



Abstract
The neurofibromatosis 2 (NF2) tumor suppressor protein, schwannomin or merlin, is commonly lost upon NF2 gene mutation in benign human brain tumors. We identified the p110 subunit of the eukaryotic initiation factor 3 (eIF3c) as a schwannomin interacting protein. The eIF3 complex consists of ∼10 subunits whose functions are only recently becoming known. Interaction between schwannomin and eIF3c suggests a role for schwannomin in eIF3c-mediated regulation of proliferation related to changes in protein translation. We found that schwannomin was most effective for inhibiting cellular proliferation when eIF3c was highly expressed. When we examined these proteins in 14 meningiomas, we observed high eIF3c abundance in those that had lost schwannomin expression but low eIF3c abundance in those retaining schwannomin. Consequently, eIF3c appears to be involved in NF2 pathogenesis and deserves to be investigated as a prognostic marker for NF2 and target for treatment of NF2 patient tumors.
INTRODUCTION
The majority of sporadic benign nervous system tumors and the inherited tumor disorder neurofibromatosis 2 (NF2) are caused by mutations in the NF2 gene and loss of the gene's protein product schwannomin (1,2). Schwannomin is a tumor suppressor of the ezrin radixin moesin (ERM) protein family involved in cytoskeletal organization with respect to the plasma membrane (3). Knockout and overexpression studies have shown that schwannomin inhibits proliferation of many cell types both in culture and in mouse models (4). However, despite efforts that have characterized several schwannomin functions, a pathway associating schwannomin to human tumor pathogenesis has not been demonstrated. Incomplete knowledge on schwannomin function continues to impede the development of therapies for NF2 patient tumors.
Schwannomin overexpression elicits changes in cellular proliferation and adhesion associated with cytoskeletal remodeling. Consistent with schwannomin's homology to ERM family proteins, schwannomin co-localizes with actin at the ruffling edge of the plasma membrane (5,6). Overexpression of schwannomin in NIH-3T3 cells and RT4-D6P2T schwannoma cells was associated with decreased proliferation (7,8) and impairment of actin cytoskeletal-mediated functions including cell spreading, attachment and motility (9). Schwannomin removal by transfection of NF2 antisense DNA reorganizes the actin cytoskeleton in STS26T Schwann-like cells, decreasing adhesion and increasing proliferation (10,11). Changes in cell adhesion may also be mediated by schwannomin regulation of adherens junction proteins including the cadherins (12) and the EBP50-interacting protein erbin (13). Schwannomin function may also relate to its phosphorylation status. Schwannomin action on proliferation and cytoskeletal remodeling is associated with Rho and Rac in schwannoma cells (14) and schwannomin is an inhibitor of Rac signaling, and Rac activation leads to schwannomin phosphorylation and inactivation (15).
Other schwannomin functions were defined by the identification of interacting proteins. The ezrin interacting proteins CD44 and EBP50/NHE-RF interact with schwannomin at the FERM domain (residues 1–305), where ERM proteins retain the greatest homology with schwannomin (16,17). Regulation of the actin cytoskeleton by schwannomin could be mediated by a direct interaction between schwannomin and actin or through the schwannomin interaction with βII-spectrin (10) and N-wasp (18). Pathogenic changes upon NF2 mutations altering cell signaling and adhesion may be associated with a complex of proteins including schwannomin and its interacting partner paxillin, as well as β-integrin and ErbB2 (19). Schwannomin action on proliferation could also be associated with its interaction with HRS (20,21). HRS traffics EGFR to the lysosome of STS26T cells, and both schwannomin and HRS inhibit signal pathways activated by EGF (22). Schwannomin also regulates proliferation through a direct interaction with the phosphatidylinositol 3-kinase effector PIKE-L (23).
We now report that schwannomin interacts with the eukaryotic initiation factor 3 (eIF3) p110 subunit (eIF3c). We demonstrated that eIF3c and schwannomin interact directly and that the FERM domain of schwannomin interacts with the C-terminal half of eIF3c. We also used various methods to show that schwannomin and eIF3c interact in vivo in cultured cell lines. We showed that increases in eIF3c expression elevate cell proliferation and that schwannomin is effective at inhibiting cellular proliferation when eIF3c abundances are at their highest. Meningiomas lacking schwannomin demonstrated higher eIF3c expression than those retaining schwannomin. Therefore, eIF3c appears to be important for mediating the pathogenesis of NF2 patient tumors.
RESULTS
eIF3c interacts with schwannomin
We used the yeast two-hybrid system (24) to identify proteins that interact with schwannomin. Previously, we demonstrated that schwannomin isoform 1 was highly expressed in human lymphocytes when compared with schwannomin isoform 2 (25). Upon screening an adult human lymphocyte cDNA library with full-length schwannomin isoform 1, we identified a plasmid encoding the eIF3c protein. This plasmid, pACT–eIF3c (68–635), included a cDNA insert of 1705 bp, from base pairs 202–1906 of the EIF3S8 gene, which encoded residues 68–635 of the 914 residues of the eIF3c protein. The screen was of moderate efficiency in which 6.1×105 colonies were screened yielding eight positives. After re-transformation of single plasmids, the number of positives was reduced to one clone, pACT–eIF3c (68–635).
Regions of interactions in schwannomin and eIF3c
We used multiple yeast two-hybrid methods to validate eIF3c as a schwannomin interacting protein and to narrow the interacting regions. Yeast two-hybrid assays showed that the affinity for eIF3c by schwannomin isoforms 1 and 2 was the same (Fig. 1A). eIF3c interacted with the FERM domain of schwannomin (residues 1–305), whereas a C-terminal domain schwannomin fragment including schwannomin isoform 1 residues 256–595 did not interact with eIF3c (Fig. 1A). To narrow the schwannomin-binding region in eIF3c, we tested schwannomin isoforms 1 and 2 and the schwannomin FERM domain for interaction with various eIF3c partial proteins. Yeast two-hybrid tests of interaction demonstrated that schwannomin binds eIF3c within amino acids 405–635 (Fig. 1B). Using a non-translation-based Ras rescue yeast two-hybrid method, we verified the interaction between the schwannomin FERM domain and the full-length eIF3c protein (Fig. 1C).
Schwannomin and eIF3c interact directly
To demonstrate that schwannomin and eIF3c interact directly and to confirm the schwannomin interacting region within eIF3c as demonstrated by the yeast two-hybrid tests, we tested eIF3c and schwannomin interaction in vitro. An eIF3c partial protein including residues 68–635 interacted with schwannomin, whereas one including only residues 68–445 did not. Another eIF3c fragment of residues 257–913 also interacted with schwannomin (Fig. 2). These data suggest that schwannomin binding by eIF3c requires eIF3c residues 446–635 and are consistent with the region of interaction determined by yeast two-hybrid interaction tests of eIF3c residues 405–635. These data demonstrate that the minimal region for schwannomin interaction within eIF3c is within eIF3c residues 405–635. However, in vitro binding assays showed that the eIF3c fragment including eIF3c residues 257–913 interacted stronger than the 68–635 residue fragment, suggesting that residues downstream of amino acid 635 may stabilize the interaction. This assay was replicated independently.
eIF3c and schwannomin co-immunoprecipitate
To demonstrate that eIF3c and schwannomin interact in vivo, we conducted co-immunoprecipitation experiments. We generated a new peptide antibody against eIF3c C-terminally located residues 880–894. This antibody, designated ab2867.2, detected a single 110 kDa band in STS26T extracts, which was not detected when the antibody was pre-absorbed with the peptide antigen (Fig. 3A). We also demonstrated that this antibody strongly detected exogenous myc-eIF3c in STS26T cells (Fig. 3B). We immunoprecipitated endogenous schwannomin in STS26T cells with anti-eIF3c ab2867.2 and observed a single band of the correct size for schwannomin detected with chicken anti-schwannomin ab2781 (Fig. 3C). In reciprocal experiments, we immunoprecipitated eIF3c with two different schwannomin antibodies in Tet-off STS26T NF2i1 cells induced to express schwannomin isoform 1 (Fig. 3D and E). Of these two, the anti-schwannomin antibody A-19 recognizes an N-terminal epitope present in both of the schwannomin isoforms, whereas antibody C-18 recognizes a C-terminal epitope present only in schwannomin isoform 1.
Endogenous schwannomin and eIF3c co-localize in STS26T cells
STS26T schwannoma cells were co-labeled with chicken anti-schwannomin and rabbit anti-eIF3c antibodies. Confocal microscopy demonstrated that schwannomin and eIF3c co-localized at perinuclear and cytoplasmic puncta. Some co-localization of both proteins was also observed at membrane structures (Fig. 4A). To further validate that schwannomin and eIF3c interact in STS26T cells, we developed a strategy for fluorescence resonance energy transfer (FRET), whereby we transfected STS26T cells with myc-eIF3c and labeled cells with anti-myc-FITC and anti-schwannomin A-19, with secondary labeling with anti-rabbit-TRITC. A strong FRET signal was observed following photobleaching of the TRITC acceptor fluorophore (Fig. 4B). A FRET efficiency map demonstrated the strongest FRET signal at perinuclear and cytoplasmic puncta (Fig. 4B). A control study conducted in the same manner but replacing myc-eIF3c with the irrelevant protein myc-PTEN showed no FRET signal, demonstrating that the FRET observed with myc-eIF3c was due to interaction between eIF3c and schwannomin and not due to non-specific antibody labeling (Fig. 4C).
Schwannomin inhibits eIF3c-mediated proliferation
We generated a new STS26T cell line that was inducible for myc-eIF3c. Myc-eIF3c was overexpressed in myc-eIF3c inducible Tet-off STS26T cells when doxycycline was removed, with minor myc-eIF3c leaky expression in the presence of doxycycline (Fig. 5A). Using this cell line, we showed that transfection of a plasmid expressing full-length schwannomin significantly reduced cell proliferation mediated by myc-eIF3c expression (Fig. 5B). The data in Figure 5B represent pooled data from two independent experiments conducted 5 months apart. Two-way ANOVA showed that the dox effect on cell abundance was not the same for all doses (P<0.0001) and that the curves were significantly different (P<0.0001). The effect of schwannomin on cell proliferation was most potent when myc-eIF3c was expressed at the highest levels (lowest dox concentrations). Regression analysis of only the first four data points in Figure 5B (dox concentrations 0, 9.4, 18.7, 37.5 ng/ml) showed that the slope of the line was not significantly different from zero for the NF2-transfected cells (P=0.958) but not for the vector-transfected cells (P<0.05) and that the slopes of the two lines were significantly different from one another (P<0.05). We then conducted the proliferation assay once more with a deletion construct of schwannomin lacking the eIF3c binding site (NF2C) (Fig. 5C). Two-way ANOVA showed that the curves were significantly different (P<0.0001). For NF2C, we expected to find a pattern of proliferation like for the vector control, which indeed we did for the extreme high and low myc-eIF3c expression conditions as demonstrated by multiple pairwise t-testing. All pairwise t-tests were significantly different (P<0.001), except for the two highest dox doses in Figure 5B and C (1 and 2 µg/ml) and the lowest dose in Figure 5C (0 µg/ml). Bonferroni post-test corrections were applied. However, cells transfected with NF2C treated with intermediate dox doses had elevated proliferations compared with the vector control, which might relate to a dominant negative action of the schwannomin fragment on endogenous schwannomin. We find this result of particular interest because we were unable to show that schwannomin overexpression altered proliferation of STS26T cells in several past but unrelated experiments using the MTT method. The ability for schwannomin to inhibit proliferation may be cell line-specific depending on the expression of other proteins including eIF3c.
Schwannomin and eIF3c expression is inversely correlated in meningiomas
To understand the significance of the relationship between eIF3c and schwannomin, we conducted immunoblotting with proteins from human meningiomas (Fig. 6). Of 14 meningiomas, eight (nos 2, 5–7, 9, 10, 12, 13) had high eIF3c but low schwannomin abundances, whereas four (nos 1, 3, 8, 11) had high schwannomin but low eIF3c abundances. One meningioma (no. 14) had loss of both and one (no. 4) had relatively high expression of both.
DISCUSSION
To identify the proteins interacting with schwannomin, we conducted yeast two-hybrid screening. We identified the eIF3 subunit eIF3c (eIF3c) as a protein that binds schwannomin and validated the interaction using multiple methods. eIF3c is a component of the eIF3 complex needed for initiating protein translation. eIF3 is required for the 40S ribosomal subunit bound to the ternary complex (eIF2–GTP–methionine) to interact with the 5′ end of the mRNA and is also needed to maintain the 40S ribosomal subunit in the dissociated state (26). We investigated the ability of schwannomin to reduce proliferation in the presence or absence of eIF3c and found that schwannomin was most potent at the highest levels of eIF3c expression. This result suggests that schwannomin may inhibit proliferation through an interaction with eIF3c and has implications to tumorigenesis.
The interaction between schwannomin and eIF3c involves the FERM domain of schwannomin (within residues 1–304), which is the domain that has the greatest homology with other proteins in the ERM family. We validated that the schwannomin FERM domain interacted with eIF3c by both GAL4- and Ras rescue-based yeast two-hybrid methods.
Both schwannomin isoforms interacted with eIF3c with similar binding affinity as determined using semi-quantitative yeast two-hybrid binding tests. Alternative splicing at the C-terminal end of schwannomin results in folding in schwannomin isoform 1, whereas schwannomin isoform 2 is thought to exist in an unfolded conformation (27). Consequently, alternative splicing between the two isoforms of schwannomin could potentially alter schwannomin affinity for proteins that bind the FERM domain as was demonstrated for the schwannomin interacting protein NHE-RF (16). However, we observed no differences in eIF3c binding between the two schwannomin isoforms suggesting that eIF3c can bind schwannomin both in its folded and unfolded conformations.
We determined that schwannomin binds eIF3c directly by using in vitro binding assays and compared the regions of interaction in yeast two-hybrid assays. In our original yeast two-hybrid screen, we obtained a fragment of eIF3c including eIF3c residues 68–635. By secondary yeast two-hybrid testing, we narrowed the interacting region to eIF3c residues 405–635. In our in vitro binding assays, we also showed that two eIF3c fragments encompassing this region interacted with schwannomin, including eIF3c fragments with residues 68–635 and 257–913. The 257–913 amino acid fragment interacted with schwannomin more efficiently than did the 68–635 amino acid fragment, suggesting that regions downstream of residue 635 may participate in or stabilize the schwannomin interaction. On the basis of both the yeast two-hybrid and the in vitro binding methods, we concluded that a minimal region for schwannomin binding resides within eIF3c residues 405–635.
We demonstrated that schwannomin and eIF3c interacted in vivo in cultured STS26T schwannoma cells by co-immunoprecipitation and confocal microscopy. Immunoprecipitation of endogenous eIF3c with endogenous schwannomin was accomplished from STS26T protein extracts. Additional immunoprecipitations were successful using two commercially available NF2 antibodies that interact with schwannomin N- and C-terminal epitopes resulting in co-immunoprecipitations of eIF3c from schwannomin inducible Tet-off STS26T cells induced to express schwannomin. Further evidence that endogenous eIF3c and endogenous schwannomin interact in vivo was obtained by confocal immunofluorescent microscopy of STS26T cells labeled with eIF3c and schwannomin antibodies. Schwannomin and eIF3c co-localized to punctate structures in the cytoplasm and at some membranous structures. FRET microscopy with a strategy using exogenously expressed myc-tagged eIF3c demonstrated that the strongest co-localization was in cytoplasmic puncta. Additional work will be required to determine whether these structures co-localize with other proteins of the translation machinery.
Because some eIF proteins including eIF3c are overexpressed in various tumors (discussed subsequently), we hypothesized that schwannomin exerts its tumor suppressor function by way of inhibiting eIF3c-mediated proliferation. To investigate this hypothesis, we established a new inducible eIF3c STS26T schwannoma cell line model system and tested proliferation mediated by regulated eIF3c expression both in the presence and absence of exogenous schwannomin and a schwannomin fragment lacking the eIF3c binding site. Expression of eIF3c in this system highly elevated proliferation. Further increases in proliferation were eliminated entirely by the expression of schwannomin at the four highest tested doses of eIF3c (Fig. 5B). Transfection of a schwannomin C-terminal fragment lacking the eIF3c binding site demonstrated no effect on reducing eIF3c-mediated proliferation (Fig. 5C).
Because schwannomin is lost by NF2 mutation in ∼50% of meningiomas (28,29), we used meningiomas to assess whether eIF3c expression might correlate to schwannomin expression. Surprisingly, we observed an inverse relationship in which eIF3c was expressed more strongly when schwannomin was lost, whereas tumors that retained schwannomin had relatively little eIF3c (Fig. 6). On the basis of our proliferation assays, we predict that loss of eIF3c regulation in tumors lacking schwannomin accounts for pathogenesis in these tumors, whereas eIF3c does not account for the pathogenesis of tumors retaining schwannomin.
The identification of schwannomin interaction with eIF3c adds to other findings that suggested a role of schwannomin in protein translation. Recently, schwannomin was shown to interact with and inhibit proliferation mediated by the trans-activation response RNA-binding protein (TRBP) (30). TRBP interacts with ribosomes, is an activator of protein translation and may function as an oncoprotein (31,32). TRBP blocks autophosphorylation of the double-strand RNA-dependent protein kinase (PKR). PKR activation causes eIF2α phosphorylation, thereby inhibiting protein translation (33). These findings suggest a model for schwannomin action in protein translation (Fig. 7). This model proposes that schwannomin inhibits proliferation by blocking the actions of both eIF3c and TRBP, which stimulate protein translation, and that eIF3c and TRBP are unregulated when schwannomin is lost by NF2 mutation.
Findings that other eIF3 subunits are overexpressed or amplified in various tumor types support that eIF3c may be significant to NF2 pathogenesis. The gene for eIF3-p40 is amplified in some breast and prostate cancers and p40 amplification reduced survival in prostate cancer patients (34,35). The largest eIF3 subunit (known as p150, p170 or p180) is amplified in breast, cervical, esophageal, stomach and lung cancers (36,37). Overexpression of eIF3-p48, an oncoprotein that interacts with eIF3c, causes malignant transformation in NIH-3T3 cells (38–41). In addition, eIF2α, which is regulated by TRBP, is overexpressed in some benign and malignant neoplasms of melanocytes and colonic epithelium (42,43).
eIF3c has also been directly implicated in tumorigenesis. Overexpression of the eIF3c protein has been associated with testicular germ cell tumors (seminomas) (44). The eIF3c gene EIF3S8 is located on chromosome 16p11.2 within an unstable region of the genome, and intact duplication of the entire EIF3S8 gene has been observed (45). Our demonstrations that schwannomin interacts with eIF3c, that schwannomin inhibits eIF3c-mediated proliferation and that eIF3c is overexpressed in meningiomas lacking schwannomin implicate eIF3c as a mediator of NF2 tumorigenesis. Our findings are consistent with schwannomin as a regulator of eIF3c-mediated protein translation and proliferation. Altered regulation of translation has also been described for the genes causing tuberous sclerosis complex (TSC), another disease in the group of the phakomatoses. The TSC1 and TSC2 proteins hamartin and tuberin interact and are jointly associated with eIF4e regulation by mTOR (46). Further analysis of eIF3c as a prognostic marker for NF2 and as a target for therapy of NF2 patient tumors is highly merited.
MATERIALS AND METHODS
Yeast two-hybrid screening
A yeast two-hybrid screen (24) of a human adult lymphocyte cDNA library cloned in GAL4 activation domain vector pACT was accomplished using pGBT9–NF2i1, encoding schwannomin isoform 1 fused to the GAL4-binding domain as previously described (10,20). We identified a plasmid containing a cDNA encoding eIF3c residues 68–635. The screen was conducted with yeast strain Y190 in which double-transformants were grown on SC media with leucine, tryptophane and histidine dropped out and with 25 mm 3-amino-1,2,4-triazole and 2% glucose. β-Galactosidase production was assayed by incubating freeze-fractured colonies on nitrocellulose in Z-buffer (60 mm Na2HPO4, 40 mm NaH2PO4, 10 mm KCl, 1 mm MgSO4, pH 7.0, 0.03 mm β-mercaptoethanol and 2.5 µm X-gal) at 37°C for 15 min to 8 h. Liquid assays for β-galactosidase were conducted by incubating yeast extracted in Z-buffer and 5% chloroform with 0.6 mg/ml o-nitrophenylgalactoside for 2 min to 1 h. β-Galactosidase units=1000×[OD420/(OD600×time×volume)].
Cloning of full-length eIF3c
A human adult brain lambda phage library was screened for sequences hybridizing with a 32P-labeled eIF3c probe made by amplification of a 1462 bp fragment of eIF3c with primers NF2BND2A1 (5′-CTGGCTGACCTAGAGGACTAT-3′) and NF2BND2B1 (5′-TGTCCTGCAAGTGGCTCATGA-3′). Plaque lifts were hybridized in 50 mm PIPES buffer with 50% deionized formamide and 0.5% sodium dodecyl sulfate (SDS). Filters were washed in 2×SSC, 0.1% SDS at 65°C, then in 0.2×SSC, 0.1% SDS at 65°C and autoradiography was performed. Plaques were replated and rescreened two additional times. Following the tertiary screen phage, DNAs were cloned and sequenced yielding four cDNAs containing eIF3c base pairs 27–2591 (type 1), 164–2813+ (type 2), 158–2837 (type 3) and 1004–2813+ (type 4). A pBluescript KS(−) construct containing eIF3c base pair 27–2924 was made by ligating fragments from the type 1 and type 3 constructs together at BspE1. The N-terminal end of the eIF3c gene was obtained by amplification using a forward vector primer (T7) and the reverse eIF3c primer CLCL2ECOB.6, (5′-AGGCGAATTCTGACATCACGAATCTTCA-3′) using lambda phage library as template. Once the 5′ sequence of the EIF3S8 gene was determined, refinement of the N-terminal fragment was accomplished by amplification with primers CL2XHOA.0 (5′-CGTACTCGAGCCGCGCCGTCGCCATGTC-3′) and CLCL2ECOB.6, and the fragment was ligated between the vector XhoI and eIF3c BspE1 sites completing the EIF3S8 gene sequence. This construct was validated by sequence analysis and named pBluescriptKS(−)–eIF3c.
Plasmid constructs
The primers EIF3CAA68A (5′-CCGGGAATTCCGTAATGCCATGAAGATTC-3′), EIF3CAA300B (5′-CCGGGAATTCTCCCACTCCCCGCCTTCAT-3′), EIF3CAA405A (5′-GAAGGAATTCGACTGCATCAATGAGCTGA-3′) and EIF3CAA635B (5′-CTCGGAATTCTGTCCAGCAGGGCGTTGTGT-3′) were used to prepare inserts for ligating into the EcoRI site of pGAD10 resulting in the constructs pGAD10–eIF3c (405–635) (EIF3CAA405A and EIF3CAA635B), pGAD10–eIF3c (68–300) (EIF3CAA68A and EIF3CAA300B). The construct pGAD10–eIF3c (68–635) was constructed by excising the eIF3c (68–635) insert from pACT–eIF3c (68–635) with XhoI and ligating it into the XhoI site of pGAD10. The constructs pGBT9–NF2i1, pGBT9–NF2i2, pGBT9–NF2N (1–304), pGBT9–NF2i2 (256–590), pcDNA3.1his–NF2 and pcDNA3.1his–NF2C (encoding schwannomin residues 304–595) were previously described (10,20,21). The primers EIF3CXHO1 (5′-AGCCCTCGAGATGTCGCGGTTTTTCACCACC-3′), EIF3CHIN2 (5′-CTCGAAAGCTTGTCCAGCAGGGCGTTGTGT-3′), EIF3CHIN3 (5′-GTTCAAGCTTAGTTAGGATGCAGCCACGGA-3′), EIF3CHIN4 (5′-AGTGAAGCTTTCAGTAGGCCGTCTGAGACT-3′), EIF3CXHO6 (5′-CAAGCTCGAGATGGTCACCAAGTGCCTGGAAGA-3′) and EIF3CXHO7 (5′-CACCCTCGAGATGGACAAGAAGGCAGCCGAGAA-3′) were used to prepare inserts for cloning into the XhoI and HindIII sites of pBluescriptKS(−) (Stratagene) resulting in the constructs pBluescriptKS(−)– eIF3c (1–445) (EIF3CXHO1 and EIF3CHIN3), pBluescriptKS(−)–eIF3c(68-635) (EIF3CXHO6 and EIF3CHIN2), pBluescriptKS(−)–eIF3c (68–445) (EIF3CXHO6 and EIF3CHIN2), pBluescriptKS(−)–eIF3c (257–635) (EIF3CXHO7 and EIF3CHIN2), pBluescriptKS(−)–eIF3c (257–913) (EIF3CXHO7 and EIF3CHIN4) and pBluescriptKS(−)–eIF3c (257–445) (EIF3CXHO7 and EIF3CHIN3). The construct pSOS–NF2N (1–304) was generated by excising the NF2N (1–304) insert from pGBT9–NF2N (10) with EcoRI and treating with T4 polymerase (Promega) to fill in the sticky ends of the restriction site and blunt-end ligated into pSOS (Stratagene) prepared by digesting with SalI followed by treating with T4 polymerase to fill in the sticky ends of the restriction site. The primers EIF3CMYRECOA (5′-CCGGGAATTCCGTAATGCCATGAAGATTCG-3′) and EIF3CMYRXHOB (5′-G CGACTCGAGGTCCAGCAGGGCGTTGTGT-3′) were used to prepare a full-length eIF3c insert for cloning into the EcoRI and XhoI sites of pMyr (Stratagene) resulting in the pMyr–eIF3c construct. The construct pRSET–NF2 was prepared by excising the insert from pcDNA3.1hisb–NF2 (20) with XhoI and SalI and ligating into the XhoI site of pRSET. The primers EIF3CECOA (5′-AGCTGAATTCGATCGCGGTTTTTCACCACCGGTTC-3′) and EIF3CXHOB (5′-CGATCTCGAGTCAGTAGGCCGTCTGAGACTGC-3′) were used to prepare a full-length eIF3c insert for cloning into the EcoRI and XhoI sites of pCMV-Myc (Clontech) resulting in the pCMV-myc–eIF3c construct. To prepare pRevTRE-myc–eIF3c, we cut out the myc-eIF3c insert from pCMV-myc–eIF3c with HpaI and ScaI, then ligated it to pRevTRE (Invitrogen) that was cut with HpaI. For all inserts that were prepared by PCR, amplification was accomplished using the Expand High Fidelity PCR System (1732641, Roche). For amplifications of eIF3c inserts, the template was pBluescriptKS(−)–eIF3c (Cloning of Full-Length eIF3c), and for amplifications of NF2 inserts, the template was pGBT9–NF2 (10). Each of the constructs was validated by sequencing.
Ras rescue yeast two-hybrid tests of interaction
Interactions were tested between the full-length eIF3c protein and the schwannomin FERM domain fragment using the Ras rescue yeast two-hybrid method using the CytoTrap Two-Hybrid System (Invitrogen). The temperature-sensitive cdc25 mutant yeast strain cdc25H was co-transformed with pSOS and pMyr constructs. Fusion proteins encoded by pMyr are under the control of the Gal1 promoter and are glucose repressible, galactose inducible. Replica stamped plates of established double transformants were grown on SD/glucose plates (synthetic dropout media with glucose, lacking uracil and leucine for plasmid selection) and SD/galactose plates (synthetic dropout media with galactose, raffinose, lacking uracil and leucine) and grown at 25°C and 37°C. Positive interactions are indicated by growth at 37°C but lack of growth at 25°C on galactose.
Purification of recombinant proteins
The NF2 full-length gene or gene fragments ligated in pRSET were transformed in DH5α and his(6x)-fusion proteins were grown in LB media overnight at 37°C. The next morning 10 ml of the culture was inoculated into 500 ml fresh media and shaken at room temperature. When the culture reached an optical density of 0.8, 0.05 mm IPTG was added and the culture was left to shake at room temperature overnight. His(6x)-fusion proteins were purified by affinity chromatography using Ni-NTA agarose following the manufacturer's protocol (Qiagen). Maltose-binding protein expressed using the vector pMAL-c2 was induced in DH5α and purified using amylose resin as previously described (10). Before use in in vitro binding assays or in vitro translation assays, proteins were dialyzed against PBS.
Antibodies
Polyclonal rabbit anti-eIF3c antibody ab2867.2 was raised against eIF3c residues 880–894 (TYGGYFRDQKDGYRK) and affinity purified. Affinity purified polyclonal chicken anti-schwannomin antibody 2781 was raised against schwannomin residues 528–541 (YMEKSKHLQEQLNE) and was described previously (21). NF2 antibodies A-19 and C-18 were purchased from Santa Cruz Immunochemicals. The myc-epitope tag encoded by pCMV-myc was detected immunofluorescently with anti-myc-FITC antibody (OP10F, Oncogene), or on immunoblots with anti-myc-HRP (Invitrogen). All secondary antibodies were from Jackson ImmunoResearch Laboratories.
Cell lines
STS26T schwannoma cells were derived from a human malignant schwannoma (47). Establishment of schwannomin inducible Tet-off STS26T cells and parental Tet-off STS26T cells was previously described (21). Myc-eIF3c inducible Tet-off STS26T cells were generated by using the RevTet-Off vector system (Clontech) as described (21). Briefly, Tet-off STS26T cells were infected by retroviruses made from the PT67 packaging cell line stably transfected with RevTRE–myc-eIF3c, and infected cells were placed under hygromycin B selection. Twenty-four colonies of myc-eIF3c inducible Tet-off STS26T cells were collected by ring cloning and tested for doxycycline inducible myc-eIF3c expression by immunoblotting with anti-myc-HRP antibody.
In vitro tests of interaction
eIF3Cc fragments ligated in pBluescript KS(−) were translated in vitro in the presence of [35S]methionine using the TnT kit (Promega). Input eIF3c proteins were first normalized by assessing band intensities on autoradiographs. Purified his(6x)-schwannomin immobilized on Ni-NTA agarose was incubated with equal amounts of eIF3c proteins for 2 h at 4°C in binding buffer (50 mm NaH2PO4, 300 mm NaCl, 10 mm imidazol, pH 8). After washing complexes in binding buffer with 0.05% Tween-20, radioactive eIF3c proteins binding the immobilized his(6x)-schwannomin were analyzed by autoradiography (output).
Co-immunoprecipitation of eIF3c and schwannomin
Either STS26T or Tet-off MEF NF2i1 cells were grown in Dulbecco's Modified Eagle's medium (DMEM) with 10% fetal bovine serum (FBS) in the presence of penicillin and streptomycin. Schwannomin in Tet-off MEF NF2i1 cells was induced for 24 h with 1 µg/ml doxycycline. Cells were homogenized in CoIP buffer (50 mm Tris pH 7.5, 150 mm NaCl, 1% Tween-20, 0.05% deoxycholic acid, 0.05% SDS, 0.05% sodium azide) containing a protease inhibitor cocktail (1836135, Roche). Cells were suspended in a glass homogenizer with 10 passes and lysates were centrifuged at 4°C for 25 min at 10 000 g. Lysates were pre-cleared with a 50:50 mixture of rabbit serum agarose (R-6755, Sigma) and rabbit IgG agarose (A-2909, Sigma) (300 µl total resin pellet per 10 ml lysate) with rotation at 4°C for 1 h. Immunoprecipitation was accomplished by adding 0.5–1.5 µg/ml antibody (either ab2867.2, A-19, C-18 or control serum) to 500 µl lysate and rotating overnight at 4°C. Antibody–protein complexes were collected on protein A-agarose (P2545, Sigma) by adding 30 µl of the washed resin and rotating for 1 h at 25°C. Immune complexes were washed four times in CoIP buffer and eluted by boiling 5 min in 50 µl of 2×SDS–PAGE buffer (50 mm Tris–HCl, pH 6.8, 20% glycerol, 2% SDS, 50 mm β-mercaptoethanol, 0.1% bromophenol blue). Proteins were analyzed by immunoblotting with separation on 4–15% gradient Tris–glycine Ready Gels (Bio-Rad).
Immunofluorescent microscopy and FRET analysis
STS26T cells were plated on polylysine-coated glass cover slips in DMEM with 10% FBS. Cells were labeled for immunofluorescence as previously described (10,21). Cells were fixed in 4% paraformaldehyde for 10 min, washed, then blocked with 3% donkey serum. The primary antibody dilution was 10 µg/ml for chicken anti-schwannomin ab2781 or rabbit anti-eIF3c ab2867.2. Primary antibodies were incubated for 60 min at 37°C. Following three washes with cold DPBS, cells were incubated with a 1:2000 dilution of rhodamine-conjugated affinity purified donkey anti-rabbit IgG or fluorescene-conjugated affinity purified donkey anti-chicken IgG (Jackson ImmunoResearch Laboratories) for 1 h at room temperature, washed six times in cold DPBS and mounted using Vectashield mounting media (Vector Laboratories). When exogenous myc-eIF3c was detected, transfection was accomplished using Superfect Transfection Reagent (Qiagen) and myc-eIF3c was detected with a 1:2000 dilution of anti-myc-FITC antibody (Oncogene). Fluorescent microscopy was performed using a Leica TCS SP confocal microscope with a Leica Plan-APO 100×/1.40 objective. Fluorescein was excited with a ArKr laser at 488 nm with emission set to a range of 503–562 nm. Rhodamine was excited with an ArKr laser at 568 nm with emission set to a range of 585–648 nm. Confocal FRET images were obtained by the method of acceptor depletion (adFRET) with irreversible acceptor photobleaching (48).
Proliferation assays
Myc-eIF3c inducible Tet-off STS26T cells were transfected with pcDNA3.1his, pcDNA3.1his-NF2 or pcDNA3.1his-NF2C. The next day, cells were removed with trypsin and distributed uniformly among the wells of a 96-well plate. An equal volume of media containing 2× doxycycline was added such that columns of eight wells had the following final concentrations of doxycycline: 0, 9.4, 18.7, 37.5, 75, 125, 250, 500, 1000, 2000 ng/ml. Cells were cultured at these conditions for 4 days and cell abundances were analyzed using the CellTiter 96® Non-Radioactive Cell Proliferation Assay Kit (Promega) following the vendor's protocol. Formazan production from MTT tetrazolium substrate was determined by measuring the absorbance at 570 nm. Values were converted to a percentage of the highest absorbance.
ACKNOWLEDGEMENTS
We thank the following contributors to the study: Duong Huynh, Pattie Figueroa, Carlos Barraza, Mercy Chen, Eric Coulsell, Daniel A. Gorelick-Feldman, Diane Ho, Sam Lam and Ping Mi. Support was provided by grant DAMD17-00-1-0553 from the Department of Defense and grant no. 220020049 from the James S. McDonnell Foundation to D.R.S. Support was also provided by the Carmen and Louis Warschaw Endowment Fund and grants RO1NS037883 and KO8NS001428 from the National Institutes of Health to S.M.P.
Conflict of Interest statement. None declared.
Present address: Women's Cancer Research Institute, Cedars-Sinai Medical Center, Room D-1089, 8700 Beverly Boulevard, Los Angeles, CA 90048, USA.
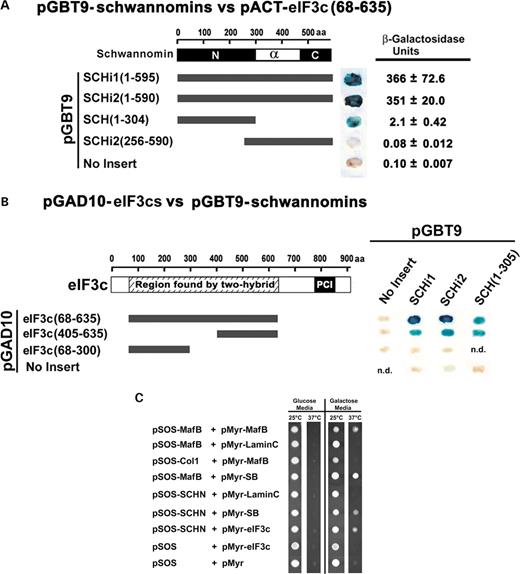
Figure 1. Yeast two-hybrid tests of interaction. (A) An eIF3c fragment of amino acids 68–635 encoded using the vector pACT was tested for interaction with the indicated schwannomin proteins encoded by pGBT9 demonstrating that eIF3c binds the N-terminal half of schwannomin, between schwannomin residues 1–305. Blue color indicates the presence of β-galactosidase and a positive test of interaction between encoded proteins in a plate assay. β-Galactosidase units are from the liquid assay of binding strength. No data (n.d.). (B) The eIF3c fragments indicated in the drawing encoded using the vector pGAD10 were tested for interaction with the indicated schwannomin proteins encoded by pGBT9. These tests narrowed the interacting region to within eIF3c amino acids 405–635. (C) Full-length eIF3c was tested for interaction with schwannomin amino acids 1–305 (SCHN) using the Ras rescue yeast two-hybrid method. Proteins encoded by pMyr are fused to a myristylation signal and are targeted to the membrane. Interactions that are accomplished by myristylated fusion proteins with the pSOS encoded bait proteins fused to SOS result in SOS being positioned at the membrane and the rescue of Ras signaling in the temperature-sensitive cdc25H yeast strain. The test was positive for schwannomin and eIF3c and for the positive controls MafB versus MafB, MafB versus SOS binding protein (SB) and schwannomin versus SB. All other tests were negative controls, including irrelevant control proteins murine type IV collagenase (ColI) and human lamin C (amino acids 67–230). Note that proteins encoded by pMyr are glucose repressible and galactose inducible. Not shown are the total of eight replicates for each interaction.
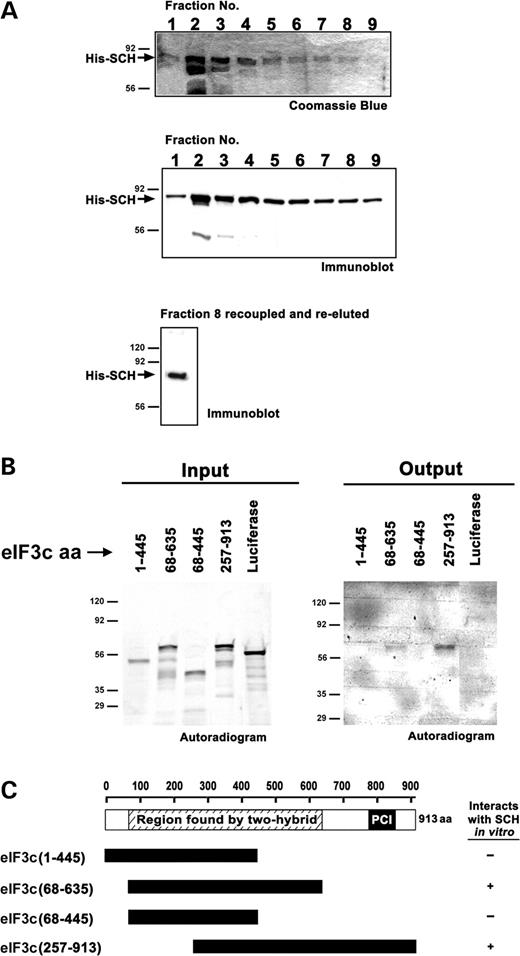
Figure 2.In vitro binding assay between schwannomin and eIF3c. (A) Recombinant his(6x)-schwannomin (His-Sch) was expressed and purified from bacteria. Fraction 8 was recoupled to resin and used for in vitro binding assays. A second elution showed that recoupling was successful. (B) The indicated eIF3c proteins generated by in vitro translation (input) were incubated with immobilized his-schwannomin. eIF3c proteins that interacted were detected by autoradiography (output). The results are consistent with two-hybrid interaction tests showing that schwannomin interacts within eIF3c residues 405–635. (C) Summary of result.
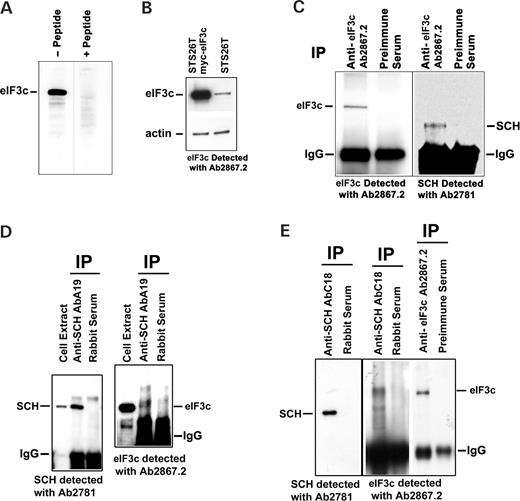
Figure 3. eIF3c antibody characterization and co-immunoprecipitation of eIF3c with schwannomin. (A) Anti-eIF3c peptide antibody ab2867.2 recognized a 110 kDa band in STS26T cell extracts, but could not recognize eIF3c after absorption with peptide antigen. This experiment was accomplished using identical adjacent strips from a blotted preparative gel to ensure equal loading. (B) Anti-eIF3c antibody ab2867.2 detected exogeneously expressed myc-eIF3c in STS26T cells. (C) Anti-eIF3c antibody ab2867.2 immunoprecipitated a 110 kDa band consistent for eIF3c that was absent when pre-immune serum for the antibody was used (left panel). Detection of the same immunoprecipitates with anti-schwannomin antibody ab2781 showed that a 66 kDa band consistent for the size of schwannomin was precipitated with ab2867.2 that was absent when pre-immune serum for the antibody was used (right panel). (D) Anti-NF2 antibody A-19 immunoprecipitated a 66 kDa band consistent for schwannomin that was absent in a control immunoprecipitation conducted with rabbit serum (left panel). Detection of the same immunoprecipitates with anti-eIF3c antibody Ab2867.2 showed that a 110 kDa band consistent for the size of eIF3c was precipitated with anti-schwannomin antibody A-19 that was absent in the control immunoprecipitation (right panel). (E) Anti-NF2 antibody C-18 immunoprecipitated a 66 kDa band consistent for schwannomin that was absent in a control immunoprecipitation conducted with rabbit serum (left panel). Detection of the same immunoprecipitates with anti-eIF3c antibody Ab2867.2 showed that a 110 kDa band consistent for the size of eIF3c was precipitated with anti-schwannomin antibody C-18 that was absent in the control immunoprecipitation (near right panel). The size of the co-immunoprecipipated eIF3c band was validated by co-migration with an eIF3c band immunoprecipipated with anti-eIF3c ab2867.2 on the same gel (far right panel) (note that all lanes in the right panel were on the same gel but the two right panel sets were contrasted differently). STS26T cells (C) and Tet-off STS26T NF2i1 cells induced to express schwannomin (D and E).
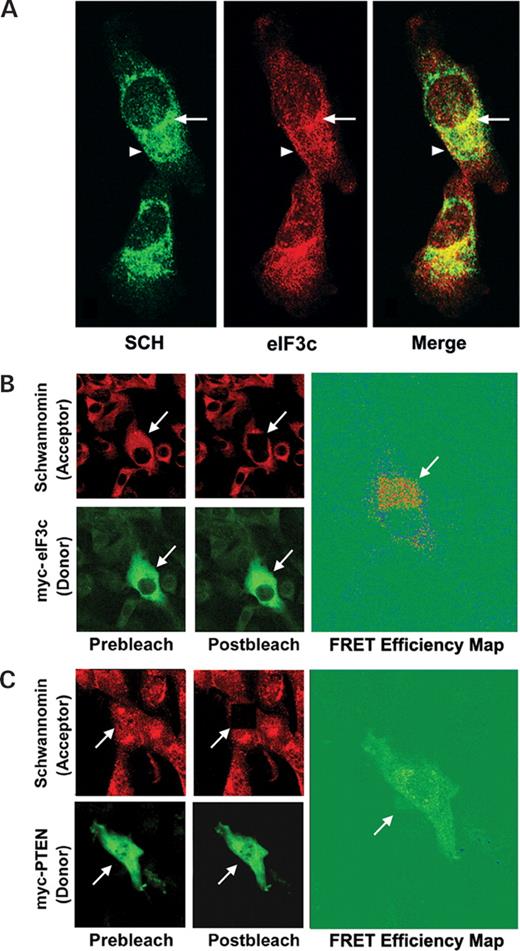
Figure 4. Co-localization of schwannomin and eIF3c in STS26T cells. (A) STS26T cells were immunofluorescently labeled with rabbit anti-eIF3c antibody 2867.2 and chicken anti-schwannomin ab2781. Confocal immunofluorescent microscopy showed that schwannomin and eIF3c co-localized at cytoplasmic, perinuclear (arrow) and plasma membrane structures (arrowhead). (B) FRET of eIF3c and schwannomin in STS26T cells. STS26T cells were transfected with myc-eIF3c and labeled with rabbit anti-schwannomin antibody A-19. Schwannomin and eIF3c localization was detected by secondary labeling with anti-myc-FITC and donkey anti-rabbit-TRITC. The acceptor (TRITC) was photobleached at the position indicated by the arrow by continuos scanning with the 568 nm confocal laser. A FRET efficiency map demonstrated the strongest FRET signal (red) at cytoplasmic puncta. (C) FRET analysis same as in (B) but replacing myc-eIF3c with myc-PTEN as an irrelevant control protein. No FRET between schwannomin and myc-PTEN was observed.
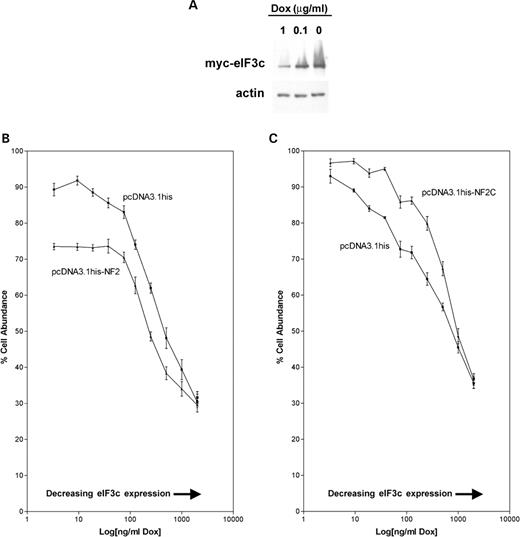
Figure 5. Schwannomin inhibits eIF3c-activated proliferation in myc-eIF3c inducible Tet-off STS26T cells. (A) Myc-eIF3c inducible Tet-off STS26T cells express myc-eIF3c in the absence of doxycycline. Slight expression of the myc-eIF3c inducible transgene was observed in the presence of 1 µg/ml doxycycline (dox). Detection conducted with anti-myc antibody. (B) Increasing myc-eIF3c expression was associated with increased proliferation in STS26T cells. Transient schwannomin expression reduced proliferation most effectively when myc-eIF3c was expressed at the highest levels. This suggests that schwannomin inhibition of proliferation is mediated by eIF3c. (C) In a control experiment where we transiently expressed only the C-terminal domain of schwannomin that lacks the eIF3c binding site, the schwannomin fragment increased proliferation at intermediate dox doses. This could reflect a dominant negative action of the C-terminal fragment on endogenous schwannomin; however, the differences were not significant for the highest and lowest dox doses. Mean values and standard deviations in (C) were calculated from eight replicates within each experiment. These values in (B) were also calculated from eight replicates, but data from two independent trials were pooled. Please see Results for statistical analysis. Note that because the log of zero is undefined the 0 µg/ml dose was assigned a value of 3.3, after statistical analysis, so it would appear on the chart.
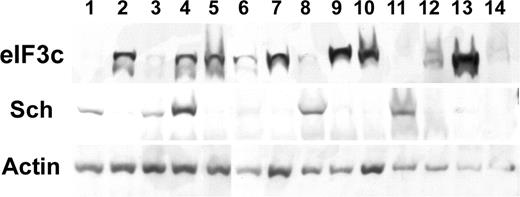
Figure 6. Expression of schwannomin and eIF3c in sporadic human meningiomas. EIF3C was detected on immunoblots with antibody 2867.2, and schwannomin was detected with antibody A-19. Loading was controlled by comparing with actin.
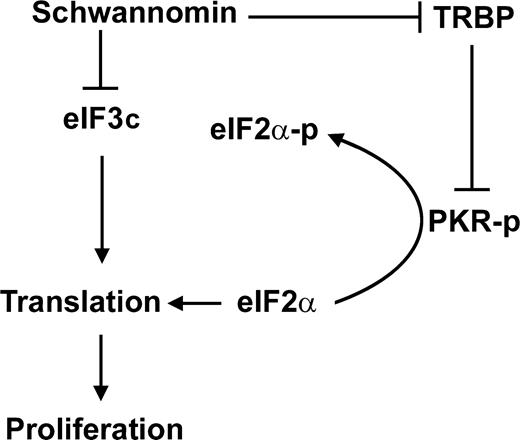
Figure 7. Model for schwannomin action involving TRBP, eIF3c, protein translation and proliferation. Schwannomin directly interacts with and inhibits proliferation mediated by the expression of both TRBP and eIF3c. TRBP inhibits the ribosome-associated protein kinase PKR, which is autophosphorylated upon binding double-stranded RNA. PKR-p phosphorylates eIF2α resulting in the inhibition of protein translation and proliferation.
References
Trofatter, J.A., MacCollin, M.M., Rutter, J.L., Murrell, J.R., Duyao, M.P., Parry, D.M., Eldridge, R., Kley, N., Menon, A.G., Pulaski, K. et al. (
Rouleau, G.A., Merel, P., Lutchman, M., Sanson, M., Zucman, J., Marineau, C., Hoang-Xuan, K., Demczuk, S., Desmaze, C., Plougastel, B. et al. (
Bretscher, A., Edwards, K. and Fehon, R.G. (
Sun, C.X., Robb, V.A. and Gutmann, D.H. (
Gonzalez-Agosti, C., Wiederhold, T., Herndon, M.E., Gusella, J. and Ramesh, V. (
Scherer, S.S. and Gutmann, D.H. (
Sherman, L., Xu, H.M., Geist, R.T., Saporito-Irwin, S., Howells, N., Ponta, H., Herrlich, P. and Gutmann, D.H. (
Lutchman, M. and Rouleau, G.A. (
Gutmann, D.H., Sherman, L., Seftor, L., Haipek, C., Hoang Lu, K. and Hendrix, M. (
Scoles, D.R., Huynh, D.P., Morcos, P.A., Coulsell, E.R., Robinson, N.G., Tamanoi, F. and Pulst, S.M. (
Huynh, D.P. and Pulst, S.M. (
Lallemand, D., Curto, M., Saotome, I., Giovannini, M. and McClatchey, A.I. (
Rangwala, R., Banine, F., Borg, J.P., and Sherman, L.S. (
Pelton, P.D., Sherman, L.S., Rizvi, T.A., Marchionni, M.A., Wood, P., Friedman, R.A. and Ratner, N. (
Shaw, R.J., Paez, J.G., Curto, M., Yaktine, A., Pruitt, W.M., Saotome, I., O'Bryan, J.P., Gupta, V., Ratner, N., Der, C.J., Jacks, T. and McClatchey, A.I. (
Murthy, A., Gonzalez-Agosti, C., Cordero, E., Pinney, D., Candia, C., Solomon, F., Gusella, J. and Ramesh, V. (
Sainio, M., Zhao, F., Heiska, L., Turunen, O., den Bakker, M., Zwarthoff, E., Lutchman, M., Rouleau, G.A., Jaaskelainen, J., Vaheri, A. and Carpen, O. (
Manchanda, N., Lyubimova, A., Ho, H.Y., James, M.F., Gusella, J.F., Ramesh, N., Snapper, S.B. and Ramesh, V. (
Fernandez-Valle, C., Tang, Y., Ricard, J., Rodenas-Ruano, A., Taylor, A., Hackler, E., Biggerstaff, J. and Iacovelli, J. (
Scoles, D.R., Huynh, D.P., Chen, M.S., Burke, S.P., Gutmann, D.H. and Pulst, S.M. (
Scoles, D.R., Nguyen, V.D., Qin, Y., Sun, C.X., Morrison, H., Gutmann, D.H. and Pulst, S.M. (
Scoles, D.R., Qin, Y., Nguyen, V., Gutmann, D.H. and Pulst, S.M. (
Rong, R., Tang, X., Gutmann, D.H. and Ye, K. (
Fields, S. and Song, O. (
Huynh, D.P., Nechiporuk, T. and Pulst, S.M. (
Seal, S.N., Schmidt, A. and Marcus, A. (
Gutmann, D.H., Haipek, C.A. and Hoang Lu, K. (
Ruttledge, M.H., Sarrazin, J., Rangaratnam, S., Phelan, C.M., Twist, E., Merel, P., Delattre, O., Thomas, G., Nordenskjold, M., Collins, V.P. et al. (
Sainz, J., Huynh, D.P., Figueroa, K., Ragge, N.K., Baser, M.E. and Pulst, S.M. (
Lee, J.Y., Kim, H., Ryu, C.H., Kim, J.Y., Choi, B.H., Lim, Y., Huh, P.W., Kim, Y.H., Lee, K.H., Jun, T.Y. et al. (
Benkirane, M., Neuveut, C., Chun, R.F., Smith, S.M., Samuel, C.E., Gatignol, A. and Jeang, K.T. (
Eckmann, C.R. and Jantsch, M.F. (
Park, H., Davies, M.V., Langland, J.O., Chang, H.W., Nam, Y.S., Tartaglia, J., Paoletti, E., Jacobs, B.L., Kaufman, R.J. and Venkatesan, S. (
Saramaki, O., Willi, N., Bratt, O., Gasser, T.C., Koivisto, P., Nupponen, N.N., Bubendorf, L. and Visakorpi, T. (
Nupponen, N.N., Isola, J. and Visakorpi, T. (
Lin, L., Holbro, T., Alonso, G., Gerosa, D. and Burger, M.M. (
Pincheira, R., Chen, Q. and Zhang, J.T. (
Mayeur, G.L. and Hershey, J.W. (
Asano, K., Merrick, W.C. and Hershey, J.W. (
Morris-Desbois, C., Bochard, V., Reynaud, C. and Jalinot, P. (
Hoareau Alves, K., Bochard, V., Rety, S. and Jalinot, P. (
Rosenwald, I.B. (
Rosenwald, I.B., Wang, S., Savas, L., Woda, B. and Pullman, J. (
Rothe, M., Ko, Y., Albers, P. and Wernert, N. (
Loftus, B.J., Kim, U.J., Sneddon, V.P., Kalush, F., Brandon, R., Fuhrmann, J., Mason, T., Crosby, M.L., Barnstead, M., Cronin, L. et al. (
Inoki, K., Corradetti, M.N. and Guan, K.L. (
Dahlberg, W.K., Little, J.B., Fletcher, J.A., Suit, H.D. and Okunieff, P. (

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}