




Abstract
Myotonic dystrophy type 1 (DM1) is a debilitating multisystemic disorder caused by a CTG repeat expansion in the DMPK gene. Aberrant splicing of several genes has been reported to contribute to some symptoms of DM1, but the cause of muscle weakness in DM1 and elevated Ca2+ concentrations in cultured DM muscle cells is unknown. Here, we investigated the alternative splicing of mRNAs of two major proteins of the sarcoplasmic reticulum, the ryanodine receptor 1 (RyR1) and sarcoplasmic/endoplasmic reticulum Ca2+-ATPase (SERCA) 1 or 2. The fetal variants, ASI(−) of RyR1 which lacks residue 3481–3485, and SERCA1b which differs at the C-terminal were significantly increased in skeletal muscles from DM1 patients and the transgenic mouse model of DM1 (HSALR). In addition, a novel variant of SERCA2 was significantly decreased in DM1 patients. The total amount of mRNA for RyR1, SERCA1 and SERCA2 in DM1 and the expression levels of their proteins in HSALR mice were not significantly different. However, heterologous expression of ASI(−) in cultured cells showed decreased affinity for [3H]ryanodine but similar Ca2+ dependency, and decreased channel activity in single-channel recording when compared with wild-type (WT) RyR1. In support of this, RyR1-knockout myotubes expressing ASI(−) exhibited a decreased incidence of Ca2+ oscillations during caffeine exposure compared with that observed for myotubes expressing WT-RyR1. We suggest that aberrant splicing of RyR1 and SERCA1 mRNAs might contribute to impaired Ca2+ homeostasis in DM1 muscle.
INTRODUCTION
Myotonic dystrophy type 1 (DM1), the most common type of muscular dystrophy in adults, is a dominantly inherited multisystemic disorder caused by unstable expansion of CTG trinucleotide repeats in the myotonic dystrophy protein kinase (DMPK) gene (1). The CTG repeats are polymorphic and can expand to any number between 50 and several thousand in DM1, compared to the normal number of between 5 and 35. The clinical expression of DM1 includes myotonia, progressive muscle weakness, cataracts, insulin resistance and cardiac conduction defects. Subsequently, myotonic dystrophy type 2 has been demonstrated to be caused by a CCTG repeat expansion located in intron 1 of the zinc finger protein 9 gene (2).
In DM, disrupted mRNA splicing has been reported in several genes. These include cardiac troponin T (cTNT), insulin receptor (IR), muscle-specific chloride channel (ClC-1) and myotubularin-related protein 1 (MTMR1) (3–6) in skeletal muscle and microtubule-associated tau, NMDA receptor 1 and amyloid precursor protein in the brain (7,8). In all cases, the developmental regulation of splicing is disrupted, resulting in preferential expression of fetal isoforms of the proteins. In addition, we previously showed significant induction of mRNA for a small-conductance Ca2+-activated K+ channel (SK3) in DM1 muscle, the expression of which is repressed in adult muscle during normal differentiation (9). A recent report suggests that CUG RNA expansions affect the distribution of transcription factors and may consequently disrupt regulation of gene expression (10). Thus, mutant DM transcripts could alter developmental regulation of both splicing and transcription.
Among the symptoms of DM, myotonia and insulin resistance are attributed to the disruption of the ClC-1 and IR alternative splicing, respectively (4,5). However, the cause of the progressive muscle wasting, which is the most disabling symptom, is less well defined. An elevated concentration of Ca2+ has been shown in cultured DM muscle cells and is suggested to be a possible cause of muscle degeneration (11). The regulation of intracellular Ca2+ homeostasis in skeletal muscle cells depends mainly on two sarcoplasmic reticulum (SR) proteins: ryanodine receptor 1 (RyR1) and sarcoplasmic/endoplasmic reticulum Ca2+-ATPase (SERCA). RyR1 releases Ca2+ from the SR, in response to plasma membrane depolarization via a mechanical interaction with voltage-gated calcium channels [excitation–contraction (EC) coupling]. SERCA transport of Ca2+ across the SR/ER membrane is coupled to ATP hydrolysis (12). As a consequence of its activity, the resting cytosolic free Ca2+ is maintained three to four orders of magnitude lower than the intra-SR/ER Ca2+ concentration. During a normal skeletal muscle contraction/relaxation cycle, Ca2+ is released from SR into the cytoplasm through RyR1, inducing muscle contraction. Ca2+ is subsequently pumped back into the lumen of SR by SERCA to allow relaxation.
There are three genes encoding RyR in mammals. RyR1 and RyR2 are expressed in skeletal muscle and heart muscle, respectively. Although RyR3 is not predominant, it is detected in immature skeletal muscles and decreases markedly at later stages of development (13). The expression of RyR1 splice variants is regulated both developmentally and in a tissue-specific manner (14). SERCAs are also encoded by three homologous genes: SERCA1, SERCA2 and SERCA3 (15). Transcripts from these genes undergo alternative splicing in a developmentally regulated and tissue-specific manner, giving rise to isoforms that differ in their C-terminal region (12). SERCA1a (adult form) and 1b (neonatal form) are mainly expressed in fast-twitch (type 2) skeletal muscle (16) and SERCA2a is expressed in slow-twitch (type 1) skeletal and cardiac muscles, whereas SERCA2b expression is widespread (17). SERCA3 is expressed in several non-muscle tissues at variable levels, co-localizing with SERCA2b (18).
As splicing and transcription of these genes are regulated developmentally, it is possible that their splicing and/or expression might be impaired in DM1 muscle and thus contribute to altered calcium homeostasis in skeletal muscle of DM1 patients. Therefore, in this study, we examined the splicing patterns and the expression levels of RyR1 and SERCAs in muscles of DM. The function of the RyR1 splice variant, which we found increased in DM muscle, was further investigated using both heterologous and homologous expression systems.
RESULTS
Splicing of RyR1
To investigate splicing isoforms, RT–PCR analysis of spliced transcripts was performed on skeletal muscles from humans with DM1 and without DM1 [non-DM: normal control, amyotrophic lateral sclerosis (ALS) and polymyositis (PM)]. Each primer set generated fragments of different length, according to the inclusion and the exclusion of each exon (Fig. 1). Futatsugi et al. (14) described several splice variants of the skeletal muscle RyR1. The variant [ASI(−)], which lacks ASI (exon 70, residues 3481–3485) constitutes 100% of RyR1 expressed in mouse embryonic skeletal muscle and 30% of those in adult muscle. Consistent with these results, we found that the ASI(+) isoform was dominantly expressed in normally developed human muscles and ASI(−) predominated in cultured myotubes (Fig. 2A). In contrast, ASI(−) was significantly increased in DM1 patients when compared with control (P=0.003) and PM (P=0.04). The degree of aberrant splicing in DM1 was variable. Some showed a splicing pattern indistinguishable from that of control, whereas others showed 100% ASI(−). The increase in ASI(−) was further investigated using transgenic mice, HSALR, in which expanded CUG repeat expression in skeletal muscle leads to a DM-like phenotype (19). The expression of ASI(−) was significantly higher in HSALR mice when compared with wild-type (WT) mice (P=0.03) (Fig. 2A), showing the expression of mRNA with expanded CUG repeats was sufficient to cause aberrant splicing of RyR1. It should be noted that the degree of aberrant splicing among HSALR mice was less variable than DM1 patients, indicating the homogeneity of samples from the mice.
The α-subunit of the IR has two spliced isoforms with different insulin sensitivity, IR-A and IR-B. It was previously reported that the muscles in DM patients predominantly expressed IR-A, which is usually expressed in immature myotubes and other tissues with low insulin sensitivity (4). To investigate a possible common mechanism of aberrant splicing, we compared the extent of abnormal splicing of ryanodine receptors with that of IRs (Fig. 2B). The result showed significant correlation between the degree of abnormal splicing in RyR1 and that of the IR (Pearson's correlation coefficient R=0.694, P=0.0374), suggesting that a common mechanism might contribute to the splicing abnormalities.
Another variant, ASII(−), which lacks ASII (exon 83, residues 3865–3870), is transiently expressed in skeletal muscle after birth and constitutes ∼10% of RyR1 in adults. According to the previous study using mice, the splicing change in ASII [at around postnatal day (P) 8] takes place earlier than that in ASI (around P16) (14). We found that ASII(+) was dominantly expressed even in human myotubes (Fig. 2C). Therefore, it is likely that the transition of ASII splicing pattern from ASII(−) to ASII(+) in human also takes place earlier than that of ASI. We did not find any change in ASII(−) expression in muscle among these groups of humans, nor between HSALR and WT mice (Fig. 2C). These results indicate that the ASII splice switch which occurs during a more developmentally immature state is not affected in DM1.
Splicing of SERCA1 isoforms
The mammalian SERCA1 has two major splice variants; SERCA1a (adult form) and SERCA1b (neonatal form) (Fig. 1). The difference between these isoforms results from the alternative splicing of exon 22 and consequently affects seven amino acids located at the C-terminal. Recently, Chami et al. (20) described another splice variant (S1T), characterized by exon 4 and/or exon 11 skipping (20). S1T transcripts were expressed in several tissues such as liver, lung and pancreas, but not in muscle. Because exon 11 skipping leads to frameshift, which results in premature stop codon in exon 12, S1T transcripts do not encode functional pumps.
We used a set of primers that co-amplify SERCA1 cDNAs with or without exon 4, exon 11 and exon 22 splicing. SERCA1b (−exon 22) was exclusively expressed with other minor alternatively spliced variants in several DM1 muscles, whereas only SERCA1a (+exon 22) was found in non-DM muscles (Fig. 3A). The proportion of SERCA1b was significantly higher in DM1 muscles than in control (P=0.005), ALS (P=0.002) and PM (P=0.01). Moreover, the relative amount of SERCA1b was higher in myotubes from DM1 patients when compared with those from normal control (Fig. 3A). In contrast, the S1T isoform was not detected in either DM1 or non-DM muscle (data not shown).
We then analyzed SERCA1 variants in skeletal muscles and myotubes in HSALR mice. As with DM1 patients (Fig. 3A), SERCA1b was exclusively expressed in muscles and myotubes of HSALR mice, except that the degree of splicing is less variable in HSALR than in DM1 patients (Fig. 3B).
Splicing of SERCA2 isoforms
It has been reported that the mammalian SERCA2 has three splice variants; SERCA2a, SERCA2b and SERCA2c, which differ in their C-terminal region as is the case in SERCA1 isoforms (Fig. 1). Although SERCA1 splicing is developmentally regulated, splicing of SERCA2 is regulated in a tissue-specific manner. SERCA2a is the major isoform in cardiac muscle and type 1 skeletal muscle. SERCA2b is expressed in smooth muscle and many other tissues as a house-keeping gene, although it has three mRNA subclasses which differ only in their 3′-untranslated region (3′-UTR) and are regulated in a tissue-dependent manner. Recently, Gelebart et al. (21) identified a variant of SERCA2a isoform containing exon 21 (SERCA2c) in human monocytes.
To analyze human SERCA2a splice variants in skeletal muscles from DM1, we performed RT–PCR using a set of primers to the flanking exon 19 and exon 21. However, neither SERCA2c nor other new splice variants were detected in either DM1 or non-DM muscles or in either HSALR or WT mouse muscles.
Then SERCA2b splice variants were investigated using a set of primers on exons 19 and 20. No splice variants were identified in HSALR or WT mice. However, we detected a novel splice variant in human skeletal muscles (Fig. 4), denoted SERCA2d. Sequence analysis revealed that SERCA2d contains the full-length of intron 19. Intron 19 consists of 82 bp and the sequence including intron 19 and its flanking exons have been found in the EST database (sequence number BG011840). Interestingly, the expression of SERCA2d was significantly lower in DM1 muscles (P<0.05).
Expression of RyR1, SERCA1 and SERCA2 mRNA and protein
During normal skeletal muscle development, the expression of RyR1, SERCA1 and SERCA2a are increased, but RyR3 is reduced (13,22). We reported increases in mRNA and protein of apamin-sensitive small-conductance Ca2+-activated K+ channel (SK3), which is normally expressed in immature skeletal muscle, in DM1 muscle (9,23). Moreover, SERCA2 mRNA stability depends on its 3′-UTR (24). To test the possibility that aberrantly spliced isoforms affect total mRNA levels or alter their own stability, we analyzed the amount of each mRNA in muscles using semi-quantitative RT–PCR. There was no significant difference in the expression levels of mRNA for RyR1, RyR3, SERCA1, SERCA2a and total SERCA2 between DM and non-DM muscle (Fig. 5A–E). However, total RyR3 transcript levels in ALS were significantly increased when compared with control (P=0.009). This increase in ALS is probably due to denervation, because denervation is known to induce several mRNAs that are normally expressed in immature skeletal muscle. In addition, the amount of mRNA for RyR1, RyR3, SERCA1 and SERCA2 was not significantly different in skeletal muscle of HSALR and WT mice (Fig. 5F).
We also used immunoblot analysis to determine the quantity of RyR1, SERCA1 and SERCA2 protein expression in mouse skeletal muscle SR. With regard to SERCA protein expression, previous studies were inconclusive; one suggested decrease in total SERCA protein in cultured DM muscles (25) and another showed increase in SERCA1 expression in DM skeletal muscles (26). We found a trend toward increased levels of SERCA1 and SERCA2 proteins in the SR fraction of HSALR muscle, but not of RyR1 (n=2, Fig. 6). Clearly, SERCA1 protein levels were not decreased, suggesting that functional differences caused by aberrant splicing of RyR1, SERCA1 and SERCA2 alone may contribute to Ca2+ dysregulation in DM.
Functional studies of ASI(+) and ASI(−)
To examine the functional difference between the ASI(+) and the ASI(−) RyR1 isoforms, we characterized [3H]ryanodine binding to microsomal vesicles of HEK293T cells transfected with RyR1 ASI(+) and ASI(−). [3H]ryanodine binding is a standard technique for assessing the open probability of RyR channels, because ryanodine binds solely to open channels and the binding is proportional to open probability (27,28). The binding of [3H]ryanodine to microsomal vesicles of transfected cells was significantly greater than that of control vesicles from non-transfected cells. Scatchard analysis showed that the affinity of [3H]ryanodine binding (Kd) to ASI(+) was higher than that to ASI(−) (P=0.0284, Fig. 7A–C and Table 1). Maximum ryanodine binding (Bmax), which reflects the RyR expression level, was not significantly different (Table 1). Immunoblot analysis also indicated similar expression levels (data not shown). These data indicate that the channel activity of ASI(−) RyR1 is less than that of ASI(+), i.e. wt-RyR1. ASI(+) and ASI(−) showed indistinguishable biphasic Ca2+ dependence of [3H]ryanodine binding (Fig. 7D).
We then determined the open probability in single-channel measurements with the planar lipid bilayer method. Figure 7E shows representative current traces of ASI(+) and ASI(−). In accordance with the results obtained from [3H]ryanodine binding, ASI(−) was less stably open than ASI(+). Open probability (Po) was significantly decreased (P=0.0005) and mean open time (To) was significantly shorter (P=0.02) in ASI(−) (n=12) than in ASI(+) (n=14) (Table 1).
We next tested whether the reduced activity of ASI(−) observed in [3H]ryanodine binding and planar lipid bilayers experiments results in observable changes in SR Ca2+ release channel function in intact muscle cells. We have previously shown that expression of full-length rabbit RyR1 [i.e. ASI(+)] in dyspedic myotubes results in restoration of bi-directional EC coupling, caffeine-induced Ca2+ release and an elevation in resting Ca2+ (9). In addition, when compared with normal myotubes, ASI(+)-expressing dyspedic myotubes exhibit an increased tendency for Ca2+ oscillations during application of 10 mm caffeine [76% in the study of O'Connell et al. (30)]. Because the ASI(−) isoform is uniformly expressed in developmentally immature normal myotubes (Fig. 2A), these results could reflect inherent differences in ASI(+)and ASI(−). Therefore, we characterized caffeine-induced Ca2+ release in ASI(+)- and ASI(−)-expressing dyspedic myotubes (Fig. 8). For these experiments, ASI(+) and ASI(−) were cloned into pCIneo and injected at the same concentration (0.5 µg/µl) in myotubes derived from RyR1-knockout (dyspedic) mice. Figure 8A shows representative indo-1 fluorescence recordings in which myotubes expressing either ASI(+) or ASI(−) were challenged with sequential additions of 10 and 30 mm caffeine. Average resting indo-1 fluorescence was slightly lower in ASI(−)-expressing myotubes (Fig. 8B), although this difference did not quite meet statistical significance (P=0.07). Peak elevations in intracellular Ca2+ were similar in ASI(+)- and ASI(−)-expressing myotubes following application of either 10 mm (Fig. 8C) or 30 mm caffeine (Fig. 8D). However, in spite of identical peak Ca2+ responses, ASI(+)-expressing myotubes exhibited a significantly higher incidence of Ca2+ oscillations during application of 10 mm caffeine (Fig. 8E). The significantly higher incidence of caffeine-induced Ca2+ oscillations and tendency for higher resting Ca2+ in ASI(+)-expressing myotubes are consistent with the increased activity of this splice variant observed in biochemical and lipid bilayers experiments (Fig. 7).
DISCUSSION
We show for the first time that alternative splicing of RyR1 and SERCA1 is dysregulated in skeletal muscle of DM1 patients and the DM mouse model (HSALR). As with cTNT, IR and ClC-1, the increase in immature isoforms of RyR1 and SERCA1 indicates dysregulation of a developmental splicing switch. We also identified a novel splicing variant of SERCA2b and its decreased expression in DM1. The total amount of mRNA for RyR1, RyR3, SERCA1, SERCA2a and total SERCA2 in DM1, and the expression levels of their proteins in HSALR mice were not significantly different. Moreover, we found that the in vitro and in situ activity of RyR1 ASI(−) was reduced compared with that of RyR1 ASI(+).
It is possible that these juvenile splicing variants are expressed in DM1 as a consequence of regeneration, as regenerating muscle typically expresses juvenile isoforms characteristic of undifferentiated myotubes. However, this possibility is unlikely because the severe degeneration and regeneration in PM is not correlated with splice variants of the RyR.
We found the abnormal splicing of only ASI exon in DM1 muscle, although the splicing of two exons in RyR1, ASI and ASII, is under developmental regulation. The intronic sequences or molecular machineries responsible for the splicing of these exons have not been revealed. However, it seems that ASI and ASII splicing is regulated by a different mechanism, because the switching of splice isoforms occurs at different developmental stages in normal muscle development. Because the switching of ASI splicing takes place later than that of ASII, our result suggest that the aberrant splicing in DM1 might arise from the dysfunction of certain splicing mechanisms essential for differentiation of skeletal muscle rather than the global derangement of nuclear environment.
Recently, substantial evidence has accumulated supporting the hypothesis that RNA containing expanded repeats account for splicing abnormalities common to both DM1 and DM2 (RNA gain of function) (31). The overexpression of CUG-BP and/or depletion of Mbnl1 might cause specific changes in alternative splicing. It has been reported that increased levels of CUG-BP promotes aberrant splicing of several mRNAs including IR (4). Indeed, we found that the degree of aberrant splicing of RyR1 was correlated with altered splicing of IR (Fig. 2B). In contrast, Fardaei et al. reported that the sequestration of Mbnl1 in the CUG and CCUG ribonuclear foci results in depletion of Mbnl1 (32). The recently developed Mbnl1-knockout mouse demonstrated myotonia, cataracts and also RNA-splicing abnormalities characteristic of DM (33). In fact, downstream from the SERCA1 splicing exon 22, there are several ‘YGCU(U/G)Y motifs’ that could potentially serve as Mbnl1 binding motifs(31). The mRNA analysis of Mbnl1-knockout mouse and transfection study with RyR1 and SERCA minigenes would clarify these two possibilities.
In addition, other nuclear RNA binding proteins could be sequestrated in RNA foci, because nuclear mRNAs exist as multimeric ribonucleoprotein (RNP) complexes rather than naked polynucleotides (8). However, Jiang et al. (8) reported that the overall distribution of hnRNP H or F did not show obvious changes in DM. Accordingly, we did not find any splicing abnormality of β-tropomyosin (data not shown), which is developmentally regulated via proteins including hnRNP H, I and A1 (34), suggesting that alterations in these RNP proteins are not directly related to aberrant splicing in DM.
Mutations of RyR1 have been linked to four genetic disorders: malignant hyperthermia (MH), central core disease (CCD), multiminicore disease and nemaline rod myopathy. The mutations are essentially clustered in three regions: the N-terminal region, a central region and a C-terminal region (35). Although ASI is not located in either of these regions, a homozygous mutation of CCD has been recently identified at an exon (P3527S, exon71) adjacent to that of ASI (exon 70) (36). Functional studies of RyR1 disease mutations support two distinct cellular mechanisms for muscle weakness in CCD. The ‘leaky channel hypothesis’ proposes that mutations in the RyR1 protein generate SR Ca2+ release channels that are excessively leaky to Ca2+ and thus result in a depleted Ca2+ reservoir available for release during muscle activation. Alternatively, the ‘EC-uncoupling hypothesis’ proposes that CCD mutations in the pore region of RyR1 result in a deficit in EC coupling, i.e. reduced Ca2+ release in the presence of normal SR Ca2+ content (35,37). In theory, aberrant splicing of RyR1 could contribute to muscle weakness in DM via either of these two mechanisms. However, it is unlikely that the ASI(−) variant results in significant SR Ca2+ leak because maximal caffeine-induced Ca2+ release is identical in ASI(+) and ASI(−) expressing dyspedic myotubes (Fig. 8). In addition, the leaky channel mutants exhibit impaired Ca2+ inhibition by high Ca2+ (35), whereas the Ca2+ dependence of inactivation is similar for ASI(−) and wt-RyR1 (Fig. 7D). Finally, although most of the leaky channel mutations in RyR1 predispose individuals to MH, whereas no link has been demonstrated between DM and MH (38). In addition, because EC coupling is similar for normal myotubes which express ASI(−) and dyspedic myotubes expressing RyR1 [i.e. ASI(+)], it seems unlikely that the ASI(−) splice variant results in EC uncoupling like that observed for CCD mutations in the pore of RyR1. However, a more definitive conclusion will require future work to carefully compare the efficacy of EC coupling in ASI(+) and ASI(−) expressing myotubes.
Mutations in SERCA1 are responsible for Brody disease, an inherited disorder characterized by painless muscle cramps and impaired muscle relaxation. Sequencing of the SERCA1 gene from Brody disease patients has revealed a number of frameshift mutations that truncate SERCA1, as well as a missense mutation, all of which lead to loss of SERCA1a function (39). The difference between SERCA1a and SERCA1b is seven amino acids at the C-terminal. Heterologous expression in COS-1 cells failed to reveal a functional difference between these two isoforms (40). Nevertheless, it is possible that this sequence difference provides binding sites or affects the binding to muscle-specific factors that influence ATPase activity and are not present in COS-1 cells. For instance, sarcolipin (SLN), which regulates SERCA1 activity, is not expressed in COS-1 cells (41). Thus, difference in C-terminal splicing may alter the interaction between SERCA1 and SLN or other muscle-specific proteins.
We identified a novel splice variant of SERCA2 (SERCA2d) in skeletal muscle and found that the proportion of SERCA2d was reduced in DM1. SERCA2d contains intron 19 and its retention leads to a frameshift encoding 27 amino acids (VSGWVGLGTSHLLPGEAGGVTRLPCVS) in the C-terminal that is followed by a premature stop codon in exon 20. Several studies have suggested that the C-terminal region of SERCA2 is a critical determinant of its functional properties. The SERCA2a and SERCA2b splice variants also differ in their C-terminal. SERCA2b exhibits a 2-fold higher apparent affinity for Ca2+, but at the same time a 2-fold lower catalytic turnover rate (18,42,43). Chimeric analysis revealed that SERCA2b acquired the functional properties of SERCA2a when the last 12 amino acids were removed (44). Future experiments will be required to determine whether the unique C-terminal tail of SERCA2d results in a unique affinity for Ca2+ and corresponding differences in catalytic turnover rate. Nevertheless, because SERCA2d is ubiquitously expressed, an altered function of this splice variant could contribute to a dysregulation of SR/ER Ca2+ reuptake in both muscle and non-muscle tissues.
An increase in resting Ca2+ concentration has been reported in cultured DM myotubes (11), but the Ca2+ concentration in intact muscle fibers of DM has not been investigated. The functional changes in the RyR1 ASI(−) isoform found in this study did not result in an increase in cytosolic Ca2+ concentration when compared with WT-RyR1 following expression in dyspedic myotubes. Indeed, there was a trend towards decreased resting Ca2+ concentrations. It will thus be important for future work to clarify the functional consequences of the co-existence of juvenile variants of both RyR1 and SERCA in muscle (e.g. HSALR muscle). Furthermore, as many other developmentally regulated proteins including the fetal isoform of myosin heavy chain and cardiac troponin are not increased in DM1 muscle fibers (4), the interaction between these two juvenile variants and adult isoforms of other skeletal muscle proteins may also be disturbed.
Although HSALR mice exhibit a consistent muscle histopathology, with increases in central nuclei and ring fibers and variability in fiber size, severe muscle weakness is not observed in these animals up to the age of 6 months (19). This illustrates that these splicing differences cannot themselves explain muscle wasting. However, the possibility that the mice might exhibit muscle wasting with aging or hormonal changes was not excluded because older mice were not examined. Thus, the development of severe muscle weakness may require other factors such as aging, hormonal changes or others yet to be identified splicing abnormalities. In fact, younger DM1 patients often exhibit a similar muscle histology in the absence of weakness and severe degeneration/regeneration which can develop later (45).
In conclusion, our work characterizes aberrant splicing of RyR1, SERCA1 and SERCA2 in DM1. Ryanodine binding, single-channel recordings and functional measurements following homologous expression in dyspedic myotubes all indicate that the juvenile RyR1 splice variant exhibits reduced activity. As RyR1 and SERCA contribute to intracellular Ca2+ homeostasis in muscle, aberrant splicing of these transcripts could contribute to altered intracellular Ca2+ homeostasis and muscle degeneration in DM. Further investigations of the functional properties of these juvenile splice variants separately and in combination are needed in order to further elucidate the pathogenesis of muscle weakness and wasting in DM.
MATERIALS AND METHODS
RNA preparation and cDNA synthesis
Human muscle tissues were obtained from 26 biopsy specimens; 10 from DM1, six from normal control, five from ALS and five from PM. All muscle specimens were frozen immediately after biopsy in a dry ice isopentane bath and stored at −80°C until analysis. Myotubes were cultured from two muscle specimens; one from DM1 and one from normal control. Primary myoblast cultures were established as described previously (46). The differentiation to myotubes was induced by reducing the concentration of fetal bovine serum to 5%. Informed consent for use of specimens for research purposes was obtained from each patient with the approval of institutional ethical committee. We also employed skeletal muscle of hind limb from five HSALR and four WT mice. The experiments using mice were performed in accordance with the protocol complying with the Guideline for the Care and Use of Laboratory Animal at the Osaka University Graduate School of Medicine. Total mRNA was extracted from each sample by ISOGEN procedure (Nippon Gene, Japan). First-strand cDNA was synthesized using 1–3 µg of total mRNA, 1 µm random hexamer RT primer, 0.5 mm dNTPs and 600 U reverse transcriptase at 42°C for 1 h. cDNA equivalent to 20 ng of total mRNA was used for PCR in the final volume of 20 µl.
PCR analysis of splicing pattern
The sequences of primers used for RT–PCR assays of splicing in human muscle RNA are listed in Supplementary Material, Table S1. PCR was repeated 30 cycles for human SERCA2a, 32 cycles for human SERCA1 and 35 cycles for the others. Assays reflect the proportion of splicing isoforms with annealing temperatures of 62°C for SERCA1, 55°C for SERCA2a/b and 60°C for the rest.
Quantitative RT–PCR study
The primers designed for semi-quantitive RT–PCR study of RyR1, RyR3, SERCA1 and SERCA2 of human are listed in Supplementary Material, Table S2. Two primer sets for human and mouse glyceraldehyde-3-phosphate dehydrogenase (G3PDH) (Clontech, Palo Alto, CA, USA) were used as an internal control. PCR was repeated 32 cycles for human SERCA1,2a/b, 28 cycles for human G3PDH and 30 cycles for the others within the range of exponential amplification, with annealing temperatures of 62°C for SERCA1, 2a/b and 60°C for the others.
The PCR product for both studies was electrophoresed on 8% polyacrylamide gels for separation. Quantitative analysis of the amplified products was performed by Fluorimager595 (Amersham Biosciences, Buckinghamshire, UK). The quality of PCR products was ascertained by sequencing representative samples. Sequencing was performed using a dye deoxy terminator cycle sequencing kit and a DNA sequencer (373A; Applied Biosystems, Foster, CA, USA).
Protein analysis
SR proteins were isolated from mouse hind limb, as previously described with minor modifications (47). Briefly, muscle tissue was added with 20 volumes of ice-cold homogenization buffer containing 300 mm sucrose, 5 mm imidazole and 1% diluted proteinase inhibitor cocktail (Sigma-Aldrich, St Louis, MO, USA) and homogenized. The homogenate was centrifuged twice for 20 min at 11 000g. The supernatant was collected, filtered through cotton gauze and centrifuged for 90 min at 110 000g. The pellets were resuspended in five volumes of homogenization buffer. Equal amounts of protein (12 µg per lane for RyR1; 3 µg for SERCA1 and 25 µg for SERCA2) were electrophoretically separated as previously described (48) and transferred to Immobilon-P (Millipore, Bedford, MA, USA) in an electrical field (3 mA/cm2) for 1 h. Blots were blocked for non-specific protein binding with 5% (w/v) non-fat milk for 1 h and then incubated at room temperature for 1 h with a 1:5000, a 1:20 000 and a 1:250 diluted monoclonal antibody against RyR1, SERCA1 and SERCA2 (Affinity Bioreagents, Golden, CO, USA), respectively. After repeated washings, the membranes were incubated at room temperature for 1 h with horse radish peroxidase-conjugated goat anti-mouse IgG (ICN Pharmaceuticals, Aurora, OH, USA) diluted 1:10 000. The membranes were then washed and developed to enhance chemiluminescence (Amersham, Brucks, UK), and X-ray film was exposed to the membranes. Densitometry was performed with FluorChem IS-8000 (Alpha Innotech, Sun Leandro, CA, USA).
Transfection and preparation of microsomal protein
The ASI(−) RyR1 variant was introduced into a rabbit RyR1 cDNA using a standard two-step site-directed mutagenesis strategy and the entire PCR-modified cDNA portion of the clone was ultimately confirmed by sequence analysis. HEK293T cells were grown in 100 mm petri dishes at 37°C, 5% CO2 in MEM complemented by 10% fetal calf serum. At 50–60% confluency, cells were transfected with a mammalian expression vector (pCIneo) encoding either ASI(+) or ASI(−) (40 µg DNA per plate) using calcium phosphate precipitation method. Cells maintained for 48 h were collected in harvest buffer (137 mm NaCl, 3 mm KCl, 8 mm Na2HPO4, 1.5 mm KH2PO4, 0.5 mm EDTA and pH 7.4 with NaOH) and centrifuged for 10 min at 100g. The pellet was resuspended in ice-cold hypotonic buffer (1 mm EDTA, 1 µg/ml leupeptin, 500 µm phenylmethylsulfonyl fluoride, 1 µg/ml pepstatin A, 1 µg/ml aprotinin, 1 mm benzamidine and 10 mm HEPES pH 7.4), homogenized on ice, added with equal volume of ice-cold 20% sucrose buffer (10 mm HEPES, pH 7.4), and centrifuged at 110 000g for 45 min at 4°C. The pellet was then resuspended in 1 ml of buffer containing 10% sucrose and 10 mm HEPES (pH 7.4) and stored under liquid nitrogen until use.
[3H]Ryanodine binding
[3H]Ryanodine binding was performed at 37° for 180 min in a medium containing 200 µg microsomal protein obtained from transfected cells. The Kd and Bmax values were determined from the results obtained in a solution containing 800 mm KCl, 200 µm free Ca2+, 20 mm PIPES pH 6.8, 1 mm EGTA, 200 µm AEBSF (4-(2-Aminoethyl)-benzenesulfonyl fluoride hydrochloride), and various concentrations (1–32 nm) of [3H]ryanodine by Scatchard plot analysis. The Ca2+ dependence of [3H]ryanodine binding was examined in a solution containing 100 mm KCl, 20 mm PIPES pH 6.8, 15 nm [3H]ryanodine, 1 mm EGTA, 200 µm AEBSF and the indicated concentrations of free Ca2+. Non-specific binding was measured in the presence of 667-fold molar excess of cold ryanodine. Binding assay was filtered through Whatmann GF/F filters, which were then washed three times with 3 ml of ice-cold solution containing 100 mm KCl, 20 mm PIPES pH 6.8, 1 mm EGTA and the indicated free Ca2+. Radioactivity bound to the filters was measured by scintillation counting.
Isolation and reconstitution of expressed RyR
The ryanodine receptor was isolated as described by Lee et al. (49). Microsomal proteins were solubilized in buffer A (1.0 m NaCl, 0.1 mm EGTA, 0.2 mm CaCl2, 5 mm AMP, 1 mm PMSF, 1 mm DTT and 20 mm PIPES pH 7.4) containing 1.5% CHAPS and 5 mg/ml phosphatydylcholine (95% soybean phosphatidylcholine) and in protein inhibitors for 1 h. Insoluble material was removed by centrifugation at 100 000g for 30 min, and supernatant was layered at the top of three gradients made up of 17 ml of a 5% (w/w) sucrose solution in buffer A containing 1% CHAPS and 5 mg/ml phosphatydylcholine and 17 ml of a 10–20% linear sucrose gradient solution in buffer A containing 0.5% CHAPS and 5 mg/ml phosphatydylcholine. The gradients were centrifuged at 4°C in a Beckman SW28 rotor at 20 000 r.p.m. (53 000g) for 16 h. Fractions were collected and those containing the RyR were identified by SDS–PAGE and reconstituted into proteoliposomes by removal of CHAPS by dialysis.
Single-channel techniques
Bilayers of phosphatidylethanolamine, phosphatidylserine and phosphatidylcholine (5:3:2 w/w) (Avanti Polar Lipids, Alabaster, AL, USA) were formed across an aperture of ∼200 µm diameter in the wall of a 1.0 ml Delrin cup (Cadillac Plastics, Australia). Purified RyR1 were added to the cis chamber. The cytoplasmic side of channels incorporated into the bilayer faced the cis solution. Bilayer potential was controlled, and single-channel currents were recorded, using an Axopatch 200 A amplifier (Axon Instruments, Foster City, CA, USA). Bilayer potential is expressed as Vcis−Vtrans (Vcytoplasm−Vlumen).
Bilayers were formed and purified RyR1 incorporated using cis solutions containing (in millimolar): 230 CsCH3O3S, 20 CsCl, 5.0 CaCl2, 10 TES and 500 mannitol (pH 7.4 with CsOH) and a trans solution containing (in millimolar) 30 CsMS, 20 CsCl, 1 CaCl2, and 10 TES (pH 7.4). Following incorporation, (a) the cis solution was replaced with an identical solution, except that the [Ca2+] was 100 µm and (b) 200 mm CsCH3O3S was added to the trans chamber for symmetry.
Analysis of channel activity
Channel activity was analyzed over one to two 30 s periods of continuous activity at +40 mV and then at −40 mV. Slow fluctuations in the baseline were corrected using an in-house baseline correction program (written by Dr D.R. Laver). Channel activity was measured using a threshold analysis with the program Channel 2, (developed by P.W. Gage and M. Smith, John Curtin School of Medical Research). Open probability (Po), mean open time (To) and mean closed time (Tc) measurements were restricted to records in which the opening of a single-channel alone could be detected. Threshold levels for channel opening and closing were set to exclude baseline noise at ∼20% of the maximum single-channel conductance.
Preparation and cDNA injections of dyspedic myotubes
Primary cultures of dyspedic myotubes were generated from skeletal muscle myoblasts obtained from neonatal dyspedic mice as previously described (50). Individual myotube nuclei were microinjected with pCIneo encoding either ASI(+) or ASI(−) (0.5 µg/µl), 5–7 days after initial plating. Coinjection of CD8 cDNA (0.1 µg/µl) was used to enable identification of expressing myotubes via incubation with antiCD8 antibody-coated beads. Experiments were carried out on the third day following microinjection.
Measurements of resting Ca2+ and caffeine-induced Ca2+ release in myotubes
Expressing myotubes grown on glass coverslips were loaded for 75 min at 37°C with 6 µm indo-1 AM (Molecular Probes, Eugene, OR, USA) in a rodent Ringer's solution consisting of (in millimolar): 145 NaCl, 5 KCl, 2 CaCl2, 1 MgCl2 and 10 HEPES (pH 7.4 with NaOH). Myotubes were then rinsed several times with dye-free Ringer's solution and incubated for an additional 20 min to allow for de-esterification of the dye. A rectangular region of indo-1-loaded myotubes was excited at 350 nm and fluorescence emission at 405 and 485 nm monitored using a 40× (1.35 NA) oil objective was collected at 100 Hz using a photomultiplier detection system. Traces are presented after offline lowpass filtering as the emission ratio of F405 and F485 (F405/F485). Caffeine-induced Ca2+ release was measured via local application of maximal activating concentrations of caffeine (10 and 30 mm) and subsequent wash out with control Ringer's solution using a rapid local perfusion system (Warner Instruments, Hamden, CT, USA). Data were analyzed using FeliX (Photon Technology International, Lawrenceville, NJ, USA) and SigmaPlot 2000 (SPSS Inc., Chicago, IL, USA) software packages. Differences in the percent of myotubes exhibiting Ca2+ oscillations during caffeine application (Fig. 8E) were evaluated for statistical significance using a χ2 test.
Statistics
Differences between two groups were evaluated by Mann–Whitney U test for the data of splicing and expression studies and by unpaired Student's t-test for the data of [3H]ryanodine binding assay, single-channel measurements and Ca2+ concentration measurements, unless otherwise indicated. Error bar indicates mean±SEM.
SUPPLEMENTARY MATERIAL
Supplementary Material is available at HMG Online.
ACKNOWLEDGEMENTS
We thank Drs Ami Mankodi and Charles Thornton for kindly providing HSALR mice. Part of this work was supported by a Research Grant (14B-4, 17A-10) for Nervous and Mental Disorders from the Ministry of Health, Labour and Welfare, Japan to S.S., Grants-in Aid for Scientific Research from the Japan Society for the Promotion of Science to M.P.T., the Australian National Health and Medical Research Council #316937 to A.F.D., NIH grant #AR44657 to R.T.D. and a University of Rochester Paul D. Wellstone Muscular Dystrophy Cooperative Research Center #AR050762.
Conflict of Interest statement. None declared.
Present address: Department of Neurology, Toneyama National Hospital, 5-1-1 Toneyama, Toyonaka, Osaka 560-8552, Japan.
The authors wish it to be known that, in their opinion, the first two authors should be regarded as joint First Authors.
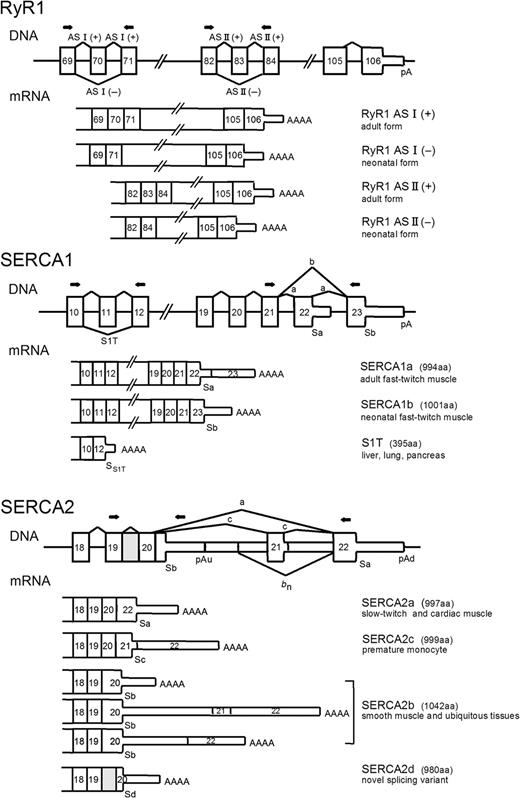
Figure 1. Exon/intron layout of human RyR1, SERCA1 and SERCA2. For each gene, partial exons/introns are given. Exons represented as thick boxes are translated segments and thin boxes indicate untranslated segments. Introns and downstream flanking regions are represented by horizontal lines. Lines with letters represent different splice modes. For SERCA2, bn indicates the removal of the optional untranslated exon specific for neuronal tissue. The different classes of mRNA are represented below the corresponding DNAs. The protein products are shown on the right. The gray box represented intron 19 of SERCA2 gene, which is included in a novel variant (SERCA2d). The relative positions of the sense and antisense PCR primers used to amplify the different RNAs are depicted as arrows. Sa–Sd: position of stop codons for the corresponding protein isoforms. pA, pAu and pAd: position of polyadenylation site (with specification of upstream or downstream in case of SERCA2).
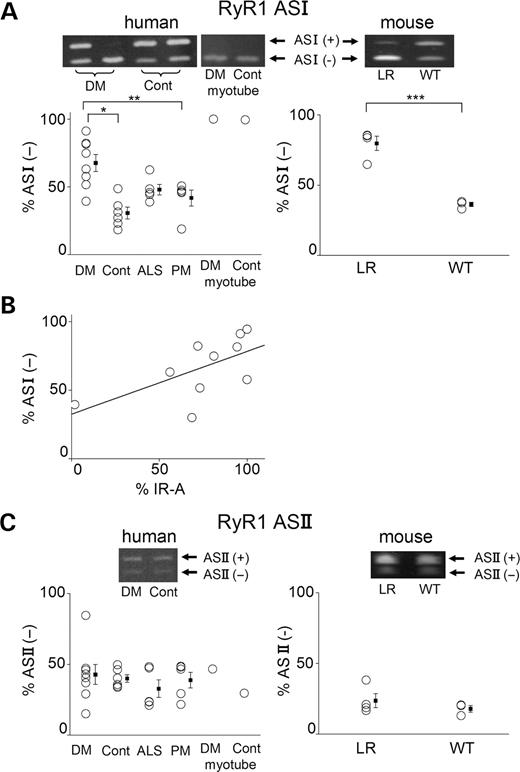
Figure 2. Analysis of RyR1 splice variants shows an increase in ASI(−) in DM1. (ALeft): RT–PCR analysis for exon ASI of RyR1 mRNA from skeletal muscle tissues of DM1 (DM, n=10), normal control (Cont, n=6), ALS (n=5) and PM (n=5) patients and myotubes cultured from DM and control patients. Top: Representative RT–PCR products. PCR products from ASI(+) (+exon70) and ASI(−) (−exon70) are 69 and 54 bp, respectively. Bottom: The mean percentages of the human RyR1 ASI(−) isoform: *P=0.003, **P=0.04. (ARight): RT–PCR analysis at ASI from skeletal muscle tissues of adult HSALR mice (LR, n=4) and adult WT mice (n=3). Top: Representative RT–PCR products. Bottom: The mean percentages of mouse RyR1 ASI(−) isoform: ***P=0.03. (B) Relationship between the proportion of RyR1 ASI(−) and that of the IR-A insulin receptor in skeletal muscle tissues from DM1 patients. The percentage of RyR1 ASI(−) (Y-axis) plotted against that of IR-A (X-axis). R=0.694, P=0.0374. (C) RT–PCR analysis for RyR1 exon ASII mRNA from skeletal muscle tissues of patients (left) and mice (right). Top: Representative RT–PCR products. Bottom: The mean percentages of RyR1 ASII(−) isoform.
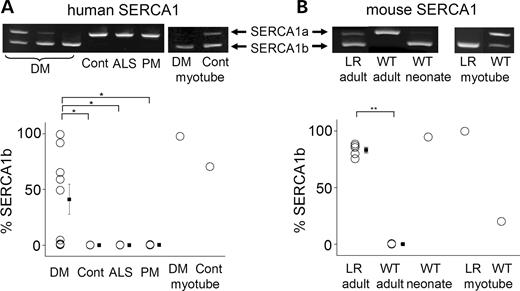
Figure 3. Analysis of SERCA1 splice variants shows an increase in SERCA1b in DM1. (A)Top: RT–PCR analysis of endogenous SERCA1 mRNA from skeletal muscle tissues of DM1 (DM), normal control (Cont), ALS and PM patients. PCR products from SERCA1a (+exon 22) and SERCA1b (−exon 22) are 259 and 217 bp, respectively. Bottom: The mean percentages of human SERCA1b isoform: *P<0.01. (B) RT–PCR analysis of endogenous SERCA1 mRNA from skeletal muscle tissues of adult and neonatal muscle obtained from WT and HSALR (LR) mice. Top: PCR products from SERCA1a (+exon 22) and SERCA1b (−exon 22) are 243 and 201 bp, respectively. Bottom: The mean percentages of mouse SERCA1b isoform: **P<0.0001.

Figure 4. No changes in SERCA2b variants but reduced expression of a novel splicing isoform, SERCA2d, in DM1 muscle. Top: Representative RT–PCR analysis of endogenous SERCA2b mRNA from skeletal muscle tissues of DM1 (DM), normal control (Cont), ALS and PM patients. RT–PCR was analyzed using a primer to the flanking exon 19 and 20. The PCR product from SERCA2b is 325 bp. A novel splicing variant (SERCA2d, with the inclusion of intron19) is 407 bp. Bottom: The mean percentages of SERCA2d isoform: *P<0.01.
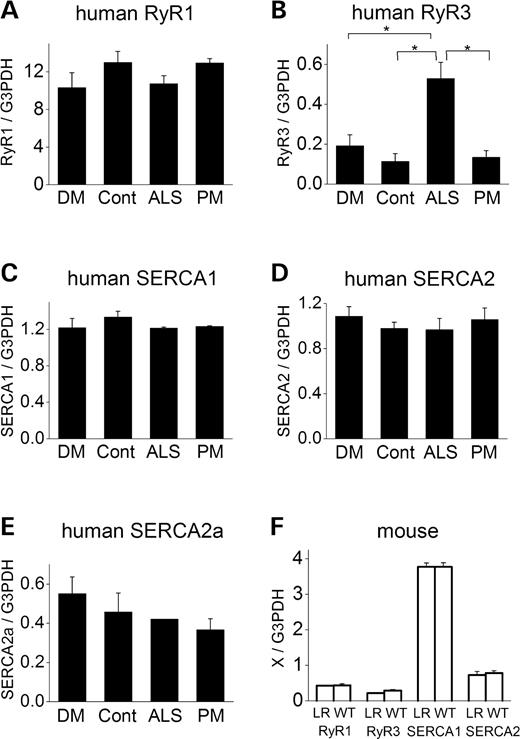
Figure 5. Analysis of the amount of mRNA using semi-quantitive RT–PCR shows no change in RyR1, RyR3, SERCAs in human DM1 or the mouse DM model. (A–E): Relative amounts of RyR1 (A), RyR3 (B), SERCA1 (C), total SERCA2 (D) and SERCA2a (E) mRNA levels in skeletal muscles from DM1 (DM), normal control (Cont), ALS and PM patients. (F) Relative amounts of RyR1, RyR3, SERCA1 and SERCA2 from HSALR (LR) and WT mice muscle. *P<0.05.
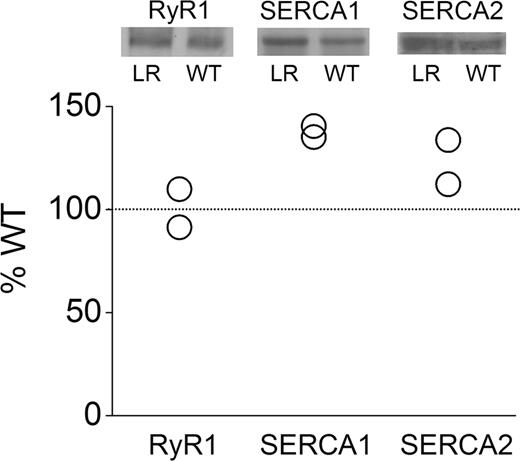
Figure 6. Immunoblot analysis shows no change in RyR1 protein expression in the mouse DM model, but an increase in SERCA1 and 2 expression. Top: Immunoblot analyses of RyR1, SERCA1 and SERCA2 in skeletal muscle from WT and HSALR mice. Bottom: The densities of immunoblot assay from HSALR normalized with that from WT are shown.
![Figure 7. [3H]Ryanodine binding to membrane preparations (A–D) and single-channel recording of purified ryanodine receptor (E) from HEK293T transfected with wt-RyR1 [ASI(+)](closed circles) and the juvenile splice variant [ASI(−)] (open circles). Ryanodine binding was carried out as described in the Materials and Methods section. (A–C): Dose-dependent binding of [3H]ryanodine (1–32 nm) to membranes. (A) Average data from eight experiments. Representative data (B) and Scatchard plot (C) from one experiment. ASI(+) and ASI(−) showed a significant difference in Kd values (Table 1, P=0.0282), but not in Bmax value (Table 1). (D) Ca2+ dependence of [3H]ryanodine binding (n=3). (E) Five second recordings (top) and 30 s all-points histograms (bottom) at +40 mV with 100 µmcis Ca2+. ASI(−) (right) demonstrated less activity with briefer openings compared with ASI(+) (left).](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/hmg/14/15/10.1093_hmg_ddi223/1/m_ddi22307.jpeg?Expires=1716335578&Signature=WMbwT7pLK7oUBzZUvGVrjIHN7oOvzUurg7YabUSTG1PXBZSvWVVYtlY63c9G~BAkE~rH-p98DmRJ7x3NAYc9rNzBqwfsGyrC8DvD0wN5bnWfDZ4hOlHui9uGUBl0iJixr~AiRJUcN~EdtUEPfAioZZ9RrEVRcz2GaBBU10ALCpN7drPZrW-EpoWMKmjGWhHvHRuCPy-OpQdEznQvjNhTtLXZ1Kagja~JtTtfd2tDeK~dgVMD8EM9jT7928jiLlkZuI3hMiNKgV8l8fQUkilzQcjIy2NoAr~7FVsgv8bqKdCi5QLY67iSQuWzvx~eje6I0Va4XnwnVRozS6oQZmhvpw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Figure 7. [3H]Ryanodine binding to membrane preparations (A–D) and single-channel recording of purified ryanodine receptor (E) from HEK293T transfected with wt-RyR1 [ASI(+)](closed circles) and the juvenile splice variant [ASI(−)] (open circles). Ryanodine binding was carried out as described in the Materials and Methods section. (A–C): Dose-dependent binding of [3H]ryanodine (1–32 nm) to membranes. (A) Average data from eight experiments. Representative data (B) and Scatchard plot (C) from one experiment. ASI(+) and ASI(−) showed a significant difference in Kd values (Table 1, P=0.0282), but not in Bmax value (Table 1). (D) Ca2+ dependence of [3H]ryanodine binding (n=3). (E) Five second recordings (top) and 30 s all-points histograms (bottom) at +40 mV with 100 µmcis Ca2+. ASI(−) (right) demonstrated less activity with briefer openings compared with ASI(+) (left).
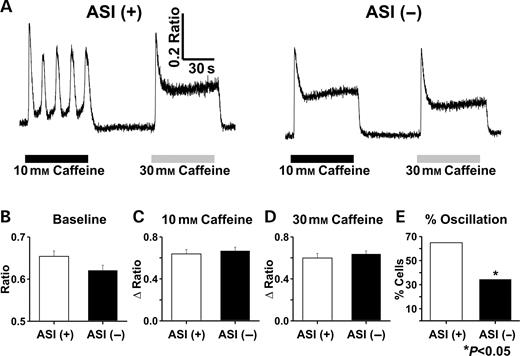
Figure 8. Caffeine-induced Ca2+ release in indo-1 AM-loaded dyspedic myotubes expressing either ASI(+) or ASI(−). (A) Representative traces for ASI(+)-expressing (left) and ASI(−)-expressing (right) dyspedic myotubes following sequential 60 s applications of 10 mm (black bars) and 30 mm caffeine (gray bars). Each drug application was followed by a 60 s wash with control Ringer's solution. Average (±SE) values of resting indo-1 fluorescence (B) peak responses to 10 mm (C) and 30 mm (D) caffeine and the percentage of myotubes exhibiting Ca2+ oscillations during application of 10 mm caffeine (E) in myotubes expressing either ASI(+) (n=37) or ASI(−) (n=29).
Properties of RyR1 ASI(+) and ASI(−)
| ASI(+) | ASI(−) | |
|---|---|---|
| [3H]Ryanodine binding | ||
| n | 8 | 8 |
| Kd (nm) | 5.4±0.42* | 8.2±1.2* |
| Bmax (pmol/mg) | 0.172±0.022 | 0.153±0.012 |
| Single-channel recording | ||
| n | 14 | 12 |
| Po | 0.83±0.03* | 0.61±0.05* |
| To (ms) | 1222±423* | 132.7±40.3* |
| Tc (ms) | 126±61 | 74.2±27.44 |
| ASI(+) | ASI(−) | |
|---|---|---|
| [3H]Ryanodine binding | ||
| n | 8 | 8 |
| Kd (nm) | 5.4±0.42* | 8.2±1.2* |
| Bmax (pmol/mg) | 0.172±0.022 | 0.153±0.012 |
| Single-channel recording | ||
| n | 14 | 12 |
| Po | 0.83±0.03* | 0.61±0.05* |
| To (ms) | 1222±423* | 132.7±40.3* |
| Tc (ms) | 126±61 | 74.2±27.44 |
*Significant difference (P<0.05) of ASI(−) from ASI(+).
Properties of RyR1 ASI(+) and ASI(−)
| ASI(+) | ASI(−) | |
|---|---|---|
| [3H]Ryanodine binding | ||
| n | 8 | 8 |
| Kd (nm) | 5.4±0.42* | 8.2±1.2* |
| Bmax (pmol/mg) | 0.172±0.022 | 0.153±0.012 |
| Single-channel recording | ||
| n | 14 | 12 |
| Po | 0.83±0.03* | 0.61±0.05* |
| To (ms) | 1222±423* | 132.7±40.3* |
| Tc (ms) | 126±61 | 74.2±27.44 |
| ASI(+) | ASI(−) | |
|---|---|---|
| [3H]Ryanodine binding | ||
| n | 8 | 8 |
| Kd (nm) | 5.4±0.42* | 8.2±1.2* |
| Bmax (pmol/mg) | 0.172±0.022 | 0.153±0.012 |
| Single-channel recording | ||
| n | 14 | 12 |
| Po | 0.83±0.03* | 0.61±0.05* |
| To (ms) | 1222±423* | 132.7±40.3* |
| Tc (ms) | 126±61 | 74.2±27.44 |
*Significant difference (P<0.05) of ASI(−) from ASI(+).
References
Liquori, C.L., Ricker, K., Moseley, M.L., Jacobsen, J.F., Kress, W., Naylor, S.L., Day, J.W. and Ranum, L.P.W. (
Philips, A.V., Timchenko, L.T. and Cooper, T.A. (
Savkur, R.S., Philips, A.V. and Cooper, T.A. (
Mankodi, A., Takahashi, M.P., Jiang, H., Beck, C.L., Bowers, W.J., Moxley, R.T., Cannon, S.C. and Thornton, C.A. (
Buj-Bello, A., Furling, D., Tronchere, H., Laporte, J., Lerouge, T., Butler-Browne, G.S. and Mandel, J.L. (
Sergeant, N., Sablonniere, B., Schraen-Maschke, S., Ghestem, A., Maurage, C.A., Wattez, A., Vermersch, P. and Delacourte, A. (
Jiang, H., Mankodi, A., Swanson, M.S., Moxley, R.T. and Thornton, C.A. (
Kimura, T., Takahashi, M.P., Okuda, Y., Kaido, M., Fujimura, H., Yanagihara, T. and Sakoda, S. (
Ebralidze, A., Wang, Y., Petkova, V., Ebralidse, K. and Junghans, R.P. (
Jacobs, A.E., Benders, A.A., Oosterhof, A., Veerkamp, J.H., van Mier, P., Wevers, R.A. and Joosten, E.M. (
MacLennan, D.H., Rice, W.J. and Green, N.M. (
Bertocchini, F., Ovitt, C.E., Conti, A., Barone, V., Scholer, H.R., Bottinelli, R., Reggiani, C. and Sorrentino, V. (
Futatsugi, A., Kuwajima, G. and Mikoshiba, K. (
Burk, S.E., Lytton, J., MacLennan, D.H. and Shull, G.E. (
Brandl, C.J., Green, N.M., Korczak, B. and MacLennan, D.H. (
Wuytack, F., Raeymaekers, L., De Smedt, H., Eggermont, J.A., Missiaen, L., Van Den Bosch, L., De Jaegere, S., Verboomen, H., Plessers, L. and Casteels, R. (
Lytton, J., Westlin, M., Burk, S.E., Shull, G.E. and MacLennan, D.H. (
Mankodi, A., Logigian, E., Callahan, L., McClain, C., White, R., Henderson, D., Krym, M. and Thornton, C.A. (
Chami, M., Gozuacik, D., Lagorce, D., Brini, M., Falson, P., Peaucellier, G., Pinton, P., Lecoeur, H., Gougeon, M.-L., le Maire, M. et al. (
Gelebart, P., Martin, V., Enouf, J. and Papp, B. (
de Smedt, H., Eggermont, J.A., Wuytack, F., Parys, J.B., Van den Bosch, L., Missiaen, L., Verbis, J. and Casteels, R. (
Kimura, T., Takahashi, M.P., Fujimura, H. and Sakoda, S. (
Misquitta, C.M., Mwanjewe, J., Nie, L. and Grover, A.K. (
Benders, A.A., Timmermans, J.A., Oosterhof, A., Ter, L.H., van, K.T., Wevers, R.A. and Veerkamp, J.H. (
Damiani, E., Angelini, C., Pelosi, M., Sacchetto, R., Bortoloso, E. and Margreth, A. (
Meissner, G. (
Dulhunty, A.F., Haarmann, C.S., Green, D., Laver, D.R., Board, P.G. and Casarotto, M.G. (
Avila, G., O′Brien, J.J. and Dirksen, R.T. (
O′Connell, K.M.S., Yamaguchi, N., Meissner, G. and Dirksen, R.T. (
Ho, T.H., Charlet, B.N., Poulos, M.G., Singh, G., Swanson, M.S. and Cooper, T.A. (
Fardaei, M., Rogers, M.T., Thorpe, H.M., Larkin, K., Hamshere, M.G., Harper, P.S. and Brook, J.D. (
Kanadia, R.N., Johnstone, K.A., Mankodi, A., Lungu, C., Thornton, C.A., Esson, D., Timmers, A.M., Hauswirth, W.W. and Swanson, M.S. (
Expert-Bezancon, A., Sureau, A., Durosay, P., Salesse, R., Groeneveld, H., Lecaer, J.P. and Marie, J. (
Lyfenko, A.D., Goonasekera, S.A. and Dirksen, R.T. (
Ferreiro, A., Monnier, N., Romero, N.B., Leroy, J.P., Bonnemann, C., Haenggeli, C.A., Straub, V., Voss, W.D., Nivoche, Y., Jungbluth, H. et al. (
Brini, M. (
White, R.J. and Bass, S.P. (
Odermatt, A., Taschner, P.E., Khanna, V.K., Busch, H.F., Karpati, G., Jablecki, C.K., Breuning, M.H. and MacLennan, D.H. (
Maruyama, K. and MacLennan, D.H. (
Odermatt, A., Becker, S., Khanna, V.K., Kurzydlowski, K., Leisner, E., Pette, D. and MacLennan, D.H. (
Verboomen, H., Wuytack, F., De Smedt, H., Himpens, B. and Casteels, R. (
Ver Heyen, M., Heymans, S., Antoons, G., Reed, T., Periasamy, M., Awede, B., Lebacq, J., Vangheluwe, P., Dewerchin, M., Collen, D. et al. (
Verboomen, H., Wuytack, F., Van den Bosch, L., Mertens, L. and Casteels, R. (
Tohgi, H., Kawamorita, A., Utsugisawa, K., Yamagata, M. and Sano, M. (
Blau, H.M. and Webster, C. (
Chu, A., Dixon, M., Saito, A., Seiler, S. and Fleischer, S. (
Matsunaga, S., Harmon, S., Gohlsch, B., Ohlendieck, K. and Pette, D. (
Lee, H.B., Xu, L. and Meissner, G. (

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}