglycan glycosylation (hyperglycosylation) and a corresponding increase in high affinity binding to several extracellular matrix ligands ( 20 ). In addition, LARGE-induced hyperglycosylation was also observed in fibroblasts from patients with WWS, MEB and FCMD and led to a restoration of extracellular matrix ligand binding. This suggests that LARGE overexpression can activate additional pathways that result in α-dystroglycan glycosylation ( 20 , 21 ). Secondly, the N-terminal domain of α-dystroglycan has very recently been shown to interact directly with LARGE and that this association is a requirement for normal glycosylation ( 22 ). However, there are still several unanswered questions regarding LARGE function. There is no information on the subcellular localization of LARGE and therefore where its association with α-dystroglycan occurs. It is also not clear whether the participation of LARGE in α-dystroglycan glycosylation is restricted to a structural interaction or whether it has an additional enzymatic function. Sequence analysis predicts LARGE to contain two catalytic domains, both possessing at least one conserved DxD (Asp-any-Asp) motif typical of many glycosyltransferases ( 23 – 26 ). The first domain is related to bacterial α-glycosyltransferases, whereas the second is most closely related to human β-1,3- N -acetylglucosaminyltransferase, required for synthesis of the poly- N -acetyllactosamine backbone (Galβ1→4GlcNAcβ1→3) n found on N - and O -glycans and glycolipids ( 27 , 28 ). The presence of two catalytic domains raises the possibility they may function independently.
We have now transiently expressed both normal and mutated LARGE constructs in a variety of cultured cell lines in order to characterize LARGE localization and function. The overexpression of LARGE localized at the Golgi apparatus in all cell lines investigated. LARGE MDC1D mutants failed to localize correctly and had no effect on α-dystroglycan glycosylation. Systematically altering the conserved DxD motifs and deleting the transmembrane region demonstrated that LARGE needs to both physically interact with α-dystroglycan and function as a glycosyltransferase in order to stimulate α-dystroglycan hyperglycosylation.
Genes encoding a closely related homologue of LARGE are present in the genomes of human ( glycosyltransferase-like 1B ; GYLTL1B ), mouse ( Glylt1b ; also known as Large-Like or LargeL ) and other vertebrate species ( 24 ). The human gene is located on chromosome 11p11.2 and encodes a 721 amino acid protein that shares 67% identity with LARGE, suggesting the two genes may have arisen by gene duplication. Like LARGE, it is predicted to have two catalytic domains, though it lacks the coiled-coiled region present in the former protein. In order to establish if GYLTL1B has a similar biochemical function to LARGE, we transiently overexpressed it in a variety of cultured cell lines and determined its effect on α-dystroglycan glycosylation. In an identical pattern to LARGE, the overexpression of GYLTL1B localized at the Golgi apparatus and stimulated α-dystroglycan hyperglycosylation. We further show that GYLTL1B-induced hyperglycosylation had a corresponding increase in its laminin binding capacity.
RESULTS
Wild-type LARGE is localized at the Golgi apparatus and stimulates the synthesis of highly glycosylated α-dystroglycan
The human LARGE coding sequence was cloned into the pcDNA3.1-TOPO V5/His expression vector (Fig. 1 ) and transfected into mouse C2C12 myoblasts, human fibroblast and Chinese hamster ovary (CHO) cell cultures. Transfection efficiency for all experiments was consistent at around 10% for myoblasts, 10% for fibroblasts and 65% for CHO cells. Transfected cultures were labelled with antibodies to the V5 epitope and either calreticulin [a molecular chaperone abundantly expressed in the endoplasmic reticulum (ER)] or GM130 (a marker for the Golgi apparatus). Co-localization with GM130 was seen in transfected C2C12 myoblast cultures (Fig. 2 ) and both fibroblast and CHO cultures (data not shown). When C2C12 transfected myoblast cultures were allowed to fuse and differentiate into myotubes, co-localization with GM130 was once more observed (Supplementary Material Fig. 1). No co-localization with calreticulin was seen in any of the cell lines (Fig. 3 B). Treatment of transfected C2C12 myoblast cultures with brefeldin A, a fungal toxin that causes the Golgi to disassemble, resulted in the relocation of LARGE expression to the ER, indicative of a Golgi resident protein (Fig. 3 A).
Double-labelling of all transfected cultures with the α-dystroglycan IIH6 antibody (a mouse monoclonal antibody raised against an unknown glycosylated epitope) and anti-V5 showed a marked increase in the immunoreactivity of all transfected cell lines, most clearly seen in CHO cultures due to low levels of endogenous IIH6 expression (Fig. 4 ). Western blotting of protein lysates from all transfected cultures demonstrated that the increase in IIH6 immunoreactivity was due to a dramatic increase in highly glycosylated α-dystroglycan, appearing as a broad smear on the polyacrylamide gel. In contrast, both the levels of expression and molecular weight of β-dystroglycan were unaltered (data not shown). Again these results were most marked in transfected CHO cultures, reflecting both low endogenous IIH6 production and a greater efficiency of transfection (Fig. 5 ).
LARGE MDC1D mutants are mislocalized to the ER and fail to stimulate the synthesis of highly glycosylated α-dystroglycan
To observe the effects of MDC1D mutations on LARGE localization and function we introduced the corresponding mutations into the wild-type LARGE–V5 construct (E509K and the 693 amino acid truncation due to 1999insT, Fig. 1 ). Both constructs no longer co-localized with GM130 in transfected C2C12 myoblast and myotube cultures, rather immunostaining with anti-V5 appeared as both perinuclear and a more diffuse pattern of cytoplasmic staining, typical of retention within the ER. (Fig. 2 ; Supplementary Material, Fig. 1). Rare co-localization (∼1%) of the G1525A mutant with GM130 was observed but was always accompanied by diffuse cytoplasmic staining (data not shown). Double-labelling of transfected cultures with anti-V5 and an antibody to calreticulin demonstrated the perinuclear and cytoplasmic staining co-localized with the ER (Fig. 3 B). Labelling LARGE MDC1D transfected CHO cultures with the IIH6 antibody detected no increase in immunoreactivity (Fig. 4 ) and western blotting analysis of protein lysates showed no increase in highly glycosylated α-dystroglycan (Fig. 5 ).
LARGE DxD→NNN mutant constructs fail to stimulate the synthesis of highly glycosylated α-dystroglycan
The DxD motifs in LARGE are indicative of the presence of glycosyltransferase activity. To assess the contribution of the three conserved DxD motifs in LARGE, we used site-directed mutagenesis to change these amino acids to NNN (Asn-Asn-Asn; Fig. 1 ). Mutating the DxD motif to NNN has previously been used to abolish the function of putative glycosyltransferases ( 29 ). The change from acidic to basic residues inhibits the binding of positively charged groups at the active site of the domain, diminishing its catalytic activity. Mutant constructs replacing the conserved DxD motif in the first catalytic domain, LARGE DxD1→NNN and LARGE DxD2→NNN, co-localized with GM130 in transfected C2C12 cultures (Fig. 2 ). Transfected cultures, however, showed no increase in IIH6 immunoreactivity (data not shown) and western blotting analysis of protein lysates similarly showed no α-dystroglycan hyperglycosylation (Fig. 5 ). The LARGE DxD3→NNN construct, replacing the DxD motif in the second catalytic domain, did not co-localize with GM130 in transfected C2C12 myoblast cultures. Rather immunostaining was observed to be both perinuclear and diffusely cytoplasmic. Double-labelling with antibodies to calreticulin and anti-V5 once more demonstrated ER localization (Fig. 3 B). Again IIH6 immunoreactivity was not increased on immunostaining (data not shown) and western blotting of protein lysates showed no α-dystroglycan hyperglycosylation (Fig. 5 ).
LARGE constructs lacking the transmembrane domain do not localize at the Golgi apparatus and fail to stimulate the synthesis of highly glycosylated α-dystroglycan
The transfection of C2C12 myoblasts with a LARGE construct lacking both its putative transmembrane and cytoplasmic C-terminal domain (LARGE ΔTMD, Fig. 1 ) failed to co-localize with either Golgi GM130 or calreticulin (Figs 2 and 3 B). Expression was relocated to the cytoplasm where it gave rise to a punctate appearance, consistent with its localization in vesicular structures. The subcellular compartment to which these vesicles belong remains unclear because double-staining failed to detect any co-localization with antibodies to either EEA1 (an early endosomal marker) or COP I (a coat protein of vesicles involved in the retrograde transport from Golgi to ER; data not shown). Moreover, the LARGE ΔTMD construct did not increase IIH6 immunoreacivity of transfected CHO cultures (data not shown) or stimulate α-dystroglycan hyperglycosylation on western blotting (Fig. 5 ).
GYLTL1B localizes to the Golgi apparatus and stimulates the synthesis of highly glycosylated α-dystroglycan in a fashion identical to LARGE
The human GYLTL1B coding sequence was also cloned into the pcDNA3.1-TOPO V5/His expression vector (Fig. 1 ) and transfected into the same cell lines as for LARGE. Transfected cultures double-labelled with anti-V5 and anti-GM130 showed co-localization in all cell lines investigated (Fig. 2 ; Supplementary Material, Fig. 1). Labelling with the α-dystroglycan IIH6 antibody showed a marked increase in immunoreactivity in all transfected cultures (Fig. 4 ) and western blotting of protein lysates demonstrated a corresponding increase in highly glycosylated α-dystroglycan (Fig. 6 A). The results of transient GYLTL1B expression were indistinguishable from those of LARGE.
GYLTL1B overexpression increases α-dystroglycan laminin binding
In order to see if GYLTL1B-induced hyperglycosylation of α-dystroglycan increased its laminin binding ability we performed a laminin-1 overlay assay. Both LARGE and GYLTL1B induced hyperglycosylation were associated with a corresponding increase in laminin binding. Moreover, there was no detectable difference between the increased laminin binding due to GYLTL1B when compared with LARGE (Fig. 6 B).
DISCUSSION
In this paper, we have demonstrated that by its co-localization with GM130, transient overexpression of LARGE is localized at the Golgi apparatus in a variety of cell types including differentiated C2C12 myotubes. Treating transfected cultures with brefeldin A confirmed this subcellular localization. Although overexpression of tagged proteins can on occasions result in mislocalization ( 30 ), the appearance of LARGE at the Golgi apparatus is consistent with the prediction that it contains a type 2 membrane spanning region, typical of glycosyltransferases located in this organelle. As previously observed, the overexpression of LARGE resulted in a dramatic increase in α-dystroglycan glycosylation ( 20 ). This was observed in transfected cell cultures from all cell lines. Constructs harbouring MDC1D mutations failed to localize correctly and expression was concentrated around the nucleus and more diffusely throughout the cell. The immunostaining co-localized with the ER marker calreticulin, consistent with a misfolded protein retained within the ER and targeted for ER-associated degradation (ERAD). The ERAD pathway retrotranslocates (dislocates) misfolded proteins back into the cytosol for proteasomal degradation ( 31 ). Overexpression of MDC1D mutated constructs had no effect on α-dystroglycan glycosylation, confirming the pathological significance of these mutations in MDC1D.
We undertook systematic mutagenesis of all three conserved DxD motifs in LARGE in order to identify those essential for catalytic activity and address whether either catalytic domain can function independently. DxD motifs were changed to NNN so they no longer bind positively charged groups essential for the catalytic mechanism. We based our analysis on the constructs' ability to hyperglycosylate α-dystroglycan. Mutation of the two DxD motifs in the first catalytic domain had no effect on their localization at the Golgi apparatus but both constructs failed to stimulate the hyperglycosylation of α-dystroglycan. This suggests that both DxD motifs may be involved in the catalytic activity of this domain. It further suggests that catalytic domain 2 does not function independently, i.e. in the absence of a functional catalytic domain 1. The LARGE DxD3→NNN construct failed to target to the Golgi apparatus, most likely due to misfolding of the protein, and hence we were unable to draw any conclusions as to the function of this motif.
Previous work has shown that forced expression of LARGE results in the hyperglycosylation of α-dystroglycan not only in control fibroblasts but also in those from patients with WWS, FCMD and LGMD2I ( 20 ). It has also been shown that the N-terminal domain of α-dystroglycan interacts with LARGE and this is a prerequisite for its proper glycosylation ( 22 ). Amongst the glycans present on α-dystroglycan are those that are linked via an unusual O -mannosyl group. One of these has been implicated as the major laminin receptor on α-dystroglycan ( 32 ). The synthesis of O -mannosyl glycans is initiated by the enzyme protein- O -mannosyl transferase 1 (POMT1) ( 4 , 32 ). In yeasts and other lower eukaryotes this enzyme resides in the ER ( 33 ) and it suggests that partially glycosylated α-dystroglycan is transported from this organelle to the Golgi where LARGE is one of a group of enzymes that elongates immature glycans. However, LARGE has no homology with any of the enzyme families expected to participate in the synthesis of the known glycans on α-dystroglycan. This raises the possibility that the role of LARGE in the glycosylation of α-dystroglycan is mediated through a physical rather than enzymatic interaction. Our results with the LARGE DxD1→NNN and LARGE DxD2→NNN constructs, which are correctly targeted to the Golgi apparatus but fail to stimulate α-dystroglycan hyperglycosylation, demonstrate that the catalytic activity is most likely essential for proper glycosylation.
The LARGE construct lacking the transmembrane domain failed to glycosylate α-dystroglycan and was not retained within the Golgi apparatus, but was located in what appeared to be secretory vesicles. Glycosyltransferase retention within the Golgi apparatus occurs through a variety of means ( 34 ). One mechanism is the use of signal sequences, most noticeably within the transmembrane domain. Other proteins are retained through being associated in macromolecular complexes, either with itself (kin recognition) or within a multienzyme complex. It has previously been shown that enzymes lacking a transmembrane domain can still be retained within the Golgi apparatus ( 34 ). The relocation of the LARGE ΔTMD construct suggests that the signal for LARGE retention within the Golgi apparatus resides within the C-terminal cytoplasmic tail or transmembrane domain. In addition, the LARGE ΔTMD construct demonstrates that LARGE has to be correctly localized in the Golgi membrane in order to interact with α-dystroglycan and presumably other proteins involved in its glycosylation.
Little is known about the LARGE homologue GYLTL1B. Inspection of EST databases (dbEST: www.ncbi.nlm.nih.gov/entrez ) and expression profiling (GEO Profiles: WWW.ncbi.nlm.nih.gov/entrez ) suggest it is widely expressed, though the level of tissue-specific transcripts is unclear. Like LARGE, GYLTL1B stimulated α-dystroglycan hyperglycosylation and increased its affinity for laminin binding. This suggests it could potentially compensate for loss of LARGE function in tissues where both enzymes are expressed. It has recently been shown that the POMT1 knockout mouse is embryonically lethal due to the lack of formation of Reichert's membrane ( 35 ), a phenotype shared with the dystroglycan null mouse ( 36 ). The myodystrophy mouse is in effect a LARGE ‘knockout’ ( 24 ), but here the phenotype is much less severe. As the proper glycosylation of α-dystroglycan apparently requires the physical interaction of LARGE and α-dystroglycan, one would expect the phenotype of a knockout mouse to be as severe as the POMT1 knockout. That this is not the case may be due to GYLTL1B compensating for the loss. GYLTL1B compensation may also underlie the tissue-specific disease pattern of the myodystrophy mouse and MDC1D.
Despite the recent advances in the field of muscular dystrophy and glycosylation disorders, there remain many other forms of CMD with an identical pattern of dystroglycan abnormalities that are genetically uncharacterized. GYLTL1B could be a highly significant candidate gene in these disorders. The ability of LARGE overexpression to stimulate α-dystroglycan hyperglycosylation in cell lines from patients with dystroglycanopathies offers the potential of a novel therapeutic approach ( 20 ). It now seems likely that GYLTL1B could potentially fulfil the same role and will be of added benefit in being effective in patients with MDC1D.
MATERIALS AND METHODS
Construction of expression plasmids
Total human brain RNA (Clontech) was reversed transcribed using Superscript III (Invitrogen) following the manufacturer's instructions. The coding sequences of LARGE (NCBI accession no. NM 004737 ) (forward primer GGATTAGGGATTGCCACTT; reverse primer ACTTCCCCTACAGCATGTC) and GLYTL1B (NCBI accession number NM 152312 ) (forward primer CCAGAAGGGACGTCAAGG; reverse primer CTGCCCAATGCTAAGATG) were amplified using Advantage HF Polymerase (Clontech). The LARGE 693 truncated mutant was amplified using primers GGATTAGGGATTGCCACTTf and TCGTACTCCGGGCAAGTCACGTCTCAr and LARGE ΔTMD using CTGTTTTCTGGGAGCTTCGAf and ACTTCCCCTACAGCATGTCr. Amplified products were cloned into the pcDNA3.1/V5-His TOPO expression vector (Invitrogen) following the manufactures' instructions. This vector directs the synthesis of a fusion protein with the V5 epitope at its C-terminal end.
Site-directed mutagenesis
PCR-directed mutagenesis was performed on plasmid DNA using overlapping mismatch primers and Advantage HF Polymerase (Clontech). Amplified products were digested with Dpn I (10 U) to remove contaminating parental plasmid DNA and subsequently transformed into One Shot TOP10 Chemically Competent Escherichia coli cells (Invitrogen). Mutations were confirmed by DNA sequencing. The primers used to make mutant constructs were (mismatches underlined):
LARGE E509Kf CTGTCAGACGCC G AGGCCCAGCAGT
LARGE E509Kr ACTGCTGGGCCT C GGCGTCTGACAG
LARGE DxD1→NNNf GTCATCGTCCTT AACAACAAC ATCACCTTTGCC
LARGE DxD1→NNNr GGCATTGAAAAT GTTGTTGTT AGCTAAGGATGT
LARGE DxD2→NNNf ATGTTCCTGTCT AACAACAAC TTCCTGCCCATG
LARGE DxD2→NNNr CATGGGCAGGAA GTTGTTGTT AGACAGGAACAT
LARGE DxD3→NNNf ACATCCTTAGCT AACAACAAC ATTTTCAATGCC
LARGE DxD3→NNNr GGCATTGAAAAT GTTGTTGTT AGCTAAGGATGT
Cell culture and transfection
C2C12 myoblast cells (ECCAC) were cultured in 20% FCS in DMEM (high glucose) supplemented with 2 m ML -glutamine. Human fibroblast cultures derived from skin biopsies were cultured in DMEM (high glucose) and CHO cells (ECACC) in Nutrient Mixture F-12 HAM (high glucose, Sigma) plus 2 m ML -glutamine. Where indicated, cells were incubated with growth medium supplemented with 10 µ g/ml of brefeldin A (Sigma) for 30 min at 37°C and fixed in 2% paraformaldehyde or in methanol–acetone (1 : 1). Transfection of cell lines was carried out using Gene Juice (Novagen) in serum free media. After 24 h cells were either fixed in 2% paraformaldehyde or methanol for immunohistochemistry or harvested in an appropriate buffer for western blotting. Transfected myotubes were generated by transferring myoblasts, 24 h after transfection, into fusion medium (DMEM supplemented with 5% horse serum). Cells were fixed after 4 days. All transfection experiments were performed at least three times.
Antibodies and immunohistochemistry
After fixation cells were permeabilized with 0.05% Triton X-100. For double-labelling, cells were first incubated for 1 h with one of the following monoclonal antibodies: α-dystroglycan IIH6 (Upstate Biotechnology), GM130 (BD Biosciences), calreticulin (BD Biosciences), EEA1 (BD Biosciences) or COP I (ABCAM). After washing, an anti-mouse conjugated to Alexa 594 was applied for 30 min. For antibody IIH6 an anti-mouse IgM biotinylated (DAKO) was used followed by streptavidin conjugated to Alexa 594 (Molecular Probe). After washing, an anti-goat V5 FITC conjugated antibody (Bethyl) was applied for 1 h.
Cell nuclei were stained with Hoechst 33342 (Molecular Probes). All dilutions and washings were made in phosphate buffered saline and incubations were performed at room temperature. Sections were mounted in aqueous mountant and viewed with epifluorescence using a Leica Diaphot microscope and digitally captured using Metamorph software (Universal Imaging Inc.).
Western blotting and overlay assays
Cell proteins were extracted in sample buffer consisting of 75 m M Tris–HCl, 1% SDS, 2-mercaptoethanol, plus a cocktail of protease inhibitors (antipain, aprotinin and leupeptin). Soluble proteins were resolved using a NuPage Pre-cast gel (4–12% Bis–Tris; Invitrogen) and then transferred electrophoretically to nitrocellulose membrane (Hybond-ECL, Amersham). Nitrocellulose strips were blocked in 3% BSA (IgG- and protease-free, Jackson Laboratories) Tris-buffered saline buffer, and then probed with IIH6 α-dystroglycan antibody (Upstate Biotechnology) at room temperature for 1 h. After washing they were incubated with an anti-mouse biotinylated IgM (DAKO) followed by HRP-streptavidin (DAKO). Both incubations were for 1 h at room temperature. After washing, membranes were visualized using chemiluminescence (ECL+Plus, Amersham).
For the laminin overlay assay, nitrocellulose membranes were blocked for 1 h in laminin binding buffer (LBB: 10 m M triethanolamine, 140 m M NaCl, 1 m M MgCl 2 , 1 m M CaCl 2 , pH 7.6) containing 5% non-fat dry milk followed by incubation with mouse Engelbreth–Holm–Swarm laminin (Gibco) overnight at 4°C in LBB. Membranes were washed and incubated with anti-rabbit laminin (Sigma) followed by HRP-anti-rabbit IgG (Jackson). Blots were visualized using chemiluminescence (ECL+Plus, Amersham).
SUPPLEMENTARY MATERIAL
Supplementary Material is available at HMG Online.
ACKNOWLEDGEMENTS
This work was funded by the Muscular Dystrophy Campaign of Great Britain and Northern Ireland and by the Medical Research Council UK. C.B. was the recipient of a University of Bologna traveling fellowship.
The authors wish it to be known that, in their opinion, the first two authors should be regarded as joint First Authors.
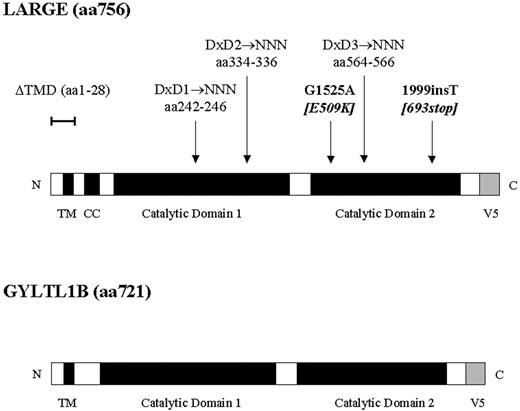
Figure 1. Schematic diagram of LARGE–V5 and GYLTL1B–V5 fusion proteins showing their domain organization. TMD, transmembrane domain, CC, coiled-coiled domain. The position of LARGE MDC1D mutations (bold), transmembrane deletion (ΔTMD) and conserved DxD motifs are also shown.
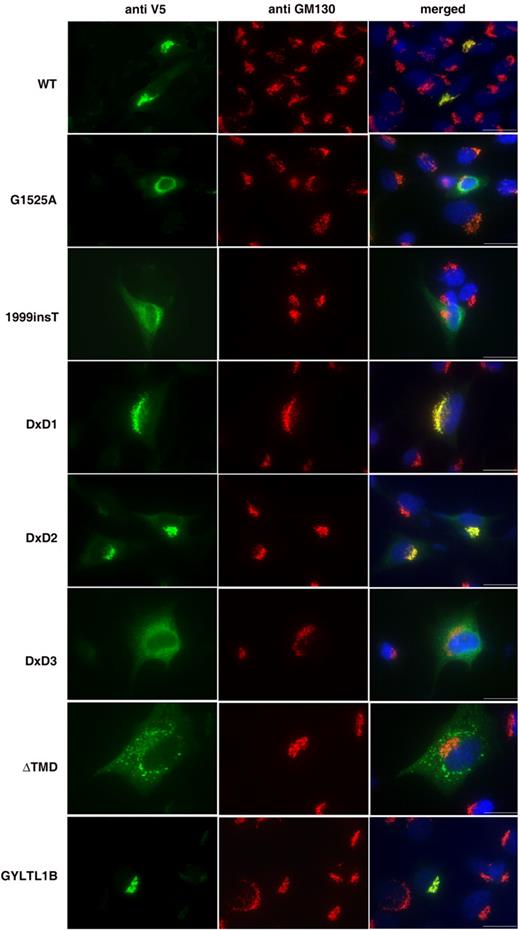
Figure 2. Localization of LARGE–V5 and GYLTL1B–V5 constructs in transfected C2C12 myoblast cultures. Cultures were double-labelled with anti-V5 (green channel) and the Golgi marker GM130 (red channel). Co-localization can be seen in the merged pictures of LARGE wild-type (WT), LARGE DxD1→NNN, LARGE DxD2→NNN and GYLTL1B constructs. No co-localization was seen with LARGE constructs harbouring MDC1D mutations (G1525A and 1999insT), LARGE DxD3→NNN or LARGE ΔTMD. Blue channel, Hoechst 33342 nuclear stain; Scale bar, 20 µm.
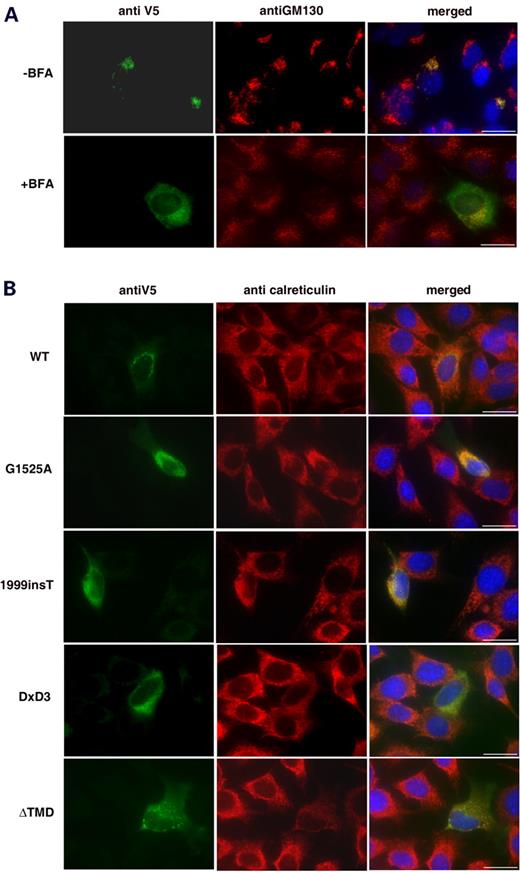
Figure 3. ( A ) Double-labelling of LARGE–V5 C2C12 transfected myoblast cultures treated with (+) and without (−) brefeldin A. Green channel, anti-V5, red channel, GM130. Brefeldin A causes both Golgi disassembly and LARGE re-localization. ( B ) Double-labelling of C2C12 transfected myoblast cultures with anti-V5 (green channel) and the ER marker anti-calreticulin (red channel). Co-localization can be seen in the merged pictures of LARGE MDC1D mutants (G1525A and 1999insT) and LARGE DxD3→NNN constructs, while LARGE ΔTMD failed to co-localize. Blue channel, Hoechst 33342 nuclear stain; Scale bar, 20 µm.
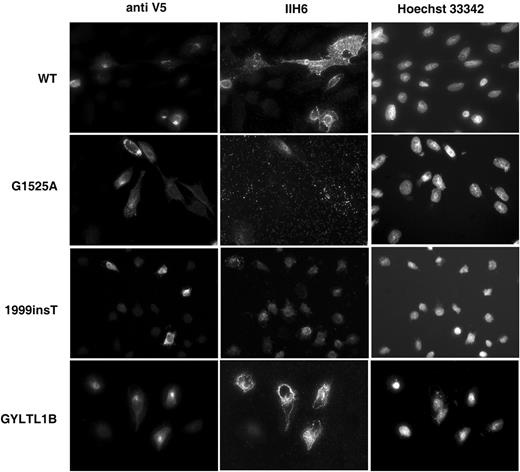
Figure 4. Double-labelling of CHO transfected cultures with anti-V5 (left hand panel) and α-dystroglycan antibody IIH6 (recognizing an unknown glycosylated epitope; middle panel). Right panel shows Hoechst 33342 nuclear staining. Increased IIH6 immunoreactivity is seen with LARGE wild-type (WT) and GLYTL1B constructs. LARGE MDC1D (G1525A and 1999insT) did not alter IIH6 immunoreactivity. Scale bar, 20 µm.
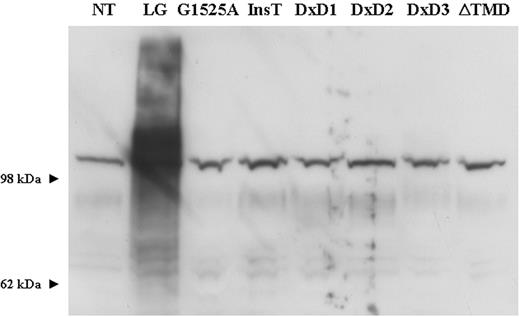
Figure 5. Western blot analysis of protein lysates from transfected CHO cultures using α-dystroglycan IIH6 antibody. The overexpression of LARGE–V5 stimulates α-dystroglycan hyperglycosylation. All other LARGE constructs had no effect on α-dystroglycan expression. NT, non-treated; LG, LARGE–V5; InsT, LARGE 1999insT.
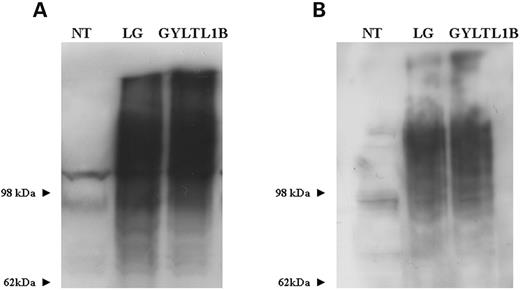
Figure 6. ( A ) Western blot analysis of protein lysates from CHO cultures transfected with LARGE and GYLTL1B using α-dystroglycan IIH6 antibody. Both constructs can be seen to be equally effective in stimulating α-dystroglycan hyperglycosylation. ( B ) Laminin-1 overlay assay of the same western blot showing LARGE and GLYLT1B-induced α-dystroglycan hyperglycosylation binds laminin. NT, non treated; LG, LARGE V5.
References
Michele, D.E. and Campbell, K.P. (
Muntoni, F., Brockington, M., Torelli, S. and Brown, S.C. (
Martin, P.T. and Freeze, H.H. (
Yoshida, A., Kobayashi, K., Manya, H., Taniguchi, K., Kano, H., Mizuno, M., Inazu, T., Mitsuhashi, H., Takahashi, S., Takeuchi, M. et al. (
Beltran-Valero de Bernabe, D., Currier, S., Steinbrecher, A., Celli, J., van Beusekom, E., van der Zwaag, B., Kayserili, H., Merlini, L., Chitayat, D., Dobyns, W.B. et al. (
Kobayashi, K., Nakahori, Y., Miyake, M., Matsumura, K., Kondo-Iida, E., Nomura, Y., Segawa, M., Yoshioka, M., Saito, K., Osawa, M. et al. (
Brockington, M., Blake, D.J., Prandini, P., Brown, S.C., Torelli, S., Benson, M.A., Ponting, C.P., Estournet, B., Romero, N.B., Mercuri, E. et al. (
Brockington, M., Yuva, Y., Prandini, P., Brown, S.C., Torelli, S., Benson, M.A., Herrmann, R., Anderson, L.V., Bashir, R., Burgunder, J.M. et al. (
Lane, P.W., Beamer, T.C. and Myers, D.D. (
Holzfeind, P.J., Grewal, P.K., Reitsamer, H.A., Kechvar, J., Lassmann, H., Hoeger, H., Hewitt, J.E. and Bittner, R.E. (
Grewal, P.K., Holzfeind, P.J., Bittner, R.E. and Hewitt, J.E. (
Longman, C., Brockington, M., Torelli, S., Jimenez-Mallebrera, C., Kennedy, C., Khalil, N., Feng, L., Saran, R.K., Voit, T., Merlini, L. et al. (
Ervasti, J.M. and Campbell, K.P. (
Holt, K.H., Crosbie, R.H., Venzke, D.P. and Campbell, K.P. (
Ibraghimov-Beskrovnaya, O., Ervasti, J.M., Leveille, C.J., Slaughter, C.A., Sernett, S.W. and Campbell, K.P. (
Ibraghimov-Beskrovnaya, O., Milatovich, A., Ozcelik, T., Yang, B., Koepnick, K., Francke, U. and Campbell, K.P. (
Durjeeb, M., Henry, M.D. and Campbell, K.P. (
Henry, M.D. and Campbell, K.P. (
Michele, D.E., Barresi, R., Kanagawa, M., Saito, F., Cohn, R.D., Satz, J.S., Dollar, J., Nishino, I., Kelley, R.I., Somer, H. et al. (
Barresi, R., Michele, D.E., Kanagawa, M., Harper, H.A., Dovico, S.A., Satz, J.S., Moore, S.A., Zhang, W., Schachter, H., Dumanski, J.P. et al . (
Muntoni, F., Brockington, M. and Brown, S.C. (
Kanagawa, M., Saito, F., Kunz, S., Yoshida-Moriguchi, T., Barresi, R., Kobayashi, Y.M., Muschler, J., Dumanski, J.P., Michele, D.E., Oldstone, M.B. and Campbell, K.P. (
Peyrard, M., Seroussi, E., Sandberg-Nordqvist, A., Xie, Y., Han, F., Fransson, I., Collins, J., Dunham, I., Kost-Alimova, M., Imreh, S. et al. (
Grewal PK and Hewitt JE. (
Busch, C., Hofmann, F., Selzer, J., Munro, S., Jeckel, D. and Aktories, K. (
Wiggins, C.A. and Munro, S. (
Heinrichs, D.E., Monteiro, M.A., Perry, M.B. and Whitfield, C. (
Sasaki, K., Kurata-Miura, K., Ujita, M., Angata, K., Nakagawa, S., Sekine, S., Nishi, T. and Fukuda, M. (
Munro, S. and Freeman, M. (
Berger, E.G. (
Hampton, R.Y. (
Gentzsch, M. and Tanner, W. (
Colley, K.J. (
Willer, T., Prados, B., Falcon-Perez, J.M., Renner-Muller, I., Przemeck, G.K., Lommel, M., Coloma, A., Valero, M.C., De Angelis, M.H., Tanner, W., et al. (

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}