




Abstract
Rett syndrome (RS) is a severe and progressive neurodevelopmental disorder caused by heterozygous mutations in the X-linked methyl CpG binding protein 2 (MeCP2) gene. MeCP2 is a nuclear protein that binds specifically to methylated DNA and functions as a general transcription repressor in the context of chromatin remodeling complexes. RS shares clinical features with those of Angelman syndrome (AS), an imprinting neurodevelopmental disorder. In AS patients, the maternally expressed copy of UBE3A that codes for the ubiquitin protein ligase 3A (E6-AP) is repressed. The similar phenotype of these two syndromes led us to hypothesize that part of the RS phenotype is due to MeCP2-associated silencing of UBE3A. Indeed, UBE3A mRNA and protein are shown here to be significantly reduced in human and mouse MECP2 deficient brains. This reduced UBE3A level was associated with biallelic production of the UBE3A antisense RNA. In addition, MeCP2 deficiency resulted in elevated histone H3 acetylation and H3(K4) methylation and reduced H3(K9) methylation at the PWS/AS imprinting center, with no effect on DNA methylation or SNRPN expression. We conclude, therefore, that MeCP2 deficiency causes epigenetic aberrations at the PWS imprinting center. These changes in histone modifications result in loss of imprinting of the UBE3A antisense gene in the brain, increase in UBE3A antisense RNA level and, consequently reduction in UBE3A production.
INTRODUCTION
The UBE3A gene that maps to the telomeric part of the 2 Mb PWS/AS imprinted domain is biallelically expressed in most human and mouse tissues, yet maternally expressed in various parts of the brain (1,2). The paternal allele is believed to be silenced by the product of UBE3A antisense that is expressed specifically in the brain (3,4). The entire 2 Mb PWS/AS imprinted domain that includes the UBE3A gene is under the coordinated control of an imprinting center (IC), composed of two sequences in humans, one at the SNRPN promoter (PWS-IC) (5) and the other 35 kb upstream (AS-IC) (6). The PWS-IC is unmethylated and has an open chromatin configuration on the paternal allele that enables it to activate paternally expressed genes. On the maternal allele, the PWS-IC is methylated with a closed chromatin conformation and thus inactivated (7–9).
Angelman syndrome (AS) is caused by repression of the maternally expressed copy of UBE3A (10), whereas Rett syndrome (RS) is a result of heterozygous mutations in methyl CpG binding protein 2 (MeCP2) (11). However, these two neurobehavioral syndromes share clinical manifestations such as mental retardation, seizures, muscular hypotonia and acquired microcephaly (12–14) and thus are sometimes misdiagnosed. Two reports have described mutations in MeCP2 in patients diagnosed with AS (12,13). This led us to hypothesize that UBE3A is affected by MeCP2 deficiency. This hypothesis was strengthened by the observation that in MeCP2 deficient mice, abnormalities are observed in the hippocampal neurons and in Purkinje cells, where the UBE3A gene is normally expressed only from the maternal allele (1,15). Here, we examine the possibility that MeCP2 deficiency downregulates UBE3A production by a mechanism that involves loss of imprinting of the UBE3Aas gene in the brain via a change in the epigenetic features at the PWS-IC locus.
RESULTS
UBE3A mRNA and protein production are reduced in RS patients and MeCP2 deficient mice
To examine the previously mentioned hypothesis, we first tested expression of the UBE3A gene at the RNA and protein levels in brains of normal individuals (N), AS patients (AS), female RS patients (RS/f), an RS male patient (RS/m) and samples of MeCP2 deficient newborn mice. The UBE3A mRNA level as measured by semi-quantitative and real-time PCR is low in AS and in the MeCP2 deficient human (RS/f and RS/m). In humans, the UBE3A mRNA level was ∼6-fold lower in brain samples of an RS/m and a AS patient, with about a 2-fold decrease in the brain of RS/f when compared with the RNA level in the normal human brain (N) (Fig. 1A and B). The expression of UBE3A and UBE3A antisense genes in human lymphoblast cell lines, in which MeCP2 is mutated on the inactive X chromosome (M10) or on the active X chromosome (M12), was also analyzed. Although UBE3A is equally expressed in both cell lines (Fig. 1B), UBE3A antisense is not expressed (data not shown). Although these cell lines obviously do not reflect the gene expression in the brain, they still can be used as controls. Western blot analysis of the UBE3A protein in human brain revealed, as expected, substantial amounts of the protein in the normal human brain (N), whereas lower amounts of the protein were observed in RS/f and no protein was detected in RS/m and in AS patients (Fig. 1C). Ube3a mRNA was similarly reduced in brains of MeCP2 deficient mice (ko/x and ko/y) as compared to normal brain (wt/y). In contrast to the observed changes in Ube3a expression, no such changes were observed when Snrpn RT–PCR products were analyzed (Fig. 1D) or when Ube3a expression was analyzed in the liver (Fig. 1E). Although substantial amounts of Ube3a protein were observed in brain of normal mice (wt/y), almost no protein is found in ko/x and no protein was observed in ko/y (Fig. 1F). It should be noted that western blot analysis is much less sensitive than the radiolabeled RT–PCR assay. Therefore, the low level of UBE3A mRNA seen in AS and RS/m (Fig. 1A) and the relative low level in ko/x and ko/y (Fig. 1D) still leaves open the possibility that small amounts of protein are produced but were not detected in western blots. Yet, the results strongly indicate that MeCP2 in the brain is associated with UBE3A production. Similar observations were reported recently by Samaco et al. (16).
The promoter region of the human UBE3A is unmethylated on both parental alleles
As differential DNA methylation patterns are characteristic of imprinted genes, where the active allele is unmethylated whereas the repressed allele is methylated (17), and as MeCP2 is a known repressor of genes that are methylated at their promoter, we asked whether the change in UBE3A expression is linked to UBE3A promoter methylation. Southern blot analyses using the methyl sensitive restriction enzymes NotI for human DNA (Fig. 2A) and CfoI for mouse DNA revealed a completely unmethylated UBE3A promoter in lymphocytes (L), Brain (Br), lymphoblasts in which MeCP2 is mutated on the inactive X chromosome (M10) and MeCP2 deficient cells in which the mutated MeCP2 is located on the active X chromosome (M12). An unmethylated UBE3A promoter was also observed in the brain of an RS/m and in the brain of an AS patient (Fig. 2A). Identical results were obtained in kidney (K), liver (L) and brain (Br) of a wild-type (WT) and a MeCP2 deficient mouse (KO/Y) (Fig. 2B). To rule out the possibility that the methylation at the restriction sites does not represent the methylation status of all CpG sites within the UBE3A promoter, we performed bisulfite methylation analysis on 34 CpG sites at positions +1 to +232 (accession no. NT_026466) (Fig. 2C). Our results clearly show that the UBE3A promoter is practically unmethylated and imply that methylation is neither involved in the normal monoallelic expression of the gene nor involved in repressing the gene in the absence of MeCP2. This is in accordance with previous observations (18).
Ube3a antisense is biallelically expressed in the brain of MeCP2 deficient mice
It has been suggested that monoallelic expression of UBE3A involves a paternally expressed antisense RNA in both human and mouse (3,4,19). This suggestion was based on the observation that paternal transmission of a PWS-IC deletion results in the lack of UBE3A antisense transcript in brain and the UBE3A gene is expressed biallelically. Biallelic expression of the UBE3A antisense results in silencing of UBE3A on both alleles (3,4). To test whether, in the absence of MeCP2, the UBE3A antisense gene maintains its monoallelic expression or becomes rather biallelically expressed, we performed reciprocal intersubspecific crosses between C57BL/6J and C57BL/6J mice congenic for the Mus musculus spretus chromosome 7 (C57BL/6.spretus.c7). Taking advantage of polymorphic GTTT repeats, five in C57BL/6.spretus.c7 (Fig. 3A) and seven in C57BL/6J (Fig. 3B), we indeed observed that the UBE3A antisense is monoallelically expressed from the paternal spretus copy in normal mice (F1 X/Y) (Fig. 3C), whereas it is expressed biallelically in brains of MeCP2 deficient mice [F1 KO/X (Fig. 3D) and F1 KO/Y (Fig. 3E)]. This is judged by the mixed sequence pattern that follows the five repeats, which are common to both alleles. As expected, the mixed pattern is more striking in the male KO/Y (Fig. 3E) than in the heterozygous female KO/X (Fig. 3D). In addition, we have performed RT–PCR with a 6FAM labeled primer. The PCR products were separated on a sequencing polyacrylamide gel that distinguishes between the maternal and paternal alleles on the basis of the size polymorphism (Fig. 3G). Primers used for RT–PCR were from one of the Ube3a antisense exons reported recently (4,19,20). Reactions without reverse transcription were carried out to exclude the possibility of DNA contamination (Fig. 3F). The observation of biallelic expression of the Ube3a antisense gene obtained by both direct sequencing and 6FAM method (Fig. 3) strengthens the possibility that MeCP2 is involved in maintaining monoallelic UBE3A expression through its effect on the UBE3A antisense gene.
MeCP2 deficiency affects the epigenetic structure of PWS-IC with no effect on SNRPN DNA methylation and expression
As deletions of the PWS-SRO in the mouse resulted in biallelic expression of Ube3a sense in brain (4), we asked whether MeCP2 regulates UBE3A expression indirectly by altering chromatin structure at the PWS-IC, consequently activating the UBE3A antisense gene on the maternal allele.
This possibility is supported by the following facts. (i) The PWS-IC is methylated on the repressed maternal allele, whereas the active paternal allele is unmethylated (21,22). (ii) MECP2 binds to methylated DNA through its methyl binding domain (MBD) and recruits histone deacetylases (HDAC1 and 2) and histone H3(K9) methylase by its transcriptional-repression domain (TRD) (23,24).
To test the possibility that MeCP2 is involved in UBE3A regulation via an effect on the epigenetic features of PWS-IC, MeCP2 binding to PWS-IC was first studied. Chromatin immunoprecipitation (ChIP) assays showed that MeCP2 in fact binds to the PWS-IC in the mouse brain and liver (compare WT and KO/Y mice) (Fig. 4A) and to the human PWS-IC in lymphoblasts preferentially on the methylated maternal allele (Fig. 4A, compare Mat with Pat). The maternal allele was distinguished using lymphoblasts of a patient, in which PWS-SRO was deleted on his paternal allele (Mat). The paternal allele was studied in lymphoblasts of the patient's father, in which the PWS-SRO deletion was on his maternal allele (Pat) (9). The ChIP assay was extended to UBE3A and Profillin2 in brain samples of normal (N) and RS/m (Fig. 4A). This rules out a genomewide unspecific binding of MeCP2 and strengthens the claim that MeCP2 does not have a direct effect on UBE3A (Fig. 4A). The low enrichment levels of Pat, M12 and mouse KO/Y testify for the specificity of the anti-MeCP2 antibody. Similar findings have been reported previously for other imprinted mouse genes (25,26).
Having observed the binding of MeCP2 to PWS-IC, we next asked whether the absence of MeCP2 had an effect on allele-specific methylation of the PWS-IC in RS/m brain (compare RS with Br) and in the MeCP2 deficient lymphoblast cell line (M12) compared with the MeCP2 producing lymphoblast cell line (M10). No effect of MeCP2 deficiency on PWS-IC methylation could be observed, yet the PWS-IC in AS brain was completely unmethylated (AS) (Fig. 4B). The parent-specific methylation of the PWS-IC in the MeCP2 deficient mice (KO/Y) was also not affected (Fig. 4C). In contrast, in the absence of the DNA methyltransferase (Dnmt1), the Snrpn promoter was completely unmethylated (Fig. 4C). It was previously reported that MeCP2 deficiency did not affect SNRPN expression pattern in human blood and brain (27). Here, we confirm this observation in the mouse (Fig. 1D).
Although MeCP2 deficiency had no effect on PWS-IC methylation or expression of SNRPN, histone modifications at the PWS-IC underwent substantial changes in the absence of MeCP2. These modifications were studied by ChIP analysis using antibodies specific to the acetylated H3, dimethylated H3(K4) and dimethylated H3(K9) on nucleosomes isolated from lymphoblasts in which the MeCP2 mutation is on the inactive X (M10), MeCP2 deficient lymphoblasts (M12) of normal individuals (N) and RS patients (RS), as well as in WT and MeCP2 deficient mice (KO/Y). As shown in Figure 4D, human histone H3 at PWS-IC was 2-fold more acetylated in cells deficient in MeCP2 (M12) when compared with M10 and 3-fold more acetylated in RS brain when compared with normal brain. Yet, no effect was observed in the acetylation of histone H3 at UBE3A. As expected, H3(K4) methylation was enriched at PWS-IC in MeCP2 deficient cells (M12) and in the brain of an RS patient (Fig. 4E). Accordingly, H3(K9) dimethylation decreased ∼2-fold in the brain of an RS patient (Fig. 4F). Similar results with respect to H3 acetylation and K4 methylation were obtained in mouse brain (Fig. 4G and H).
Taken together, the results described earlier clearly show that UBE3A gene expression in brain is not regulated by DNA methylation or histone modification on its promoter but rather indirectly by the effect of MeCP2 on PWS-IC, which in turn effects UBE3A monoallelic expression.
DISCUSSION
The present communication is one of the first demonstrations of a biologically relevant protein (UBE3A) that is affected by MeCP2 in vivo. Other proteins such as xhairy2a (28) and BDNF (29,30) have also been reported to be affected by MeCP2. The experiments described here suggest a mechanism by which MeCP2 affects UBE3A expression. This proposed mechanism may explain the similar clinical manifestations in RS and AS. We show here that MeCP2 deficiency causes an epimutation at the PWS-IC region, which consequently affects UBE3A expression by disrupting the imprinted state of the UBE3A antisense gene. As described in Figure 5, the paternal allele of PWS-IC is unmethylated and exists in an open chromatin configuration. This epigenetic status is believed to spread to all paternally expressed genes along the entire PWS/AS domain, including the UBE3A antisense gene. The UBE3A antisense RNA that is expressed primarily on the paternal allele in brain (3) is presumed to control the monoallelic expression of the UBE3A gene by silencing the gene on the paternal allele (Fig. 5A). On the maternal allele, MeCP2 binds to the methylated PWS-IC and recruits HDACs that deacetylate histone H3 and a histone methyltransferase to methylate H3(K9). This causes heterochromatinization and silencing of all paternally expressed genes across the entire PWS/AS domain, including the UBE3A antisense gene on the maternal allele (Fig. 5B). As demonstrated here, MeCP2 deficiency causes an epimutation at the maternal PWS-IC allele which is manifested in an open chromatin structure that is associated with increased histone H3 acetylation, H3(K4) methylation and decreased H3(K9) methylation (Fig. 4). This change in chromatin structure results in UBE3A antisense gene expression on the maternal copy leading to silencing in cis of the maternal copy of the UBE3A gene (Fig. 5C). Although the UBE3A antisense promoter has not been defined yet, it was suggested that in humans it resides within PWS-IC (31) and in mice it maps to sequences upstream to Snrpn (4,20). The activity of both regions is believed to be determined by the chromatin structure of PWS-IC, which spreads to the entire PWS/AS domain (9). In any event, it appears that the activity of the UBE3A promoter is independent of the activity of the SNRPN promoter. This conclusion is in accord with a previous report (27) and with the observations made here that the SNRPN promoter on the maternal allele remains methylated (Fig. 4B and C); thus, no change in SNRPN expression is observed in response to MeCP2 deficiency (Fig. 1B). This indicates that although changes in histone modifications at PWS-IC are sufficient to activate the UBE3A antisense gene on the maternal allele, demethylation at PWS-IC is required for activation of the SNRPN maternal copy. Accordingly, in AS patients, alleviation of repression of SNRPN on the maternal allele is always associated with demethylation at the PWS-IC (32).
In addition, it is striking that the UBE3A promoter shows no differential epigenetics in contrast to other genes in the domain, such as ZNF127 (33) and NDN (34). Thus, it appears that the differential expression of UBE3A in the brain is solely dependent on the differential expression of UBE3A antisense, which is controlled by the epigenetics at PWS-IC.
MeCP2 was originally believed to be a general transcriptional repressor, which binds to methylated DNA with no obvious sequence preference. However, subsequent experiments in human lymphoblasts using microarray analyses could not confirm this concept [(35) and our unpublished data]. Even in mice displaying overt disease symptoms, MeCP2 deficiency led to subtle gene expression changes in mutant brains (36). It could, therefore, be presumed that only a small subset of genes is affected by MeCP2 deficiency and some of these genes are associated with the RS phenotype. The reduced expression of UBE3A in the brain of RS patients as shown here is the first example that the production of a biologically relevant protein is affected by MeCP2 deficiency leading to the RS phenotype. As the phenotype of RS patients is complex, it is not impossible that a few proteins, other than UBE3A, are affected by MeCP2 deficiency and are involved in the RS symptoms.
MATERIALS AND METHODS
Mouse strains, cell lines and human brain tissue
Mouse strains used were C57BL/6J, C57BL/6J congenic for the M. musculus spretus chromosome 7 (C57BL/6.spretus.c7) and C57BL/6J females that carry a mutation in the MeCP2 gene (37) (Jackson laboratory). M10 and M12 cell lines are subclones of a lymphoblast cell line cloned from a blood sample of an RS patient (prepared in our laboratory). Mutation in MeCP2 is an insertion of adenine at position 23909 (accession no. AF030876). This mutation creates a stop codon that results in a truncated protein lacking MBD and TRD. M10 carries this mutation on the inactive X chromosome and M12 on the active X chromosome, therefore, it is MeCP2 deficient. Lymphoblasts of the PWS patient and his father, which were used to distinguish their maternal and paternal alleles, were generously obtained from Buiting et al. (6). Human brain tissue, normal and patients, was provided by the Brain and Tissue Bank at the University of Maryland, Baltimore, MD, USA. RS patients all had classical RS: 18-month-old RS/m (UMB no. 1238), 2-year-old RS/f (UMB no. 448) and 20-year-old-RS/f (UMB no. 1420). The AS patient (UMB no. 1494) was a 42-year-old female. Normal brain tissue was obtained from a 1-year-old male (UMB no. 135) and a 15-year-old male (UMB no. 388).
Antibodies for western blotting and ChIP
Antibodies for western blotting and ChIP are Anti-MeCP2 rabbit polyclonal IgG (Upstate) cat no. 07-013 Lot 23449, Anti-acetylated H3 rabbit polyclonal IgG (Upstate) cat no. 06-866 Lot 20667, Anti-K4 dimethylated H3 rabbit antiserum IgG (Upstate) cat no. 07-030 Lot 22672, Anti-K9 dimethylated H3 rabbit polyclonal IgG (Abcam), Anti-E6-AP (UBE3A) goat polyclonal IgG (Santa Cruz) cat no. sc-12380 Lot no. B201, Anti β-actin rabbit polyclonal IgG (Santa Cruz) cat no. sc-7210 Lot no. J2902 and Anti α-tubulin rat polyclonal IgG2a (Serotec) cat no. MCAP77 Lot no. 0400.
Western blot analysis
Identical protein samples (100 µg) were loaded on SDS–PAGE (10%) gels, which were electrophoresed, blotted and exposed to the polyclonal goat anti-human UBE3A antibody (Santa Cruz). β-Actin and α-tubulin (Serotec) antibodies were used as load controls.
Chromatin immunoprecipitation
ChIP was performed as described previously (38) with slight modifications. Lysates prepared from cross-linked lymphocytes or brain tissue were sonicated in a cell disruptor (Heat systems, Ultrasonic 350). Debris was pelleted, a 1/50 input fraction was withdrawn and supernatants were incubated overnight at 4°C shaking with anti-MeCP2, rabbit polyclonal anti-acetylated H3 or anti-dimethylated H3(K4) rabbit antiserum or rabbit polyclonal anti-dimethylated H3(K9) after pre-incubation with protein A agarose beads prior to incubation. Beads were then washed eight times and incubated for 15 min at 65°C with continuous shaking and spun down. The supernatant bound fraction was withdrawn. The bound and input fractions were incubated overnight at 65°C for reverse cross-linking. DNA was purified by phenol–chloroform extractions and two aliquots of 1 and 4 µl were used for amplification by PCR using the following conditions: 27–35 cycles of 4 min at 94°C, 2 min at 55°C, 2 min at 72°C then 30 s at 94°C, 30 s at 55°C, 30 s at 72°C in the presence of 10 µCi/µl of [α-32P]dCTP with the corresponding primers. PCR products were run on acrylamide gels and autoradiographs subjected to phosphoimaging (BAS-IIIs FUJI). Relative enrichment is expressed as the bound/input ratio calibrated against the ratio of the GAPDH positive control or β-globin positive control [with H3(K9)] in human or β-actin in mouse, which represents 100% value.
RT–PCR
Total RNA was isolated from cells or tissues with TriPure isolation reagent (Roche), treated with 250 ng DNase I (Promega) and converted to cDNA using the Moloney murine leukemia virus reverse transcriptase (Promega) and random hexanucleotide primers (Pharmacia) in a volume of 20 µl reaction mixture under conditions recommended by the manufacturer. cDNA was diluted (1:5) and 1 and 3 µl aliquots were used for PCR reactions (95°C for 1 min, 55°C for 1 min, 72°C for 1 min) in the presence of [α-32P]dCTP (Amersham) using appropriate primer pairs (sequences available upon request). These primers were designed to span intron–exon junctions to distinguish between cDNA and genomic DNA. RT–PCR fragments were separated on 7% polyacrylamide gels and exposed to autoradiography. The level of UBE3A transcription was assessed by its subjection to phosphoimager analysis normalizing for β-actin.
Quantitative real-time PCR
Real-time quantitative PCR (qPCR) was performed as previously described (39). Total RNA was extracted from human and mouse brains using High Pure RNA tissue kit (Roche) and treated with DNase I according to the manufacturer's protocol. RT–qPCR analysis was carried out using SYBR Green RT–PCR Master Mix (Applied Biosystems). Total RNA (10–30 ng) was added to a 20 µl reaction mixture with sequence-specific primers (300 nM). Sequences of primers will be provided upon request. qPCR assays were carried out in triplicates on an ABI Prism 7000 sequence detection system (Applied Biosystems). Thermocycling conditions were as follows: 95°C for 10 min followed by 40 cycles at 95°C for 15 s (denaturation) and 60°C for 60 s (annealing and extension). The threshold cycle number (Ct) was determined for all PCR reactions. The threshold was manually adjusted within the logarithmic curve above the background level and below the plateau phase. A comparative Ct method was used to calculate the relative gene number. The starting copy number of the unknown samples was determined in comparison with the known copy number of the calibrator sample using the following formula: ddCt=[dCt control gene (normal sample) −dCt target gene (normal sample)] – [dCt control gene (MeCP2 mutated sample) – dCt MeCP2 (MeCP2 mutated sample)]. The control genes were β-actin or dhfr. The relative gene copy number was calculated by the expression 2−ddCT.
Bisulfite method
DNA samples from normal human lymphoblasts and brain were prepared by standard phenol-chloroform extraction and were subjected to the bisulfite treatment as described (40). Following treatment, DNA was amplified using primer sets that encompass part of the CpG island present in exon 1 of UBE3A. Following treatment, DNA was subjected to PCR amplification. PCR products were further amplified in a semi-nested fashion. PCR products were then sequenced and analyzed for the methylation status of individual CpGs. PCR primers are available upon request.
Reaction conditions for the first PCR were five cycles at 95°C for 1 min, 53°C for 3 min, 72°C for 3 min followed by 35 cycles at 95°C for 45 s, 53°C for 1 min, 72°C for 1 min. Conditions for the semi-nested PCR were 40 cycles at 95°C for 45 s, 54°C for 1 min, 72°C for 1 min, followed by 7 min at 72°C.
ACKNOWLEDGEMENTS
We thank Dr Broria Ben Zeev and Dr Eva Gak for providing blood samples from a Rett syndrome patient. We also thank Karin Buiting and Bernhard Horsthemke for lymphoblasts of PWS patients. This work was supported by grants from the RS Research Foundation in the USA and in Israel, the Israel Science Foundation, the NIH and by the March of Dimes Birth Defects Foundation for inborn disease in the USA.
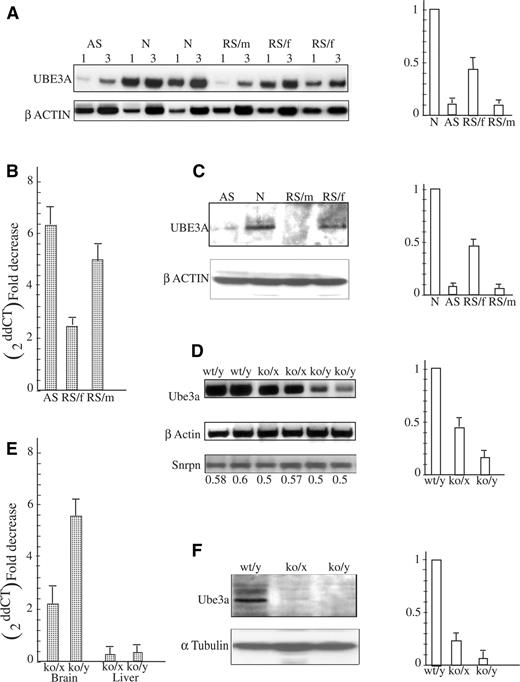
Figure 1. UBE3A expression on the transcriptional and translational level in the brain of RS patients and MeCP2 deficient mice. (A) Semi-quantitative RT–PCR results (a representative experiment). Aliquots of 1 and 3 µl cDNA were used to analyze UBE3A mRNA levels by RT–PCR (see Materials and Methods) in brains of AS patient (AS), normal individuals (N), RS male and female patients (RS/m and RS/f, respectively). UBE3A band intensities were normalized to β-actin. Right panel is a histogram of combined results of four different RT–PCR analyses. (B) The same brain RNA samples and M10 and M12 RNA (see Material and Methods) were analyzed by real-time PCR and expressed as fold decrease (2−ddCT) with respect to normal brain sample. (C) Protein extracts prepared from the brain of an AS patient (AS), normal individual (N), RS male patient (RS/m) and RS female patient (RS/f) were subjected to anti-UBE3A and anti-β-actin. Histograms on the right represent combined results of four different western blot experiments. The UBE3A band intensities were normalized to β-actin. (D) RT–PCR analysis was performed on mouse brain RNA samples of each normal (wt/y), heterozygote female (ko/x) and MeCP2-null (ko/y) mice. PCR products were analyzed as described in the Materials and Methods. Histogram on the right represents combined results of five different experiments performed with five different litters of newborns. (E) Results of real-time PCR analysis of the same samples are expressed as fold decrease (2−ddCT) with respect to normal brain or liver. (F) Brain extracts of wild-type (wt/y), heterozygote female (ko/x) and MeCP2-null (ko/y) mice were subjected to western blot analysis. Ube3a band intensities were normalized to α-tubulin. Histogram on the right represents combined results performed on three different mouse litters.
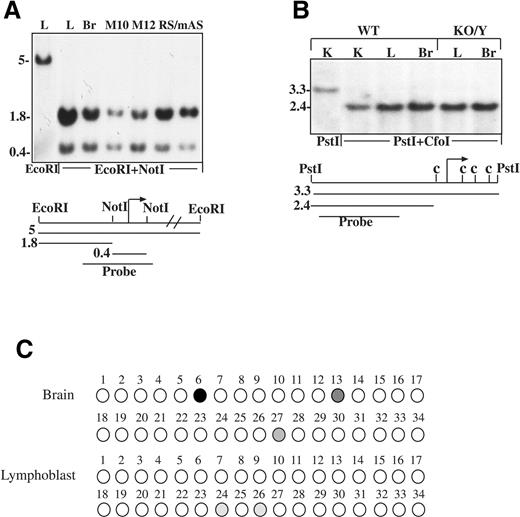
Figure 2. Methylation pattern of the mouse and human UBE3A promoters. (A) DNA was extracted from leukocytes (L), brain (Br), lymphoblast cell line that carries the mutation in MeCP2 on the inactive X (M10), lymphoblast cell line that carries the mutation in the MECP2 gene on the active X (M12), brain of an RS male patient (RS/m) and brain of an AS patient (AS). DNA was digested with EcoRI and EcoRI plus NotI, Southern blotted and probed with the radiolabeled 0.9 kb probe. (B) DNA samples isolated from kidney (K), liver (L) and brain (Br) of normal and KO/Y mice were digested with PstI and PstI plus CfoI, Southern blotted and hybridized with the 1.5 kb radiolabeled probe (c represents CfoI sites). (C) Methylation analysis of the UBE3A promoter in brain and lymphoblasts by the bisulfite method. Each circle represents a CpG site. Open circles, unmethylated; filled circles, methylated and gray circles, partially methylated.
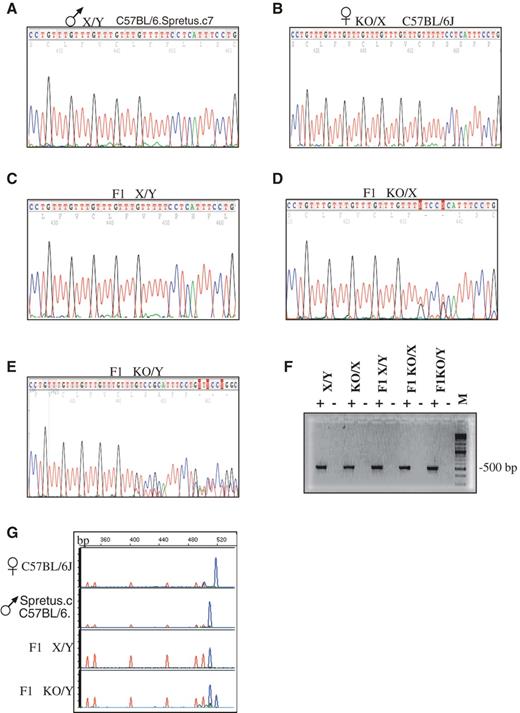
Figure 3. Biallelic expression of the Ube3a antisense gene. Total brain RNA was prepared from both C57BL/6J (J) and C57BL/6J mice congenic for the M. musculus spretus chromosome 7 (C57BL/6.spretus.c7) and from F1 offspring of a mating between a C57BL/6J KO/X female and a C57BL/6.spretus.c7 normal male. RT–PCR was performed using primers corresponding to Ube3a exons 5 and intron 6 (4,19) and the resulting products were sequenced. (A) The C57BL/6.spretus.c7 cDNA contains five GTTT repeats. (B) The C57BL/6J strain cDNA contains seven GTTT repeats. (C) The normal F1 (X/Y) apparently expresses only the paternal Ube3a antisense as judged by the presence of five GTTT repeats. (D) The F1 heterozygote female mouse (F1 KO/X) and (E) the F1 male (F1 KO/Y) MeCP2 deficient mouse show a mixed pattern following the five repeats which are common to both alleles. Forward primer: 5′CTGAGGGTCAGTTTACTCTG; reverse primer: 5′TCTCTCCATGATTGTGGCAG. (F) Minus RT control. (G) RT–PCR with 6FAM labeled primer. A cDNA containing seven GTTT repeats produces a PCR product of 517 bp, whereas a cDNA containing five GTTT repeats produces a product of 509 bp. 6FAM-label produces blue fluorescent products whereas the red peaks are ROM-labeled size markers.
![Figure 4. Changes in epigenetic modifications at the PWS/IC region due to MeCP2 deficiency. (A) MECP2 binding to mouse and human PWS-IC and other promoters like the human UBE3A and Profillin2 promoters. ChIP analysis was carried out using anti-MeCP2 antibody on brain and liver nucleosomes of WT, MeCP2 deficient (KO/Y) mice, M10 and M12 lymphoblast cell lines, nucleosomes of lymphoblasts of a PWS patient with a PWS-SRO deletion on the paternal allele (Mat) and of his father carrying the deletion on the maternal allele (Pat). Similar ChIP assays were performed at PWS/IC, UBE3A and Profillin2 promoters in brain samples of normal (N) and RS male patients (RS/m). Semi-quantitative PCR in the presence of [α-32P]dCTP (Amersham) on bound and input DNA using three different numbers of cell cycles. PCR products were run on 7% acrylamide gels, autoradiographed and subjected to phosphoimaging. Relative enrichment is expressed as the bound/input (B/In) ratio calibrated against the ratio of the positive controls of GAPDH and β-globin or mouse β-actin (see Materials and Methods). (B) Methylation pattern of the human SNRPN promoter. DNA samples were isolated from normal human brain (Br), human lymphoblast cell line M10, M12, brain of RS male patient (RS/m) and brain of AS patient (AS). DNA was digested with BglII (B) and BglII plus NotI (B+N) then hybridized to the 1 kb radiolabeled probe. (C) Methylation pattern of the mouse Snrpn promoter. Brain DNA from WT mouse, KO/Y mouse and a methyltransferase deficient mouse (Dnmt1−) were digested with PstI (P), PstI plus MspI (P+M) and PstI plus HpaII (P+H), (Southern blotted and probed with the radiolabeled probe. (D–H) Histone H3 modifications at the human PWS-IC and UBE3A promoter and at the mouse PWS-IC in MECP2 deficient cells and tissues. (D) The levels of histone H3 acetylation (Ac-H3), (E) H3 Lys4 methylation (Me-H3(K4), (F) histone H3 Lys 9 methylation (Me-H3(K9) in the PWS-IC and the UBE3A promoter were determined by ChIP analysis. Analyses were carried out for PWS-IC promoter on the ‘normal’ human lymphoblast cell line (M10) and the lymphoblast cell line deficient of MeCP2 (M12), brain of a normal individual (N) and brain of a RS male patient (RS) using antibodies against acetylated H3, dimethylated H3(K4) and dimethylated H3(K9). This was followed by semi-quantitative PCR on bound and input DNA fractions using three different numbers of cell cycles in the presence of [α-32P]dCTP. (G and H) Similar experiments were performed with WT mouse brain and MeCP2 deficient mouse brain (KO/Y). Relative enrichment is expressed as the bound/input (B/In) ratio calibrated against the ratio of the positive controls, GAPDH and β-globin and β-actin (see Materials and Methods).](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/hmg/14/8/10.1093/hmg/ddi097/2/m_ddi09704.jpeg?Expires=1716265191&Signature=pK-AC0JHDA6f1FqnwgigXlk-m8HEzyrg32ErOU4OmlmYV1evs1vqVFH66mY6fkaK49SXR9sLVr~~crQBQJp-cFPIVRjV1jJza9hMwgIRdlwmmGTkKTFmY4ipLweOTr8P2wVAEjzZXl1wU58FrBRw~7JOKTieViCKbM7uQpPsYuyqqOU9N12NHHN~5hSQjvILw5KNtuXzn2pCZIaaoLrVtE61h5d6TMv4zjCOgrpMtDO0e4VJXYXQsm7r7OAK-gk8HJEusFZc2lQwoDWwoCtXM9F0GgH7vwuwmRQYBNtV6hVUcdOEuh2PpKRyjiN1kYsCYbHL6esdbIp9I0zJsXXshw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Figure 4. Changes in epigenetic modifications at the PWS/IC region due to MeCP2 deficiency. (A) MECP2 binding to mouse and human PWS-IC and other promoters like the human UBE3A and Profillin2 promoters. ChIP analysis was carried out using anti-MeCP2 antibody on brain and liver nucleosomes of WT, MeCP2 deficient (KO/Y) mice, M10 and M12 lymphoblast cell lines, nucleosomes of lymphoblasts of a PWS patient with a PWS-SRO deletion on the paternal allele (Mat) and of his father carrying the deletion on the maternal allele (Pat). Similar ChIP assays were performed at PWS/IC, UBE3A and Profillin2 promoters in brain samples of normal (N) and RS male patients (RS/m). Semi-quantitative PCR in the presence of [α-32P]dCTP (Amersham) on bound and input DNA using three different numbers of cell cycles. PCR products were run on 7% acrylamide gels, autoradiographed and subjected to phosphoimaging. Relative enrichment is expressed as the bound/input (B/In) ratio calibrated against the ratio of the positive controls of GAPDH and β-globin or mouse β-actin (see Materials and Methods). (B) Methylation pattern of the human SNRPN promoter. DNA samples were isolated from normal human brain (Br), human lymphoblast cell line M10, M12, brain of RS male patient (RS/m) and brain of AS patient (AS). DNA was digested with BglII (B) and BglII plus NotI (B+N) then hybridized to the 1 kb radiolabeled probe. (C) Methylation pattern of the mouse Snrpn promoter. Brain DNA from WT mouse, KO/Y mouse and a methyltransferase deficient mouse (Dnmt1−) were digested with PstI (P), PstI plus MspI (P+M) and PstI plus HpaII (P+H), (Southern blotted and probed with the radiolabeled probe. (D–H) Histone H3 modifications at the human PWS-IC and UBE3A promoter and at the mouse PWS-IC in MECP2 deficient cells and tissues. (D) The levels of histone H3 acetylation (Ac-H3), (E) H3 Lys4 methylation (Me-H3(K4), (F) histone H3 Lys 9 methylation (Me-H3(K9) in the PWS-IC and the UBE3A promoter were determined by ChIP analysis. Analyses were carried out for PWS-IC promoter on the ‘normal’ human lymphoblast cell line (M10) and the lymphoblast cell line deficient of MeCP2 (M12), brain of a normal individual (N) and brain of a RS male patient (RS) using antibodies against acetylated H3, dimethylated H3(K4) and dimethylated H3(K9). This was followed by semi-quantitative PCR on bound and input DNA fractions using three different numbers of cell cycles in the presence of [α-32P]dCTP. (G and H) Similar experiments were performed with WT mouse brain and MeCP2 deficient mouse brain (KO/Y). Relative enrichment is expressed as the bound/input (B/In) ratio calibrated against the ratio of the positive controls, GAPDH and β-globin and β-actin (see Materials and Methods).
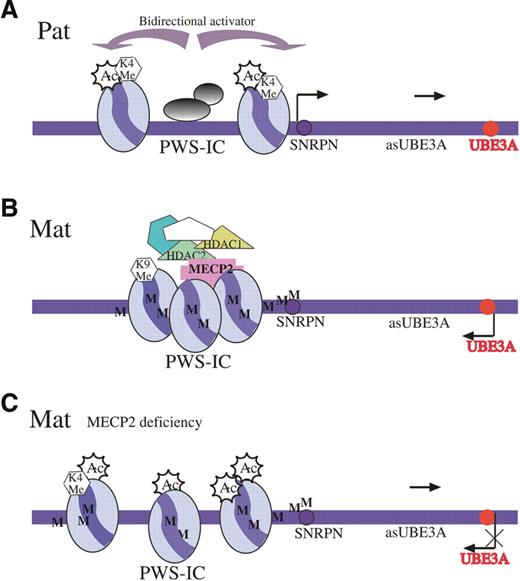
Figure 5. (A) The PWS-IC locus on the paternal allele (Pat) is unmethylated and in an open chromatin configuration. This chromatin status spreads bi-directionally to all imprinted genes on the paternal allele in the entire PWS/AS domain, including the UBE3A antisense (asUBE3A) gene. This antisense RNA is believed to suppress UBE3A gene expression. (B) PWS-IC on the maternal allele (Mat) is methylated (M) and binds to MeCP2 and recruits HDACs 1 and 2 that deacetylate histone H3 and histone methyltransferase to methylate H3(K9), causing heterochromatinization and silencing of all paternally expressed genes, including UBE3Aas on the maternal allele. (C) In the absence of MeCP2 (MeCP2 deficiency), PWS-IC on the maternal allele remains methylated (M), but a repressory complex cannot be established. Therefore, no heterochromatinization occurs, histone H3 is acetylated (Ac) and H3(K4) is methylated (K4 Me). The open chromatin structure is sufficient to direct maternal transcription of the UBE3A antisense leading to repression of the maternal UBE3A gene.
References
Albrecht, U., Sutcliffe, J.S., Cattanach, B.M., Beechey, C.V., Armstrong, D., Eichele, G. and Beaudet, A.L. (
Rougeulle, C., Glatt, H. and Lalande, M. (
Rougeulle, C., Cardoso, C., FontÈs, M., Colleaux, L. and Lalande, M. (
Chamberlain, S.J. and Brannan, C.I. (
Ohta, T., Gray, A., Rogan, P.K., Buiting, K., Gabriel, J.M., Saitoh, S., Muralidhar, B., Bilienska, B., Krajewska-Walasek, M., Driscoll, D.J. et al. (
Buiting, K., Lich, C., Cottrell, S., Barnicoat, A. and Horsthemke, B. (
Saitoh, S. and Wada, T. (
Fulmer-Smentek, S.B. and Francke, U. (
Perk, J., Makedonski, K., Lande, L., Cedar, H., Razin, A. and Shemer, R. (
Clayton-Smith, J. and Laan, L. (
Neul, J.L. and Zoghbi, H.Y. (
Watson, P., Black, G., Ramsden, S., Barrow, M., Super, M., Kerr, B. and Clayton-Smith, J. (
Imessaoudene, B., Bonnefont, J.P., Royer, G., Cormier-Daire, V., Lyonnet, S., Lyon, G., Munnich, A. and Amiel, J. (
Hitchins, M.P., Rickard, S., Dhalla, F., de Vries, B.B., Winter, R., Pembrey, M.E. and Malcolm, S. (
Chen, R.Z., Akbarian, S., Tudor, M. and Jaenisch, R. (
Samaco, R.C., Hogart, A. and Lasalle, J.M. (
Lossie, A.C., Whitney, M.M., Amidon, D., Dong, H.J., Chen, P., Theriaque, D., Hutson, A., Nicholls, R.D., Zori, R.T., Williams, C.A. et al. (
Yamasaki, K., Joh, K., Ohta, T., Masuzaki, H., Ishimaru, T., Mukai, T., Niikawa, N., Ogawa, M., Wagstaff, J. and Kishino, T. (
Landers, M., Bancescu, D.L., Le Meur, E., Rougeulle, C., Glatt-Deeley, H., Brannan, C., Muscatelli, F. and Lalande, M. (
Glenn, C.C., Saitoh, S., Jong, M.T., Filbrandt, M.M., Surti, U., Driscoll, D.J. and Nicholls, R.D. (
Shemer, R., Birger, Y., Riggs, A.D. and Razin, A. (
Nan, X., Ng, H.-H., Johnson, C.A., Laherty, C.D., Turner, B.M., Eisenman, R.N. and Bird, A. (
Lehnertz, B., Ueda, Y., Derijck, A.A., Braunschweig, U., Perez-Burgos, L., Kubicek, S., Chen, T., Li, E., Jenuwein, T. and Peters, A.H. (
Gregory, R.I., Randall, T.E., Johnson, C.A., Khosla, S., Hatada, I., O'Neill, L.P., Turner, B.M. and Feil, R. (
Fournier, C., Goto, Y., Ballestar, E., Delaval, K., Hever, A.M., Esteller, M. and Feil, R. (
Balmer, D., Arredondo, J., Samaco, R.C. and LaSalle, J.M. (
Stancheva, I., Collins, A.L., Van den Veyver, I.B., Zoghbi, H. and Meehan, R.R. (
Chen, W.G., Chang, Q., Lin, Y., Meissner, A., West, A.E., Griffith, E.C., Jaenisch, R. and Greenberg, M.E. (
Martinowich, K., Hattori, D., Wu, H., Fouse, S., He, F., Hu, Y., Fan, G. and Sun, Y.E. (
Runte, M., Kroisel, P.M., Gillessen-Kaesbach, G., Varon, R., Horn, D., Cohen, M.Y., Wagstaff, J., Horsthemke, B. and Buiting, K. (
Ohta, T., Buiting, K., Kokkonen, H., McCandless, S., Heeger, S., Driscoll, D.J., Cassidy, S.B., Horsthemke, B. and Nicholls, R.D. (
Jong, M.T., Gray, T.A., Ji, Y., Glenn, C.C., Saitoh, S., Driscoll, D.J. and Nicholls, R.D. (
Lau, J.C., Hanel, M.L. and Wevrick, R. (
Traynor, J., Agarwal, P., Lazzeroni, L. and Francke, U. (
Tudor, M., Akbarian, S., Chen, R.Z. and Jaenisch, R. (
Guy, J., Hendrich, B., Holmes, M., Martin, J.E. and Bird, A. (
Simon, I., Barnett, J., Hannett, N., Harbison, C.T., Rinaldi, N.J., Volkert, T.L., Wyrick, J.J., Zeitlinger, J., Gifford, D.K., Jaakkola, T.S. et al. (
Ariani, F., Mari, F., Pescucci, C., Longo, I., Bruttini, M., Meloni, I., Hayek, G., Rocchi, R., Zappella, M. and Renieri, A. (

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}