



Abstract
Multiple mitochondrial DNA deletions are associated with clinically heterogeneous disorders transmitted as mendelian traits. Dominant missense mutations were found in the gene encoding the heart and skeletal muscle-specific isoform of the adenine nucleotide translocator (ANT1) in families with autosomal dominant progressive external opthalmoplegia and in a sporadic patient. We herein report on a sporadic patient who presented with hypertrophic cardiomyopathy, mild myopathy with exercise intolerance and lactic acidosis but no ophthalmoplegia. A muscle biopsy showed the presence of numerous ragged-red fibers, and Southern blot analysis disclosed multiple deletions of muscle mitochondrial DNA. Molecular analysis revealed a C to A homozygous mutation at nucleotide 368 of the ANT1 gene. The mutation converted a highly conserved alanine into an aspartic acid at codon 123 and was absent in 500 control individuals. This is the first report of a recessive mutation in the ANT1 gene. The clinical and biochemical features are different from those found in dominant ANT1 mutations, resembling those described in ANT1 knockout mice. No ATP uptake was measured in proteoliposomes reconstituted with protein extracts from the patient's muscle. The equivalent mutation in AAC2, the yeast ortholog of human ANT1, resulted in a complete loss of transport activity and in the inability to rescue the severe Oxidative Phosphorylation phenotype displayed by WB-12, an AAC1/AAC2 defective strain. Interestingly, exposure to reactive oxygen species (ROS) scavengers dramatically increased the viability of the WB-12 transformant, suggesting that increased redox stress is involved in the pathogenesis of the disease and that anti-ROS therapy may be beneficial to patients.
INTRODUCTION
The accumulation of mtDNA large-scale deletions in post-mitotic tissues is the molecular hallmark of mendelian disorders caused by mutations in nuclear genes compromising the stability of the mitochondrial genome. The most common clinical presentation of these syndromes is adult-onset progressive external ophthalmoplegia (PEO) due to weakness of the external eye muscles. PEO is accompanied by a proximal myopathy with ragged-red fibers and mild reduction in the activities of respiratory-chain enzymes. The disorder is usually transmitted as an autosomal dominant trait (adPEO). Most of the adPEO families carry heterozygous mutations in either one of three genes: adenine nucleotide translocator (ANT1), encoding the muscle–heart specific mitochondrial ANT (1) (OMIM no. 609283); Twinkle, encoding a mtDNA helicase (2) (OMIM no. 606075) and POLG1, encoding the catalytic subunit of the mtDNA-specific polymerase gamma (3) (OMIM no. 157640). Mutations in both POLG1 alleles can be also found in autosomal recessive PEO sibships with multiple affected members and in apparently sporadic cases (4).
Although rare when compared with POLG1 and Twinkle mutations, various ANT1 mutations have been associated with adPEO in unrelated families from Europe and Japan (1,5–8). The course of the ANT1-related adPEO is relatively benign, symptoms being restricted to skeletal muscle. This is explained by the observation that ANT1 is the main isoform of the ADP/ATP carrier in skeletal muscle mitochondria. In humans, ANT1 accounts for 90% of the total ANT mRNA in skeletal and heart muscles; it is also present in the brain (30%) and, to a lower extent (20%), in kidney and liver, whereas in other human tissues, its expression is low relative to other ANT isoforms (9). Like the other members of the ANT family, ANT1 works as a homodimer embedded in the mitochondrial inner membrane, its physiological role being to transport ADP inside mitochondria in exchange for matrix ATP (10,11). ANT1 is believed to maintain close contact, and possibly functional relationship, with a number of mitochondrial proteins involved in apoptosis including VDAC, Bax and Bcl-II (12). The mechanism by which ANT1 mutations cause the mtDNA rearrangements is unknown. ANT1 knockout (KO) mice were shown to accumulate multiple mtDNA deletions in skeletal and cardiac muscles (13) and exhibited mitochondrial myopathy and hypertrophic cardiomyopathy (14).
We have previously documented that the expression of variants of AAC2, the yeast ortholog of ANT1, corresponding to the human dominant mutations, fails to restore oxidative phosphorylation (OXPHOS)-dependent growth in AAC2-defective haploid Saccharomyces cerevisiae strains. In heteroallelic yeasts, characterized by the presence of both wild-type (wt) and mutant genes, accumulation of mtDNA rearrangements was observed and consequent increased formation of petite colonies (15), validating S. cerevisiae as a suitable model for studying the functional and cellular consequences of ANT1 mutations.
We report here on the identification and characterization of the first ANT1 recessive mutation in a young man. Despite the drastic biochemical defects and the massive accumulation of mtDNA large-scale deletions found in the skeletal muscle of our proband, as well as in a yeast recombinant system, the clinical picture was that of childhood onset, slowly progressive myopathy with no PEO and, similar to the ANT1 KO mouse model, hypertrophic cardiomyopathy. To gain insight into how ANT1 impairment cause damage to mtDNA, we have exploited genetic analysis in S. cerevisiae. Our results indicate that oxidative DNA damage causes mtDNA instability, suggesting that anti-ROS therapy may be beneficial to patients.
RESULTS
Clinical findings
This 25-year-old male patient of Slovenian origin was born at term after a normal pregnancy. The birthweight and length were 3800 g and 52 cm, respectively. His psychomotor development proceeded normally, with no delay in the main milestones, including independent ambulation at the age of 14 months. However, a cardiac systolic murmur was detected at the age of 18 months as an incidental finding at a routine general examination, but at that time it was no longer investigated. The patient's mother, now 45 years old, and his older stepbrother are healthy. The patient's father is unavailable but reportedly normal. No neuromuscular symptoms were reported in the family. Since early childhood, the patient complained of easy fatigability and muscle pain. He was unable to walk for >1 km and physical exhaustion ensued by climbing the two-floor stair to his apartment. Slow recovery, lasting 1 or 2 days, and muscle pain followed the episodes of exercise intolerance. Frequent headache episodes often accompanied by nausea and vomiting were additional complaints. No seizures, migraine, hearing loss, vision abnormalities or unsteadiness were reported during the course of the disease. At physical examination at 25 years, neither muscle wasting nor weakness was detected; PEO was absent. The deep tendon reflexes were normal. He had mild contractures of the Achilles' tendons, with high arched feet and hammer-shaped toes. Sensation was normal. In addition, the fundus oculi was normal, and there were no signs of pyramidal tract or cerebellar involvement. The cardiac rhythm was regular, with mild sinusal tachycardia (100 b.p.m). A IVth sound and 2/6 ejection murmur were present at the apex and the left sternal border. Blood pressure was normal (115/70 mmHg). No signs of cardiac failure could be detected. An ultrasound examination revealed a left ventricular hypertrophy: the thickness of the left ventricular septum was 15–18 mm (n.v. <11 mm) and that of the posterior wall was 14 mm (n.v. <10 mm). The size of the left ventricular chamber was normal with no reduction of the ejection fraction: the end-diastolic diameter was 44 mm, and the end-systolic diameter was 24 mm. The values obtained for the same parameters at the age of 14 and 17 years indicate slow progression of the disease. The cardiac valves were structurally and functionally normal. No abnormality of the cardiac rhythm was recorded during a 24 h Holter monitoring. An ECG showed overload of the right atrium and left ventricle, inversion of the T wave in D3, AVF, and V6 and biphasic T waves in D2 and V5. The fasting serum lactate at rest was high (9.0 mm, normal values up to 2.4 mm). Serum creatine kinase (CK) levels were moderately elevated. The cerebrospinal protein concentration was within normal limits. Results of the motor and sensory conduction studies were normal, as well. A needle EMG examination showed a moderate myopathic pattern. EEG, brain MRI and audiogram were normal. The patient is not receiving any specific treatment at the present time.
Morphological and biochemical analyses in muscle
Figure 1A–C shows the results of the histological examination of a muscle biopsy taken from the left vastus lateralis muscle at 17 years of age. Numerous ragged-red fibers were present at the modified trichrome Gomori staining, a very few of which stained negative to the reaction to cytochrome c oxidase (COX). Ultrastructural examination showed the accumulation of abnormally shaped mitochondria, some of which contained highly ordered paracrystalline inclusions. Table 1 reports the activities of the mitochondrial respiratory-chain complexes in muscle homogenate. The activities of complex I, complex III and complex IV, three mtDNA-dependent respiratory-chain enzymes, were partially reduced, whereas the activity of complex II, which is entirely nuclear encoded, was normal. Consistent with the observed mitochondrial proliferation, a dramatic increase in the activity of citrate synthase (a marker enzyme of the matrix compartment) was measured (Table 1).
Molecular genetic studies
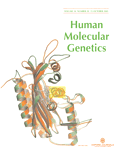
Southern blot analysis on PvuII-linearized muscle mtDNA is shown in Fig. 2A. In addition to the hybridization band corresponding to the 16.5 kb wt species, numerous additional bands corresponding to mtDNA deleted species are present in the patient's DNA sample. No reduction of wt mtDNA copy number was found by both Southern blot and real-time PCR analyses (data not shown). Nucleotide sequence analysis of the four exons of the ANT1 gene showed the presence of a homozygous C→A transversion at nucleotide position 268 of the ANT1 cDNA (Fig. 2B), predicting the replacement of a highly conserved A residue at position 123 with a D residue (A123D) (Fig. 2C). The mother was heterozygous for the same mutation (data not shown). The mutation was absent in 500 DNA samples belonging to control individuals from Northern Italy (n. 450) and Slovenia (n. 50).
ANT1 function is impaired in the patient's skeletal muscle
Western blot analysis of muscle homogenates revealed no significant decrease in the ANT1 content when compared with control samples (Fig. 3A). To directly test its function, ANT was solubilized from the skeletal muscle homogenate of the patient and three independent control subjects and reconstituted into phospholipid vesicles. As shown in Figure 3B, proteoliposomes reconstituted with protein extracts from the controls catalyzed an efficient uptake of labeled ATP. This transport activity was carried out by ANT proteins because it was nearly completely (>99%) abolished by the pre-incubation with 10 µm carboxyatractylate and 5 µm bongkrekic acid (data not shown), which are specific inhibitors of the mitochondrial ADP/ATP carrier (16). Similar results but somewhat lower specific transport activities were found with ADP instead of ATP (data not shown). In contrast, virtually no ATP (Fig. 3B) or ADP (data not shown) was taken up by proteoliposomes reconstituted with the Triton extract from skeletal muscle homogenate of the patient. The uptake of oxoglutarate, which is catalyzed by the oxoglutarate/malate carrier (17), another transport protein of the inner mitochondrial membrane, was comparable in the patient and in the control samples (Fig. 3C). No significant variation of the transcript levels of the four ANT isoforms (9) as well as that of the three isoforms of the ATP/Mg-Pi transporter (18) was observed in the skeletal muscle of the patient by real-time RT–PCR (data not shown). Taken together, these findings indicate that the A123D change is a loss-of-function ANT1 mutation.
Analysis of the aac2 A137D mutant allele equivalent to ANT1A123D
The A123 residue in the ANT1 protein sequence is conserved through eukaryotes and corresponds to the A137 residue in the yeast AAC2 protein (Fig. 2C). We introduced the A137D amino acid change in the AAC2 protein, by site-directed mutagenesis of a recombinant AAC2 cDNA cloned in the centromeric pFL38 vector (15,19). The GCT codon was changed to GAC, the preferred codon for aspartate in S. cerevisiae (20). The AAC2 mutated allele was then introduced into the S. cerevisiae WB-12 strain, which carries a double deletion abolishing both AAC1 and AAC2 genes, and transformants were analyzed for growth on three non-fermentable carbon sources: lactate, ethanol and glycerol. The growth was totally prevented on all the substrates. In Figure 4A, the growth phenotype on glycerol is shown as an example. These results indicate that the A137D mutation causes a drastic alteration of the AAC2 protein function. Measurement of the mitochondrial cytochrome content is an index of the structural integrity of the respiratory-chain complexes. We analyzed the cytochrome profile in the WB-12 strain transformed with pFL38 without insert, with the wt AAC2 allele or the aac2A137D allele. The strain transformed with the aac2A137D allele displayed a significant reduction of the cytochromes b and aa3 but not of the cytochrome c (Fig. 4B). In contrast, the inner membrane potential of the aac2A137D mutant mitochondria measured in vitro was comparable to that of wt mitochondria, indicating that the aac2A137D mutant allele did not induce significant membrane damage (data not shown). In the same transformants, the respiratory activity paralleled the cytochrome profile, the function of the respiratory chain being reduced to 59% for the null allele and to 18% for the mutant allele. Similar to the results obtained with muscle protein extracts, proteoliposomes reconstituted with extracts from the wt WB-12 transformant catalyzed an active ATP/ATP homoexchange, whereas the WB-12 aac2A137D showed virtually no ATP homoexchange activity (Fig. 5A). Interestingly, the amount of wt AAC2 and AAC2A137D mutant proteins was comparable, as estimated by western blot analysis (Fig. 5B). Therefore, the absence of ATP uptake in the aac2A137D is due to impaired catalytic activity, rather than decreased content, of the AAC2A137D mutant protein in mitochondria.
Construction and analysis of the heteroallelic AAC2/ aac2A137D strain
To study whether the aac2 A137D allele behaves as a dominant or a recessive trait, the yeast strain W303-1B AAC2 was transformed with a plasmid carrying either the AAC2 or the aac2A137D allele. The resulting heteroallelic strains mimic the diploid genetic organization of human somatic tissues. Both the AAC2/AAC2 homoallelic and the AAC2/aac2 A137D heteroallelic strains were analyzed for the growth on YNB medium supplemented with either 2% lactate or 2% ethanol or 2% glycerol. The growth phenotype of the heteroallelic and homoallelic strains was indistinguishable in all substrates (data not shown). In agreement with this result, the cytochrome content of the AAC2/aac2A137D heteroallelic strain was virtually the same as the cytochrome content of control strain (data not shown), indicating that the aac2A137D mutation behaves as a recessive trait in yeast. However, as shown in Figure 5C, proteoliposomes reconstituted with mitochondrial extracts from the heteroallelic strain took up ATP at a lower rate and to a lower maximal extent than proteoliposomes reconstituted with extracts from the homoallelic strain. A decrease in AAC content as estimated by western blot analysis (Fig. 5D) can at least partly account for the observed decrease in transport activity (see Discussion).
Cell viability analysis
In a previous report (15), we showed that adPEO-associated ANT1 mutations introduced at equivalent positions in AAC2 caused dominant effects on mtDNA mutability in the yeast S. cerevisiae. This result indicates that the isogenic WB-12/W303-1B strains represent a useful model to study the consequences of ANT1 mutations on mtDNA maintenance. Partial (rho−) or total (rho°) loss of mtDNA produces a lethal phenotype in the WB-12 strain (21), thus resembling petite-negative yeasts (22). To further characterize the cellular effects of the aac2A137D allele, we measured the effect of its expression on WB-12 viability. In strains transformed with either the empty pFL38 vector or the wt AAC2, as well as in the parental W303-1B strain, ∼80% of colonies were viable on glucose. In contrast, only 23% of the cells derived from WB-12/aac2A137D were able to form viable colonies (Fig. 6). The heteroallelic strain W303-1B/aac2A137D (AAC2/aac2A137D) is petite positive, i.e. it is viable in the presence of multiple deletions or even complete loss of mtDNA. Petite-positive strains carrying mtDNA deletions are able to grow on fermentative carbon sources but not on respiratory substrates. This enabled us to analyze whether the aac2A137D mutation can affect the mitochondrial mutability by measuring the formation of respiratory-deficient (RD) mutants. To evaluate a possible accumulation of mtDNA rearrangements in single clones, the heteroallelic strain AAC2/aac2A137D and the homoallelic strain AAC2/AAC2 were grown for approximately 15 generations and plated onto YNB medium supplemented with 2% glucose. Then, the segregation of petite mutants was estimated by the triphenyltetrazolium chloride (TTC) overlay assay and by the glycerol-negative phenotype. The accumulation of RD mutants observed with the heteroallelic strain was virtually the same as that observed with the homoallelic strain AAC2/AAC2, indicating that also for this phenotype the mutation behaved as recessive.
Role of ROS in mtDNA mutability
Increased reactive oxygen species (ROS) production was associated to an increase in the accumulation of mtDNA rearrangements in ANT1-deficient mice (13). To evaluate whether the observed reduction of cell viability could be due to an increase of oxidative stress, we analyzed the effect of two ROS scavengers, dihydrolipoic acid (30 µm) and N-acetyl cysteine (5 mm), on the viability of our yeast WB-12/aac2A137D strain. Exposure to dihydrolipoic acid increased the survival to 66% (Fig. 6), whereas N-acetyl cysteine induced a 2-fold survival increase (data not shown).
DISCUSSION
Heterozygous ANT1 mutations are a rare cause of adPEO in adults. The clinical phenotype of our A123D homozygous mutant patient is the first example of a recessive trait associated with ANT1. Evidence that this mutation is indeed pathogenic is provided by the following facts. First, like other pathogenic mutations in the ANT1 gene, the A123D mutation is associated with the accumulation of multiple mtDNA deletions in the skeletal muscle of the patient. Secondly, the mutation involves a highly conserved A residue and was absent in 1000 chromosomes from 500 consecutive control individuals, indicating a frequency of this allele <0.1% in Caucasians. Thirdly, the activity of the ADP/ATP carrier was virtually absent in muscle homogenate from the patient upon reconstitution into liposomes, indicating that A123D is a loss-of-function mutation. Finally, the A137D mutation in AAC2, which corresponds to the human A123D mutation in ANT1, leads to transport inactivation and inability to complement the OXPHOS-dependent growth phenotype in an AAC2 null yeast strain. In contrast with the other ANT1 mutations reported in humans, the A123D is a recessive mutation. This is supported by the observations that (i) the heterozygous mother is normal, with no sign of skeletal or cardiac myopathy, (ii) no neuromuscular or cardiac abnormality is reported for other members of the family and (iii) the yeast A137D mutation equivalent to the human A123D mutation behaves as a recessive trait in a heteroallelic strain. However, no muscle tissue is available from the patient's mother, thus the presence of subclinical amounts of multiple deletions cannot be ruled out in this individual. No DNA sample is available from other members of the family.
Some of the clinical features of our ANT1A123D mutant patient are intriguing. Although later development of additional symptoms cannot be excluded in this relatively young man, he has no PEO, which is an invariant feature of dominant ANT1 mutations, and in general, of the syndromes associated with the accumulation of multiple mtDNA deletions. Truly enough, the patient is still young, so this symptom could still ensue later in life. What is more surprising, however, is that the muscle symptoms are relatively mild, when compared with the dramatic drop of mitochondrial adenine nucleotide translocation in vitro. Exercise intolerance and muscle pain were the main complaints, with no muscle wasting, muscle weakness or marked elevation of CK. Likewise, his hypertrophic cardiomyopathy, although slowly worsening with time, is still in a condition of functional compensation, with no sign of heart failure. Interestingly, the clinical picture of our patient resembles that of ANT1 KO mice. These mice develop an early-onset, progressive mitochondrial myopathy and cardiomyopathy, with multiple OXPHOS deficiencies, and show time-dependent accumulation of multiple deletions in affected tissues. Similar to the mouse model, our patient developed a mitochondrial myopathy and hypertrophic cardiomyopathy diagnosed for the first time during puberty. Like in the mouse model, the absence of ANT1 activity in our patient is compatible with adult life: ANT1 KO mice survive >24 months, in spite of deterioration of motor and cardiac functions. Metabolic compensation of the mitochondrial ATP/ADP transport block through glycolysis may contribute to attenuate the shortage in ATP supply in skeletal muscle. In the heart, the activity of ANT2 and ANT3, which is three times more expressed in this tissue than in skeletal muscle may sustain a significant residual transport capacity (9,23). In addition, the conditions for the in vitro transport assays were optimized to measure specifically the activity of ANT. As different mitochondrial carriers have different set-up requirements (e.g. lipid composition, detergent and pH) for optimal functional reconstitution into liposomes, we cannot exclude that other transport activities located in the inner mitochondrial membrane, which may have not been detected by our measurements in vitro, can sustain a residual capacity of ADP/ATP exchange in vivo. For instance, in the yeast S. cerevisiae, overexpression of Sal1p complements AAC2 deficiency (24). Interestingly, the human ortholog of Sal1p has been recently identified as the mitochondrial ATP/Mg-Pi carrier (APC) (18). However, we found no increase in the transcript levels of the three APC isoforms as well as of the four ANT isoforms in our patient by real-time RT–PCR.
Hypertrophic cardiomyopathy is a common feature of several mitochondrial disorders, including, among others, syndromes associated with mutations in mtDNA genes, in nuclear genes encoding subunits of complex I in factors involved in the assembly of COX, and also in Sengers' syndrome. Sengers' syndrome (OMIM no. 103220) is a rare mitochondrial disease of unknown cause, characterized by a combination of hypertrophic cardiomyopathy, congenital cataract and, more variably, lactic acidemia. Interestingly, a profound reduction of ANT1 cross-reacting material and ATP uptake were recently documented in two non-related subjects, affected by either early-onset or adult-onset forms of Sengers' syndrome (23). No mutation in ANT1 was found in these patients, and the ANT1 locus was excluded by linkage analysis on each family. Taken together, these observations and our own case suggest that, irrespective of the presence of deleterious mutations in its coding sequence, hypertrophic cardiomyopathy and lactic acidemia are consistently associated with profound impairment of ANT1 activity.
The A123 residue in the hANT1 protein sequence is conserved through eukaryotes and is located in the third transmembrane domain of ANT1 (25), within the dimerization consensus sequence ‘GXXXG’. This sequence is thought to be involved in high affinity association between transmembrane domains (26,27) and is conserved among ANT carriers (Fig. 2C). Dimerization of carrier proteins has been shown to be essential for import into the inner mitochondrial membrane (28) and for transport catalysis (10,11,29). Using specific biochemical assays, we showed that in the WB-12 strain carrying the mutant allele aac2A137D, the ADP/ATP carrier was present in normal amount but had no catalytic activity (Fig. 5A and B). Accordingly, we found reduced ATP uptake in proteoliposomes reconstituted with mitochondrial extracts from the heteroallelic strain W303-1B/aac2A137D (Fig. 5C), in which co-expression of wt and mutant alleles is expected to result in a mixed population of homo- and heterodimers. Protein analysis demonstrated a decrease in immunoreactivity of AAC (Fig. 5D), suggesting reduced import efficiency and/or instability in the mitochondrial membrane of the heterodimers. Furthermore, among the homodimers, only the wt/wt homodimers would contribute to transport activity. Collectively, these effects may account for the observed reduction in transport activity. However, it should be noted that the observed transport alterations did not affect oxidative growth, the cytochrome profile and the rho-mutability, suggesting that the transport activity, though reduced, sustains a sufficient level of OXPHOS activity. In contrast, the WB-12/aac2A137D strain, similar to the AAC1/AAC2 defective strain, displayed a negative phenotype of growth on non-fermentable carbon sources, and its respiratory activity was severely affected. The analysis of the cytochrome profile revealed that the mutation exerted a drastic effect on the cytochrome b of complex III and on the cytochrome aa3 of COX, but not on the cytochrome c. Interestingly, both cytochrome b and three of the seven subunits of yeast COX are encoded by the mtDNA, whereas the cytochrome c is encoded by nuclear DNA. This result is in agreement with our previous findings on adPEO-associated mutations (15) and indicates that the mitochondrial ADP/ATP carrier activity affects the assembly of the respiratory complexes. However, the effect of the A137D mutation cannot be simply due to the absence of ADP/ATP translocation activity, because the aac1aac2 null mutant, which has no ADP/ATP carrier, displayed a cytochrome profile that was significantly less affected than that of aac2A137D mutant. Further investigation is required to gain insight into the multiple cellular interactions of the mitochondrial ADP/ATP carrier in vivo. In this context, the S. cerevisiae mutant strain carrying the aac2A137D allele represents a potentially useful tool as it enables us to search for extragenic suppressors.
The double mutation aac1aac2 makes the strain WB-12 petite negative; in fact, in this genetic background, mtDNA deletions are lethal. In S. cerevisiae, petite mutations occur spontaneously giving rise to RD cells with a frequency of approximately 10−2. Conditions that cause increased mtDNA mutability leads to an increase of RD cells due to rapid conversion into homoplasmic mutant strains, because the heteroplasmic status is always transient in S. cerevisiae (30). Under the assumption that a lethal phenotype could result from deletion(s) of mtDNA, we analyzed the cell viability of the aac1aac2 strain carrying the mutant allele aac2A137D. We found a dramatic loss of cell viability that dropped to 20% when compared with 80% in the AAC1AAC2 wt strain. Neither loss of cell viability nor increased formation of petite cells was observed in the heteroallelic strain, indicating that mtDNA mutability associated to the A137D–A123D mutation is a recessive trait in both yeasts and humans.
Similar to ANT1 KO mice, as well as adPEO patients associated with heterozygous mutations of ANT1, our patient with the recessive A123D mutation has multiple deletions of mtDNA in skeletal muscle. Although the loss of ANT1 function can directly impair OXPHOS by, for instance, blocking the ADP supply to the system, the damage on mtDNA can further contribute to the functional decline of mitochondrial OXPHOS by compromising the synthesis of specific respiratory subunits.
Several mechanisms could explain the effect of ANT1 dysfunction on mtDNA stability, including imbalance of the intra-mitochondrial dNTP pool, caused by a relative shortage of the dATP supply needed for mtDNA synthesis (1), damage on mtDNA by overproduction of ROS (13) or instability of the mitochondrial permeability transition pore and apoptosis (31).
Interestingly, it has been clearly demonstrated that increased production of ROS occurs in ANT1 KO mice (13). Likewise, on the basis of our own experiments with antioxidants on yeast, we also propose that oxidative stress plays a major role in the pathogenesis of mtDNA damage. We think that reduced availability of matrix ADP, due to loss of ADP/ATP translocation activity, could inhibit the ATP synthase (complex V), thereby blocking the proton influx into the matrix mediated by the ATP synthase proton-conducting activity. As a consequence, the electrochemical gradient can increase up to a level at which respiration-linked proton pumping and electron flow are both stalled. This can in turn lead to increased production of ROS, which are known to determine mitochondrial genomic instability (32,33). Indeed, in the presence of ROS scavengers, we observed a significant increase in cell viability of the WB-12 aac2A137D mutant cells, which strongly indicates that oxidative stress is one of the detrimental effects of the mutation.
ROS are known to be powerful modulators of programmed cell death (apoptosis). However, we observed neither chromatin condensation nor other ultrastructural features of apoptosis in our patient. Likewise, no morphological features of apoptosis were detected in the muscle biopsy of adPEO patients (34), including TUNEL, and no increase in apoptosis was reported in the ANT1 KO mice. Furthermore, the expression of the aac2A137D mutant allele in yeast did not cause mitochondrial permeabilization or cytochrome c release, which are common early events in the apoptotic cascade. Taken together, these results make it unlikely a major role of apoptosis in the development of the ANT1A123D associated muscle damage.
As heteroplasmy is not maintained in the yeast S. cerevisiae, we propose that the increase in the rate of unviable colonies observed in our petite-negative mutant strains could be a yeast-specific phenotype, caused by the same instability of the mitochondrial genome which leads to the accumulation of multiple mtDNA rearrangements in humans. Our data on the increased survival rate of yeast mutants exposed to anti-ROS scavengers usher in the development of a rational anti-ROS strategy for patients with ANT1 mutations.
MATERIALS AND METHODS
Histochemical and biochemical studies
Muscle sample was obducted from the patient's left vastus lateralis muscle with informed consent. Morphological analysis of skeletal muscle and biochemical assays of the individual respiratory-chain complexes on muscle homogenate were carried out as described earlier (35). Enzymatic activity of each complex was normalized to that of citrate synthase. Western blotting of muscle homogenate was performed using polyclonal antibodies raised against an ANT1-specific oligopeptide antigen (NH2-MGDQALSFLKDFLAG) or monoclonal antibodies against subunit A of human succinate dehydrogenase (Molecular Probes). Solubilization from muscle homogenates and functional reconstitution of ANT were performed as previously described (23).
DNA sequencing, real-time RT–PCR and Southern blotting of mtDNA
Molecular analysis was performed on genomic DNA extracted from skeletal muscle or lymphocytes. Southern blot analysis of PvuII-linearized mtDNA was performed as described earlier (35). The four exons of the ANT1 gene were PCR-amplified and the products were sequenced according to Kaukonen et al. (1). For real-time PCR on mtDNA, the procedure that described by Ferrari et al. was performed (36). For real-time PCRs on ANT and APC transcripts, mRNA was isolated and reverse transcribed according to standard procedures. Specific primers and probes based on the cDNA sequences of ANT1-4 and APC1-3 were used as described previously (9,18).
Yeast strains and culture media
Yeast strains were: W303-1B: MATα ade2-1 leu2-3,112 ura3-1 his3-22,15 trp1-1 can1-100 AAC1 AAC2 and its isogenic aac1 aac2 mutant WB-12: MATα ade2-1 trp1-1 ura3-1 can1-100 aac1::LEU2 aac2::HIS3 (37). Transformation of yeast strain with the centromeric vector pFL38 harboring wt or mutagenized aac2 allele was performed as described earlier (15). Cells were cultured in YNB medium, comprised of 0.67% yeast nitrogen base without amino acids (Difco), supplemented with the appropriate amino acids and bases to a final concentration of 40 µg/ml. Various carbon sources were added at 2% (w/v). Lactate was a commercial racemic product (Fluka). For the isolation of mitochondria cells were grown to mid-log phase in SC medium (38) supplemented with 2% galactose.
Characterization of yeast strains
Cytochrome spectra and oxygen consumption in intact yeast cells were recorded as described previously (15). To measure cell viability, a single fresh colony was inoculated in YNB medium supplemented with 2% glucose and cultivated for about 15 generations. After this time, 100 cells/plate of the culture were transferred to fresh YNB glucose medium by microdissection and analyzed for the ability to form colony after 3 days. TTC overlay on colonies was used to analyze the respiratory competence of cells (39). Isolation of mitochondria, membrane potential determination, western blotting, solubilization and incorporation into liposomes of Aac2p were carried out as previously described (15). For transport measurements, external substrate was removed from proteoliposomes on a Sephadex G-75 column pre- equilibrated with a buffer containing 50 mm NaCl and 10 mm HEPES, pH 7.5. Transport at 25°C was started by adding 0.1 mm14CATP (Amersham) or 14CADP (NEN) to the proteoliposomes and terminated at predetermined time intervals by addition of 30 mm pyridoxal 5′-phosphate and 10 mm bathophenanthroline [the ‘inhibitor-stop’ method (40)]. The external radioactivity was removed on Sephadex G-75 and the internal radioactivity was measured. In controls, transport was inhibited by the addition of the inhibitors together with the labeled substrate. The transport activity was the difference between experimental and control values. No activity was observed in the absence of added protein or internal substrate.
ACKNOWLEDGEMENTS
We thank Dr T. Hatanaka (Tokushima University) for the strain WB-12. This work was funded by grants from the Ministero Università e Ricerca Scientifica e Tecnologica (MIUR)—PRIN 2003, Centro di Eccellenza Geni in campo Biosanitario ed Agroalimentare (CEGBA), Fondazione Telethon-Italy no. GGP030039, Ricerca Finalizzata Ministero della Salute RF-2003 and the European Community's sixth Framework Program for Research (contract number LSHM-CT-2004-503116).
Human skeletal muscle samples were obtained from the Biobank of the University of Ljubljana Medical faculty, a partner of Eurobiobank (www.eurobiobank.org) funded by the EU within the 5th framework (QLRT-2001-02769).
Conflict of Interest statement. None declared.
Present address: Department of Neurology, University of Miami School of Medicine, Miami, FL, USA.

Figure 1. Histological examination of muscle biopsy. (A) Modified trichrome Gomori staining of skeletal muscle showed the presence of numerous ragged-red fibers. (B) COX histochemical reaction showed the presence of a COX-depleted fiber. (C)° Electron microscopy showed numerous abnormal mitochondria containing paracrystalline inclusions.
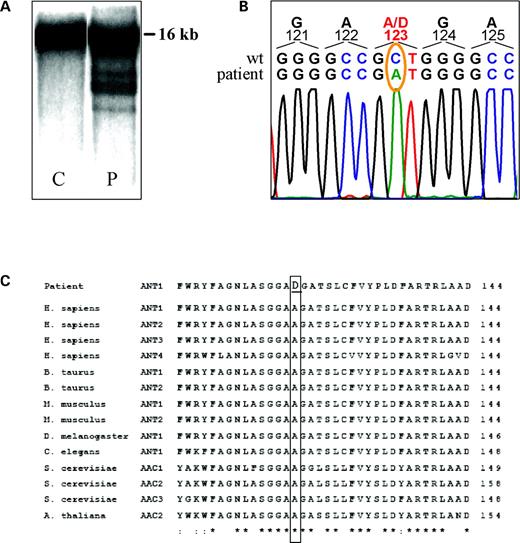
Figure 2. Molecular findings. (A) Southern blot of PvuII-linearized mtDNA from skeletal muscle. lane C: control sample; Lane P: patient's sample. In both lanes, a 16.5 kb band corresponds to wt mtDNA; in the patient's sample, additional smaller bands correspond to deletion-containing mtDNA molecules. (B) Sequence analysis of the DNA region encompassing the homozygous 268C→A transversion in the ANT1 gene of the patient. (C) Alignment of the primary structure of known mitochondrial ADP/ATP carriers. The conserved alanine mutated in the patient is boxed. For sequence alignment version 1.4 of ClustalW was used with default parameters. Asterisks denote conserved residues. Double dots indicate conservatively substituted residues.
![Figure 3. Mitochondrial ADP/ATP carrier content and activity in the patient muscle. (A) Muscle homogenate (corresponding to 20 mU of citrate synthase) from the patient (lane P) or a control subject (lane C) were separated by SDS–PAGE, transferred to nitrocellulose and immunodecorated with specific antibodies against ANT1 or subunit A of succinate dehydrogenase (SDH-A). (B and C) Transport of proteoliposomes reconstituted with mitochondrial extracts from the skeletal muscle of the patient (closed triangle) and of three control subjects (closed circle, closed square, closed diamond). 0.1 mm [14C] ATP (B) or 0.1 mm [14C] to proteoliposomes containing 20 mm ATP or 20 mm oxoglutarate, oxoglutarate (C) was added, respectively. The exchange reactions were terminated at the indicated time intervals by adding 30 mm pyridoxal 5′-phosphate and 10 mm bathophenanthroline. Similar results were obtained in three independent experiments.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/hmg/14/20/10.1093/hmg/ddi341/2/m_ddi34103.jpeg?Expires=1716360392&Signature=VpBLKqVdv-z22U90Mfv3EvRQoj-1cYugAEc2hZ9pEn0ukervCaMknsoxhNT8NixD3DvFRUSaP72154qljiENQSYcavgUaF4BHxrMq0evdhphDUUdBH1L7fF~UEBQIAKY53XwsExwf9ay6v1tuA6UIZjJDkbfNTa-hZVOpPCdbwgp5FDpvOTS4df5Xx5fvB2oHPOrMm4Q80mzhKAlIiWP6g4OgKo3yBRoPINy8tWic3Nsh3aylJp-HDHwnoRI~OP3LnzCiF98-taaVABC~Q9iAFT-4tlYvo47jAfbYdqryn52IKYdlnYNOKWBVDknOV0uvJ7IgWpu9Eye8ibUfpzWQg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Figure 3. Mitochondrial ADP/ATP carrier content and activity in the patient muscle. (A) Muscle homogenate (corresponding to 20 mU of citrate synthase) from the patient (lane P) or a control subject (lane C) were separated by SDS–PAGE, transferred to nitrocellulose and immunodecorated with specific antibodies against ANT1 or subunit A of succinate dehydrogenase (SDH-A). (B and C) Transport of proteoliposomes reconstituted with mitochondrial extracts from the skeletal muscle of the patient (closed triangle) and of three control subjects (closed circle, closed square, closed diamond). 0.1 mm [14C] ATP (B) or 0.1 mm [14C] to proteoliposomes containing 20 mm ATP or 20 mm oxoglutarate, oxoglutarate (C) was added, respectively. The exchange reactions were terminated at the indicated time intervals by adding 30 mm pyridoxal 5′-phosphate and 10 mm bathophenanthroline. Similar results were obtained in three independent experiments.
Figure 4. Oxidative growth phenotype and cytochrome spectra of WB-12 strain, carrying double aac1 and aac2 deletion, transformed with pFL38, wt AAC2 and aac2A137D. (A) Equal amounts of serial dilutions of cells from exponentially grown cultures (105, 104, 103 and 102 cells) were spotted onto YNB plates supplemented with either 2% glucose or 2% glycerol. The growth was scored after 5 days of incubation at 28°C. (B) Cytochrome spectra of glucose-grown cells were recorded at room temperature.
![Figure 5. Mitochondrial ADP/ATP carrier content and activity in homoallelic and heteroallelic yeasts. (A and C) Kinetic of [14C] ATP/ATP exchange in proteoliposomes reconstituted with mitochondrial extracts from the WB-12 strain (A) and from the W303-1B strain (C) transformed with the wt AAC2 (closed circle, closed square) or the aac2A137D mutant allele (open circle, open square). To proteoliposomes containing 20 mm ATP, 0.1 mm [14C] ATP was added. The exchange reactions were terminated at the indicated time intervals by adding 30 mm pyridoxal 5′-phosphate and 10 mm bathophenanthroline. Similar results were obtained in three independent experiments. (B and D) Mitochondria isolated from the WB-12 strain (2 µg) (B) and W303-1B strain (4 µg) (D) transformed with the indicated alleles were subjected to SDS–PAGE, transferred to nitrocellulose and immunodecorated with polyclonal antibodies against Aac2p.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/hmg/14/20/10.1093/hmg/ddi341/2/m_ddi34105.jpeg?Expires=1716360392&Signature=y0J0lFaT6puJoJwfGVaA9IUf0L3TSdnBOfEhhgYeT7y-FhbmajWmWTWTBwJPCIdZxRiWjsSJsS-rKNKD07RAQr0ChZ0UJ0UwRWPDdeDLWv0dvZd71NbQ~zPHRCD0AD8goL~-gw~NSD1rOIWwcOuOIKJJT~5Ojt~oHxT8CWi4b9dkMXLh-Xgz6CDnMU~BUuRm52TiLEOGGHOTQyR3p5UkvwtcLejrMrNgxsnF32lJFtnFNm5LaAG7jvXkgwV01g~Sh~4zS5xS6xLvzxzuADkHkYLKnmmpIbKool8YdV6-hrhgMCVGXVYMf5jPYJpJ3xgCN2K4hccSJzqh026HT3yPlA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Figure 5. Mitochondrial ADP/ATP carrier content and activity in homoallelic and heteroallelic yeasts. (A and C) Kinetic of [14C] ATP/ATP exchange in proteoliposomes reconstituted with mitochondrial extracts from the WB-12 strain (A) and from the W303-1B strain (C) transformed with the wt AAC2 (closed circle, closed square) or the aac2A137D mutant allele (open circle, open square). To proteoliposomes containing 20 mm ATP, 0.1 mm [14C] ATP was added. The exchange reactions were terminated at the indicated time intervals by adding 30 mm pyridoxal 5′-phosphate and 10 mm bathophenanthroline. Similar results were obtained in three independent experiments. (B and D) Mitochondria isolated from the WB-12 strain (2 µg) (B) and W303-1B strain (4 µg) (D) transformed with the indicated alleles were subjected to SDS–PAGE, transferred to nitrocellulose and immunodecorated with polyclonal antibodies against Aac2p.

Figure 6. Cell viability analysis. Microdissection of yeast cells showing the formation of colonies in WB-12 strain expressing the aac2A137D mutation. Cell viability was tested after growth of 15 generations in the absence (untreated) or in the presence of 30 µm dihydrolipoic acid (DHPLA treated).
Activities of the mitochondrial respiratory-chain complexes in patient's muscle homogenate
| Enzyme | Value | Range of controls |
|---|---|---|
| Complex I | 11 | 15–33 |
| Complex II | 16 | 15–28 |
| Complex III | 31 | 70–150 |
| Complex IV | 14 | 80–180 |
| Complex V | 67 | 75–200 |
| Citrate synthase | 418 | 80–210 |
| Enzyme | Value | Range of controls |
|---|---|---|
| Complex I | 11 | 15–33 |
| Complex II | 16 | 15–28 |
| Complex III | 31 | 70–150 |
| Complex IV | 14 | 80–180 |
| Complex V | 67 | 75–200 |
| Citrate synthase | 418 | 80–210 |
Activities of respiratory-chain complexes are normalized to the citrate synthase activity. The activity of citrate synthase (and the relevant range of controls) is given in nmol/min/mg of total homogenate protein.
Activities of the mitochondrial respiratory-chain complexes in patient's muscle homogenate
| Enzyme | Value | Range of controls |
|---|---|---|
| Complex I | 11 | 15–33 |
| Complex II | 16 | 15–28 |
| Complex III | 31 | 70–150 |
| Complex IV | 14 | 80–180 |
| Complex V | 67 | 75–200 |
| Citrate synthase | 418 | 80–210 |
| Enzyme | Value | Range of controls |
|---|---|---|
| Complex I | 11 | 15–33 |
| Complex II | 16 | 15–28 |
| Complex III | 31 | 70–150 |
| Complex IV | 14 | 80–180 |
| Complex V | 67 | 75–200 |
| Citrate synthase | 418 | 80–210 |
Activities of respiratory-chain complexes are normalized to the citrate synthase activity. The activity of citrate synthase (and the relevant range of controls) is given in nmol/min/mg of total homogenate protein.
References
Kaukonen, J., Juselius, J.K., Tiranti, V., Kyttälä, A., Zeviani, M., Comi, G.P., Keränen, S., Peltonen, L. and Suomalainen, A. (
Spelbrink, J.N., Li, F.Y., Tiranti, V., Nikali, K., Yuan, Q.P., Tariq, M., Wanrooij, S., Garrido, N., Comi, G., Morandi, L. et al. (
Van Goethem, G., Dermaut, B., Lofgren, A., Martin, J.J. and Van Broeckhoven, C. (
Lamantea, E., Tiranti, V., Bordoni, A., Toscano, A., Bono, F., Servidei, S., Papadimitriou, A., Spelbrink, H., Silvestri, L., Casari, G. et al. (
Napoli, L., Bordoni, A., Zeviani, M., Hadjigeorgiou, G.M., Sciacco, M., Tiranti, V., Terentiou, A., Moggio, M., Papadimitriou, A., Scarlato, G. et al. (
Siciliano, G., Tessa, A., Petrini, S., Mancuso, M., Bruno, C., Grieco, G.S., Malandrini, A., De Florio, L., Martini, B., Federico, A. et al. (
Deschauer, M., Hudson, G., Muller, T., Taylor, R.W., Chinnery, P.F. and Zierz, S. (
Komaki, H., Fukazawa, T., Houzen, H., Yoshida, K., Nonaka, I. and Goro, Y. (
Dolce, V., Scarcia, P., Iacopetta, D. and Palmieri, F. (
Klingenberg, M. (
Fiore, C., Trezeguet, V., Le Saux, A., Roux, P., Schwimmer, C., Dianoux, A.C., Noel, F., Lauquin, G.J., Brandolin, G., and Vignais, P.V. (
Bauer, M.K.A., Schubert, A., Rocks, O. and Grimm, S. (
Esposito, L.A., Melov, S., Panov, A., Cottrell, B.A. and Wallace, D.C. (
Graham, B.H., Waymire, K.G., Cottrell, B., Trounce, I.A., MacGregor, G.R. and Wallace, D.C. (
Fontanesi, F., Palmieri, L., Scarcia, P., Lodi, T., Donnini, C., Limongelli, A., Tiranti, V., Zeviani, M., Ferrero, I. and Viola, A.M. (
Kramer, R. and Klingenberg, M. (
Fiermonte, G., Walker, J.E. and Palmieri, F. (
Fiermonte, G., De Leonardis, F., Todisco, S., Palmieri, L., Lasorsa, F.M. and Palmieri, F. (
Bonneaud, N., Ozier-Kalogeropoulos, O., Li, G.Y., Labouesse, M., Minvielle-Sebastia, L. and Lacroute, F. (
Kovácova, V., Irmlerová, J. and Kovác, L. (
Bulder, C.J.E.A. (
Jordens, E.Z., Palmieri, L., Huizing, M., van den Heuvel, L.P., Sengers, R.C., Dorner, A., Ruitenbeek, W., Trijbels, F.J., Valsson, J., Sigfusson, G. et al. (
Chen, X.J. (
Pebay-Peyroula, E., Dahout-Gonzalez, C., Kahn, R., Trezeguet, V., Lauquin, G.J. and Brandolin, G. (
MacKenzie, K.R., Prestegard, J.H. and Engelman, D.M. (
Curran, A.R. and Engelman, D.M. (
Dyall, S.D., Agius, S.C., De Marcos Lousa, C., Trezeguet, V. and Tokatlidis, K. (
Schroers, A., Burkovski, A., Wohlrab, H. and Kramer, R. (
Dujon, B. (
Marzo, I., Brenner, C., Zamzami, N., Jurgensmeier, J.M., Susin, S.A., Vieira, H.L., Prevost, M.C., Xie, Z., Matsuyama, S., Reed, J.C. and Kroemer, G. (
McKenzie, M., Liolitsa, D. and Hanna, M.G. (
Doudican, N.A., Song, B., Shadel, G.S. and Doetsch, P.W. (
Fagiolari, G., Sciacco, M., Chiveri, L., Lamperti, C., Comi, G.P., Scarlato, G., Moggio, M. and Prelle, A. (
Tiranti, V., Galimberti, C., Nijtmans, L., Bovolenta, S., Perini, M.P. and Zeviani, M. (
Ferrari, G., Lamantea, E., Donati, A., Filosto, M., Briem, E., Carrara, F., Parini, R., Simonati, A., Santer, R. and Zeviani, M. (
Hashimoto, M., Shinohara, Y., Majima, E., Hatanaka, T., Yamazaki, N. and Terada, H. (
Ogur, M., St. John, R. and Nagai, S. (

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}