




Abstract
Neonatal diabetes is a genetically heterogeneous disorder with nine different genetic aetiologies reported to date. Heterozygous activating mutations in the KCNJ11 gene encoding Kir6.2, the pore-forming subunit of the ATP-sensitive potassium (KATP) channel, are the most common cause of permanent neonatal diabetes. The sulphonylurea receptor (SUR) SUR1 serves as the regulatory subunit of the KATP channel in pancreatic beta cells. We therefore hypothesized that activating mutations in the ABCC8 gene, which encodes SUR1, might cause neonatal diabetes. We identified a novel heterozygous mutation, F132L, in the ABCC8 gene of a patient with severe developmental delay, epilepsy and neonatal diabetes (DEND syndrome). This mutation had arisen de novo and was not present in 150 control chromosomes. Residue F132 shows evolutionary conservation across species and is located in the first set of transmembrane helices (TMD0) of SUR1, which is proposed to interact with Kir6.2. Functional studies of recombinant KATP channels demonstrated that F132L markedly reduces the sensitivity of the KATP channel to inhibition by MgATP and this increases the whole-cell KATP current. The functional consequence of this ABCC8 mutation mirrors that of KCNJ11 mutations causing neonatal diabetes and provides new insights into the interaction of Kir6.2 and SUR1. As SUR1 is expressed in neurones as well as in beta cells, this mutation can account for both neonatal diabetes and the neurological phenotype. Our results demonstrate that SUR1 mutations constitute a new genetic aetiology for neonatal diabetes and that they act by reducing the KATP channel's ATP sensitivity.
INTRODUCTION
Neonatal diabetes is usually diagnosed in the first 3 months of life and may be permanent and require insulin treatment for life, or transient in which case the diabetes remits, although it may frequently relapse. All cases of neonatal diabetes are likely to result from a single-gene disorder because markers of autoimmunity associated with polygenic type 1 diabetes are rare in patients diagnosed before 6 months (1,2). Neonatal diabetes can occur as an isolated phenotype or as part of a syndrome and exhibits considerable genetic heterogeneity with mutations described in eight genes to date: KCNJ11, GCK, IPF1, EIF2AK3, PTF1A, GLUT2, HNF1beta (TCF2) and FOXP3; reviewed by Slingerland and Hattersley (3). Understanding the genetic causes of neonatal diabetes has given important insights into our understanding of the pancreatic beta cell.
The commonest cause of permanent neonatal diabetes mellitus (PNDM) are heterozygous activating mutations in the KCNJ11 gene encoding Kir6.2, the pore forming subunit of the KATP channel in the pancreatic beta cell (4). KCNJ11 mutations account for 26–64% of cases of PNDM (4–8). Mutations in KCNJ11 can also occasionally result in transient neonatal diabetes mellitus (TNDM) (9), although most TNDM cases result from an abnormality of the imprinted region on chromosome 6q24 (10). To date, at least 63 patients with activating mutations in Kir6.2 have been described, comprising 21 different mutations in 49 families (8,11). The majority of patients have a de novo mutation, but each of their offspring will have a 50% risk of neonatal diabetes.
Structurally, KATP channels are hetero-octameric complexes of four pore-forming Kir6.2 subunits and four regulatory SUR subunits (12). KATP channels link cellular metabolism to electrical activity of the plasma membrane and are found in a diverse range of tissues, including endocrine cells, neurones and cardiac, skeletal and smooth muscles (13). Both Kir6.2 and SUR subunits participate in the metabolic regulation of channel activity by nucleotides, with binding of ATP to Kir6.2 closing the channel and binding of Mg-nucleotides (MgATP, MgADP) to the nucleotide-binding domains (NBDs) of SUR stimulating channel opening (14). Consequently, metabolic inhibition leads to KATP channel opening, cessation of electrical activity and suppression of cellular responses such as insulin secretion and muscle contraction, whereas enhanced cellular metabolism promotes KATP channel closure and stimulates insulin secretion and contraction (13,15). Sulphonylureas, such as glibenclamide and tolbutamide, by-pass cell metabolism and promote insulin secretion by binding directly to SUR and inhibiting KATP channel activity (16). Many patients with activating Kir6.2 mutations have already been able to transfer from insulin to sulphonylurea tablets and achieve improved glycaemic control (5,17–19).
In addition to neonatal diabetes, neurological features are found in ∼20% of patients with activating Kir6.2 mutations. The main symptoms are developmental delay, muscle weakness, facial dysmorphism and epilepsy (4,8,20), a spectrum of symptoms that have been referred to as DEND syndrome (Developmental delay, Epilepsy and Neonatal Diabetes) (11). Kir6.2 is expressed in muscle, nerve and brain as well as in the beta cell and the neurological features are likely to reflect altered KATP channel activity in these tissues.
Functional studies have shown that mutations in Kir6.2 produce neonatal diabetes by reducing the sensitivity of the KATP channel to inhibition by MgATP (4,21–23). As a consequence, the KATP channel remains open even in the presence of glucose, keeping the beta cell hyperpolarized and preventing insulin secretion. A genotype–phenotype relationship is evident, with the functional severity of the Kir6.2 mutation being correlated with the clinical presentation. The majority of mutations in patients with isolated neonatal diabetes affect residues within the ATP-binding site, and those associated with remitting diabetes have a smaller reduction in ATP sensitivity compared with those that result in permanent diabetes (9). In contrast, the mutations reported in patients with DEND syndrome are located distantly from the ATP-binding site and are found mainly within the slide helix (Q52R, V59G) or in other regions involved in channel gating (C166F, I296L) (11). This is consistent with the fact that DEND syndrome mutations stabilize the open state of the channel, thereby reducing ATP inhibition indirectly (22,24). DEND syndrome mutations also show the greatest reduction in ATP inhibition (22,24).
Kir6.2 is a member of the inwardly rectifying K+ channel family and functions as a tetrameric channel pore permitting transmembrane flux of K+ ions. SUR belongs to the ABCC-subfamily of ATP-binding cassette (ABC) transporter proteins, which includes the multidrug resistance-related proteins (MRP) (25). Similar to MRP, SUR has 17 transmembrane helices arranged in groups of 5, 6 and 6 (transmembrane domains TMD0, TMD1 and TMD2; Fig. 1C). TMD0 interacts both physically and functionally with Kir6.2 to modulate opening and closing of the pore (26–28). The large cytosolic domains following TMD1 and TMD2 contain the nucleotide-binding domains NBD1 and NBD2, respectively, which cooperate in nucleotide binding and hydrolysis (29,30). There are two isoforms of SUR, SUR1 and SUR2. SUR1 is found in pancreatic beta cells and neurones. SUR2 has several splice variants, the most common being SUR2A, which is found in cardiac and skeletal muscles, and SUR2B, which is present in smooth muscle and certain neurones (13).
Recessive loss-of-function mutations in both Kir6.2 and SUR1 lead to congenital hyperinsulinism (31) and gain-of-function mutations in Kir6.2 cause neonatal diabetes (4). We therefore hypothesized that gain-of-function mutations in the ABCC8 gene encoding SUR1 might also cause neonatal diabetes. Here, we show that this is indeed the case and present the first SUR1 mutation to cause permanent neonatal diabetes.
RESULTS
Mutational analyses
The entire coding region and conserved splice sites of the ABCC8 gene were sequenced in three patients with permanent neonatal diabetes in whom KCNJ11 mutations and other genetic causes of PNDM had been excluded. One patient (ISPAD68) was heterozygous for a novel mutation (F132L) in the ABCC8 gene encoding SUR1. A previously unreported heterozygous variant in the promoter (c.−8G>T) was detected in a second subject (ISPAD112), but this was also found in his unaffected mother and so was not associated with neonatal diabetes. In addition, multiple polymorphisms were detected including the following exonic SNPs: P69P (c.207T>C), H562H (c.1686T>C) and R1274R (c.3822G>A).
The novel F132L mutation results in the substitution of leucine for phenylalanine at residue 132 (p.Phe132Leu) in exon 3 of the ABCC8 gene (c.394T>C; Fig. 1A). This mutation arose de novo, as analysis of DNA from the unaffected parents showed neither had the mutation and microsatellite analysis confirmed family relationships. The F132L mutation was not found in 150 normal chromosomes. Figure 1B shows that F132 is conserved across a range of species, from humans to zebrafish. A phenylalanine is also found at this position in SUR2, but the equivalent residue in the related human multidrug resistance protein MRP1 is valine rather than phenylalanine. This may be significant because unlike SUR1 and SUR2, MRP1 does not associate with Kir6.2. Residue F132 is located with the first set of transmembrane domains (TMD0) of SUR1. Specifically, it lies within the short cytosolic loop that links transmembrane domains 3 and 4 (Fig. 1C). The region on either side of F132L is also highly conserved (Fig. 1B).
Partial sequencing of SUR1 (exons 1–6 of the ABCC8 gene that encode TMD0) in an additional 15 probands with diabetes diagnosed before 6 months and neurological features did not reveal any further mutations.
Clinical characteristics of patient with F132L SUR1 mutation
ISPAD68 was a boy born in 1978 with a birth weight of 2200 g to a 29-year-old mother who had previously given birth to two healthy older children. There was no parental history of diabetes and the mother had a normal oral glucose tolerance test 2 years prior to delivery and had no glycosuria in pregnancy.
The first indication of hyperglycaemia was mild glycosuria without ketonuria from the seventh day of life, and this gradually increased until it was 20 g/24 h at the age of 13 weeks. A formal diagnosis of diabetes was made at 13 weeks on the basis of an oral glucose tolerance test (1.75 g/kg) which showed glucose rising from a fasting level of 8 to 25.4 mmol/l at 2 h. At that time, the patient weighed 3400 g (less than −2SD), with a length of 53 cm (less than −2 SD) and a head circumference of 34.2 cm (less than −2 SD). Insulin values measured every 30 min during the OGTT were all less than the detection limit of the assay (7 pmol/l). Prior to treatment, random blood glucose levels varied from 9–38 mmol/l, and urinary ketones were intermittently present at low levels. A 24 h urine analysis revealed that amino acids and oligosaccharides were normal, except for glucose. Chromosome analysis was normal (46, XY). Epiphyseal dysplasia, cystic fibrosis, exocrine pancreas dysfunction and coeliac disease were all excluded. Thyroid function, liver and kidney function, blood gases, electrolytes and haematology were all unremarkable.
Insulin treatment was started at the age of 15 weeks in single or multiple dosages of ∼0.7 U/kg/day, using mixtures of short- and intermediate-acting insulin. This resulted in poor glycaemic control, with glycosylated haemoglobin varying between 10–14% (normal range 6–8%). Three admissions with ketoacidosis occurred in the first 2 years precipitated by respiratory tract infections. Islet-cell antibodies, thyroid antibodies, adrenal antibodies and gastric antibodies were not detected when first tested at 8 months or subsequently. The patient's HLA genotype was protective for type 1 diabetes with HLA-DR1 and HLA-DR5.
There were clear neurological features, with the patient showing marked motor and social developmental delay. He had muscle weakness with hypotonia, which was apparent by 1 year. During his second year, there were intermittent fine distal and athetoid involuntary movement disorders lasting 5–10 min and later he developed severe muscle spasms. Electroencephalography revealed non-specific generalized epileptiform activity, and phenobarbitone and phenytoin were prescribed. A CT scan of the brain was normal. At 5 years, the child still had severe developmental delay with greatly reduced social (not talking) and motor (not standing) milestones and he required special schooling and institutional support. He was short and had a small head.
At 27 years, he has insulin-dependent diabetes, severe physical and social problems and is consequently unable to live independently. He is unable to speak and has difficulty standing unaided owing to muscle spasms in his limbs. He remains on insulin treatment and high doses of anti-convulsive medication.
SUR1 mutant KATP channels are not closed by resting ATP levels
To analyse the functional effects of the SUR1-F132L mutation, we studied recombinant KATP channels expressed in Xenopus oocytes. We first compared the effects of metabolism on wild-type and mutant KATP whole-cell currents.
When expressed in Xenopus oocytes, wild-type KATP channels are normally closed but they are activated by metabolic inhibitors such as azide, which lower intracellular ATP (Fig. 2). In contrast, significant whole-cell K+ currents were present in the absence of metabolic inhibition (resting currents) in oocytes expressing either homomeric or heterozygous SUR1-F132L mutant channels (Fig. 2). This result suggests that basal cellular metabolism causes less block of mutant KATP channels than wild-type channels. Both homomeric and heterozygous mutant channel currents were increased by 3 mmol/l azide, indicating that they can be further activated by metabolic inhibition.
All three types of channel were blocked by 0.5 mmol/l tolbutamide (Fig. 2), a concentration that fully saturates the high-affinity-binding site for sulphonylureas (32). However, the potency of block was significantly less for mutant channels, being 98±1% (n=7) for wild-type, 72±2% (n=4) for hetF132L and 57±5% (n=6) for homF132L channels (Fig. 2).
SUR1-F132L KATP channels have reduced ATP sensitivity
To explore the molecular basis of the reduced metabolic sensitivity of mutant KATP channels, we tested the ability of ATP to block wild-type and mutant channels in inside-out patches. Mutant channels were substantially less sensitive to ATP than wild-type channels (Fig. 3A). In addition, in some patches, homF132L channels showed a time-dependent increase in KATP current in the presence of ATP (Fig. 3A, right). This was more marked for the homozygous than heterozygous channels. Similar results have been observed for other PNDM mutations (23). We measured the extent of ATP block as G/Gc, where G is using the steady-state value in the presence of ATP, and the control value (Gc) is the mean amplitude of the current before and after ATP application.
The concentration of ATP required to half-maximally inhibit the channel (IC50) increased from 18±3 µmol/l (n=6) for wild-type channels to 129±31 µmol/l (n=6) for hetF132L and 940±240 µmol/l (n=7) for homF132L channels (Fig. 3B). There was no marked change in the Hill coefficient (slope factor), which was 1.0±0.2 (n=6), 0.78±0.07 (n=6) and 0.79±0.13 (n=7) for wild-type, hetF132L and homF132L channels, respectively. Importantly, however, there was a marked increase in the amplitude of the unblocked current in the presence of 1 mm ATP, which was 2±2% (n=6) for wild-type, 24±5% (n=7) for hetF132L and 45±7% (n=6) for homF132L. These results indicate that the F132L mutation in SUR1 markedly reduces the ability of ATP to inhibit the KATP channel.
DISCUSSION
We report a novel heterozygous mutation, F132L, in the ABCC8 gene encoding SUR1 in a patient with DEND syndrome. This mutation has arisen de novo in a child born to unaffected parents. The novel promoter variant (c.−8G>T) found in the second proband has not been reported previously (33), but is believed to be a rare polymorphism because it was also present in his unaffected mother. Genetic evidence for the pathogenicity of the F132L mutation is strong; it is a spontaneous mutation, absent from 150 normal chromosomes and affects a residue that shows evolutionary conservation across species.
Functional studies also support the pathogenicity of the F132L mutation by revealing that the mutation dramatically reduces the inhibitory potency of MgATP. This leads to an increase in the whole-cell KATP current when heterologously expressed in Xenopus oocytes. In pancreatic beta cells, a similar increase in the KATP current would be expected to cause membrane hyperpolarization and reduce or abolish the membrane depolarization evoked by glucose. This would prevent electrical activity, calcium influx and insulin secretion and can thereby account for the diabetic phenotype of the patient.
Functional effect of the mutation
Residue F132 lies within the short cytosolic loop (CL2) that links transmembrane domains 3 and 4 of TMD0 (Fig. 1C). The three-dimensional structure of this region is unknown, as no ABC transporter that possesses TMD0 has been crystallized. However, recent structural studies suggest that TMD0 lies at the interface between adjacent SUR1 subunits and that it also interacts directly with Kir6.2 (34). This is in agreement with the previous work showing that TMD0 of SUR1 physically interacts with Kir6.2 (27,28) to modulate the gating kinetics of the KATP channel (26,27). Our results suggest that this physical interaction may be mediated, in part, via the second cytosolic loop of SUR1 and that the F132L mutation influences the interaction of SUR1 with Kir6.2.
Two mutations in TMD0 (A116P and V187D), which cause congenital hyperinsulinism, abrogate the association of SUR1 and Kir6.2 and lead to loss of KATP channel function (27,35). Our studies demonstrate that gain-of-function mutations in TMD0 are associated with the opposite phenotype of neonatal diabetes.
The F132L mutation causes a dramatic decrease in the ability of ATP to inhibit the activity of the KATP channel in both the homozygous and simulated heterozygous states. Of particular significance is the fact that the current amplitude at physiological ATP concentrations was increased. At 3 mmol/l ATP, which is likely to reflect beta cell ATP concentrations in the presence of glucose, the estimated current amplitude for hetF132L channels was 13% of maximal. For comparison, mutations in Kir6.2 that cause DEND syndrome have currents that range from 27 (I296L) to 40% (V59G) of maximum, whereas those that cause TNDM or PNDM are smaller and range from 4 (G53S) to 8% (R201H) of maximum (21). Mutations that cause neonatal diabetes with less severe neurological symptoms, or neurological symptoms in some patients only, have current amplitudes that more closely resemble those of hetF132L channels, being 13% for V59M and 15% for R201C (21). Thus, the ATP sensitivity of hetF132L channels is actually lower than that seen for Kir6.2 (KCNJ11) mutations that cause epilepsy. This may reflect the genetic background of the patient or the fact that Kir6.2 has a wider tissue distribution (it can couple also to SUR2A and SUR2B).
Origin of DEND syndrome
The fact that a mutation in SUR1 is associated with the phenotype of DEND syndrome offers new insight into the basis of the extra-pancreatic symptoms characteristic of this syndrome. To date, only mutations in Kir6.2 have been shown to cause DEND syndrome, or intermediate DEND syndrome (11). Because Kir6.2 is expressed in multiple tissues, including neurones and muscle, it is not clear whether the muscle weakness and delayed motor development found in DEND syndrome result from expression of mutant Kir6.2 in the motor neurones or in the skeletal muscle. However, SUR1 is not expressed in skeletal muscle. Thus, the motor problems of the patient we describe must be of neuronal origin. This may be either central or peripheral, as SUR1 is expressed in both brain neurones and motor nerve endings (36,37). It also suggests that the neurological problems may result from enhanced activity of Kir6.2/SUR1 neurones. This is consistent with the lower metabolic sensitivity of Kir6.2/SUR2 channels in neurones which are not activated as readily on metabolic inhibition as Kir6.2/SUR1 channels (37). Indeed, coexpression of Kir6.2 carrying PNDM mutations with SUR2A causes a much smaller shift in MgATP sensitivity than when coexpressed with SUR1 (38).
Predicted response to sulphonylurea treatment
In the presence of the sulphonylurea tolbutamide (0.5 mmol/l), heterozygous F132L SUR1 channels were blocked by 72%. This value is intermediate between the Kir6.2 mutations that cause isolated neonatal diabetes, which respond well to sulphonylurea therapy (blocked by ∼89% in vitro (4,22), and those that result in DEND syndrome, which is refractory to treatment with sulphonylureas (blocked by 41–65% in vitro (22,24). These results suggest that a trial with sulphonylurea tablets for our patient should not be discounted, although the probability of a successful outcome is low.
SUR mutations and human disease
Prior to this study, disease-causing mutations that have been identified in the ABCC8 gene encoding human SUR1 are loss-of-function mutations that cause hyperinsulinaemia of infancy (15,31). These mutations are usually homozygous or compound heterozygous and act either by reducing the density of KATP channels in the plasma membrane or by impairing the ability of Mg–nucleotide interactions with the NBDs of SUR1 to stimulate channel activity. Consequently, KATP currents are reduced, causing beta cell depolarization and insulin secretion even in the face of high blood glucose levels. Our results now demonstrate that gain-of-function mutations in SUR1 may result in the opposite phenotype of neonatal diabetes. Theoretically, mutations in SUR1 that reduce ATP inhibition of the KATP channel could act in at least three ways: by increasing the open-state stability of the channel, by enhancing the stimulatory effects of Mg–nucleotide interactions with the NBDs of SUR1 or by abrogating the increased ATP-sensitivity conferred by coassembly of Kir6.2 with SUR1. Given the location of residue F132 in TMD0, a region known to regulate KATP channel gating (26,27), it seems reasonable to speculate that the F132L mutation acts by stabilizing the channel open state, thus indirectly reducing its ATP sensitivity. Several Kir6.2 mutations associated with DEND syndrome have been shown to work this way (22,24). While we expect that other SUR1 mutations that act by a similar mechanism will be found, it seems possible that mutations which influence nucleotide interactions with the SUR1 NBDs might also lead to diabetes. Interestingly, gain-of-function mutations in the second NBD of the related SUR, SUR2A, cause dilated cardiomyopathy by disrupting its catalytic function (39). This translates into a reduced sensitivity of the cardiac KATP channel to ATP inhibition.
Conclusion
We report a new genetic aetiology for neonatal diabetes. Activating mutations in the ABCC8 gene encoding SUR1 are likely to be rarer than activating Kir6.2 mutations, but the genomic structure of the ABCC8 gene compared with KCNJ11 (39 exons encoding 1581 amino acids versus 1 exon encoding 390 residues) means that far fewer patients have been screened for SUR1. Further studies are required to determine the frequency of SUR1 mutations in patients with diabetes associated with neurological symptoms (DEND or intermediate DEND syndrome) and isolated neonatal diabetes. Functional analysis demonstrates that the F132L SUR1 mutation acts in a similar way to Kir6.2 mutations causing DEND syndrome by producing a marked reduction in the ability of ATP to block the KATP channel.
MATERIALS AND METHODS
Subject and clinical studies
Three probands (ISPAD 17, 68 and 112) were chosen for full sequencing of ABCC8 because they had neonatal diabetes and either neurological features or an autosomal dominant family history, but did not have a KCNJ11 mutation. Prior to testing known causes of neonatal diabetes, mutations in TCF2 (HNF1beta), GCK, IPF1 and PTF1A, had been excluded by sequencing. Following our initial finding, 15 more patients with diabetes diagnosed before 6 months of age and neurological features but no mutation in KCNJ11 were chosen for sequencing of SUR1 TMD0 (ABCC8 exons 1–6).
Molecular genetics
The coding region and conserved splice sites of the ABCC8 gene were amplified from genomic DNA by the polymerase chain reaction using 38 pairs of primers (sequences given in Supplementary Material, Table S1). The products were sequenced using Dye Terminator chemistry on an ABI 3100 (Applied Biosystems, Warrington, UK) and analysed using Staden Analysis Software (www.staden.sourceforge.net). All sequence information is based on GenBank reference sequence NM_000352.2 incorporating the alternate exon 17 (GenBank accession nos L78208, L78224) with numbering based on +1 as the A of the major start codon of exon 1. Family relationships were confirmed by testing four microsatellites on chromosome 20 (D20S107, D20S171, D20S481 and D20S851).
Recombinant expression studies
Human Kir6.2 (Genbank accession no. NM000525 with E23 and I337) and rat SUR1 (GenBank accession no. L40624) were used in this study. Site-directed mutagenesis of SUR1 was performed using the QuickChange™ XL system (Stratagene, La Jolla, CA, USA). Wild-type and mutant cDNAs were cloned in the pBF vector, and capped mRNA prepared using the mMESSAGE mMACHINE large-scale in vitro transcription kit (Ambion, Austin, TX, USA). The whole gene sequence was checked following site-directed mutagenesis to ensure only the intended sequence change had been introduced.
Female Xenopus laevis were anaesthetized with MS222 (2 g/l added to the water). One ovary was removed via a mini-laparotomy, the incision sutured and the animal allowed to recover. Once the wound had completely healed, the second ovary was removed in a similar operation and the animal was then killed by decapitation while under anaesthesia. Immature stage V–VI oocytes were incubated for 60 min with 1.0 mg/ml collagenase (Sigma, type V) and manually defolliculated. Oocytes were either injected with ∼4 ng of wild-type or mutant SUR1 mRNA and ∼0.8 ng Kir6.2 mRNA. The final injection volume was 50 nl/oocyte. To simulate the heterozygous state, Kir6.2 was coexpressed with a 1:1 mixture of wild-type and mutant SUR1 (24). Isolated oocytes were maintained in Barth's solution and studied 1–7 days after injection (40). All procedures used conformed to UK Home Office regulations and the University of Oxford ethical committee guidelines.
Electrophysiology
Currents were recorded from X. laevis oocytes 1–3 days after injection. Whole-cell currents were recorded from intact oocytes, using the two-electrode voltage-clamp method, filtered at 1 kHz and digitized at 4 kHz. Oocytes were constantly perfused at 20–22°C with a solution containing (in mmol/l) 90 KCl, 1 MgCl2, 1.8 CaCl2 and 5 HEPES (pH 7.4 with KOH). Metabolic inhibition was produced by 3 mm Na-azide. Whole-cell currents were monitored in response to voltage steps of ±20 mV from a holding potential of −10 mV. Tolbutamide was used to test sulphonylurea sensitivity.
SUPPLEMENTARY MATERIAL
Supplementary Material is available at HMG Online.
ACKNOWLEDGEMENTS
We thank the patients and their referring clinicians, especially Dr T. Barrett and Dr V. Mathew. We are grateful to Dr A.L. Gloyn for contributing primer sequences. Financial support was provided by the Wellcome Trust, the Royal Society and the Research & Development Directorate at the Royal Devon and Exeter NHS Foundation Trust. F.M.A. is a Royal Society Research Professor and A.T.H. is a Wellcome Trust Clinical Research Leave Fellow.
Conflict of Interest statement. None declared.
The authors wish it to be known that, in their opinion, the first 3 authors should be regarded as joint First Authors.
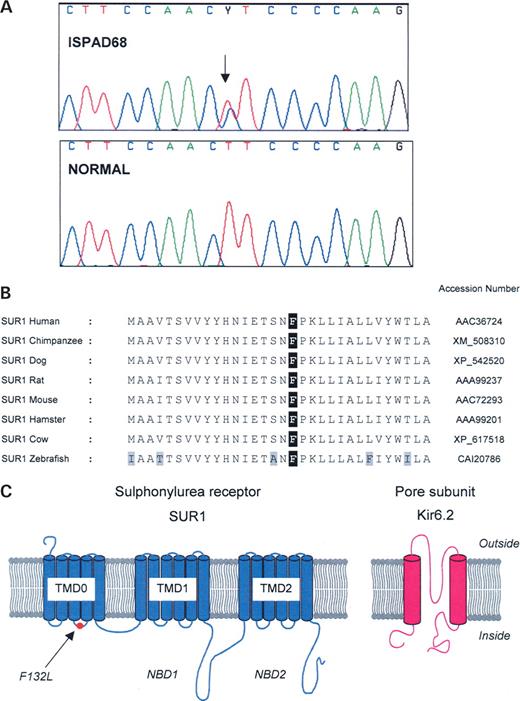
Figure 1. F132L mutation, conservation of F132 across species and location of F132L in SUR1. (A) Sequencing of ISPAD68 showing a heterozygous mutation (c.394T>C) resulting in the substitution of phenylalanine (TTC) by leucine (CTC) at residue 132 (F132L) of the ABCC8 gene (arrow). The normal sequence from his unaffected father is shown for comparison. (B) Comparison of partial TMD0 sequences for various species. The NCBI accession numbers are given on the right. F132 is indicated in black. Non-conservative substitutions are given in grey. (C) Schematic of the proposed membrane topologies of SUR1 and Kir6.2 showing the location of F132L (arrowed) in SUR1. NBD, nucleotide-binding domain; TMD, transmembrane domain.
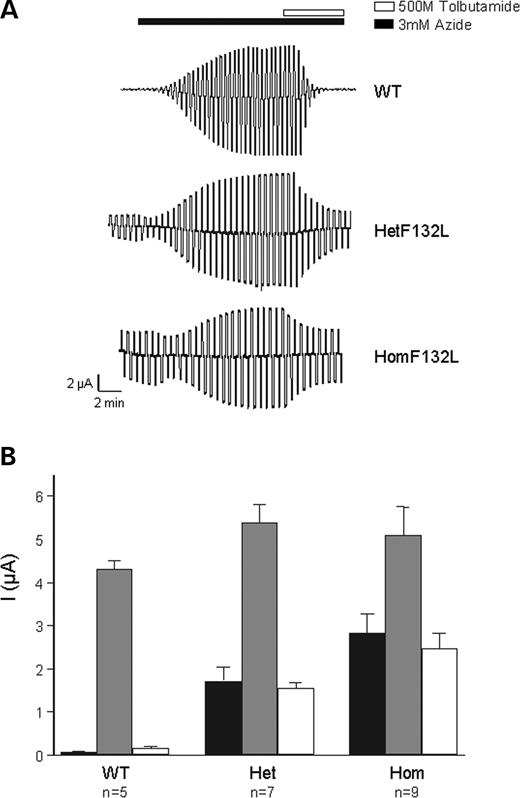
Figure 2. Comparison of wild-type and mutant whole-cell KATP currents. (A) Whole-cell currents recorded from Xenopus oocytes coexpressing Kir6.2 and either SUR1 (WT) or SUR1-F132L (homF132L) or both SUR1 and SUR1-F132L (hetF132L) in response to voltage steps of ±20 mV from a holding potential of −10 mV. Bars indicate the times of application of 3 mmol/l azide or 0.5 mmol/l tolbutamide. (B) Mean steady-state whole-cell KATP currents (as indicated) evoked by a voltage step from −10 to −30 mV before (black bars) and after application of 3 mmol/l azide (grey bars) and in the presence of 3 mmol/l azide plus 0.5 mmol/l tolbutamide (white bars). The number of oocytes is given below the bars.
![Figure 3. MgATP concentration–response relations. (A) KATP currents recorded in inside-out patches excised from oocytes coexpressing Kir6.2 and either SUR1 or SUR1-F132L (Kir6.2/SUR1-homF132L) or both SUR1 and SUR1-F132L (Kir6.2/SUR1-hetF132L). Currents were recorded in response to successive voltage ramps from −110 to 100 mV in an inside-out patch. The dashed line indicates the zero current level. Currents were recorded in the presence of 2 mmol/l Mg2+. (B) Mean relationship between [ATP] and KATP conductance (G), expressed relative to the conductance in the absence of nucleotide (Gc) for Kir6.2/SUR1 (open circles, n=6), and heterozygous (solid circles, n=6) or homomeric (solid squares, n=7) Kir6.2/SUR1-F132L channels. Currents were recorded in the presence of 2 mmol/l Mg2+. The smooth curves are the best fit of Eq. (1) to the mean data. For wild-type channels, IC50=16 µmol/l, h=0.93 and a=0. For hetF132L, IC50=102 µmol/l, h=0.75 and a=0.06. For homF132L, IC50=448 µmol/l, h=0.85 and a=0.18.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/hmg/15/11/10.1093_hmg_ddl101/1/m_ddl10103.jpeg?Expires=1716579095&Signature=gGjJEsjMFVBdYxBx7653AXP4Ay2oj152WeQevBxz5MZ5U4dBRi0~ZCVREDEFwKAUyS7pK5WB8ZUV7cakbecXaKwnA5B~jVqnS8RFi68zZGAe35ZjUf2ZDuUYJAGMo47vxdsA7pB5cDoLzFLoEq27kH5ZTAfRYWoW9OAdyzY7INnn4fp2fhQ39k1KISQa3J3GvITnJAsyG2oWO0MwWNM7xWYzWyZFsezH4lPow8R-LvY-xnc1iSDwcWfXoKlHoDarcQVyaCfMLdXAVP1ovfuCrygWsVEInsD1E3Gwrgdw9Qu92Ql1oRn1iWvz6sU6BsUz8oMhcdGZSwK3gdRzjOW-Kg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Figure 3. MgATP concentration–response relations. (A) KATP currents recorded in inside-out patches excised from oocytes coexpressing Kir6.2 and either SUR1 or SUR1-F132L (Kir6.2/SUR1-homF132L) or both SUR1 and SUR1-F132L (Kir6.2/SUR1-hetF132L). Currents were recorded in response to successive voltage ramps from −110 to 100 mV in an inside-out patch. The dashed line indicates the zero current level. Currents were recorded in the presence of 2 mmol/l Mg2+. (B) Mean relationship between [ATP] and KATP conductance (G), expressed relative to the conductance in the absence of nucleotide (Gc) for Kir6.2/SUR1 (open circles, n=6), and heterozygous (solid circles, n=6) or homomeric (solid squares, n=7) Kir6.2/SUR1-F132L channels. Currents were recorded in the presence of 2 mmol/l Mg2+. The smooth curves are the best fit of Eq. (1) to the mean data. For wild-type channels, IC50=16 µmol/l, h=0.93 and a=0. For hetF132L, IC50=102 µmol/l, h=0.75 and a=0.06. For homF132L, IC50=448 µmol/l, h=0.85 and a=0.18.
References
Edghill, E.L., Dix, R., Flanagan, S.E., Bingley, P.J., Hattersley, A.T., Ellard, S. and Gillespie, K.M. (
Iafusco, D., Stazi, M.A., Cotichini, R., Cotellessa, M., Martinucci, M.E., Mazzella, M., Cherubini, V., Barbetti, F., Martinetti, M., Cerutti, F. et al. (
Slingerland, A.S. and Hattersley, A.T. (
Gloyn, A.L., Pearson, E.R., Antcliff, J.F., Proks, P., Bruining, G.J., Slingerland, A.S., Howard, N., Srinivasan, S., Silva, J.M., Molnes, J. et al. (
Sagen, J.V., Raeder, H., Hathout, E., Shehadeh, N., Gudmundsson, K., Baevre, H., Abuelo, D., Phornphutkul, C., Molnes, J., Bell, G.I. et al. (
Vaxillaire, M., Populaire, C., Busiah, K., Cave, H., Gloyn, A.L., Hattersley, A.T., Czernichow, P., Froguel, P. and Polak, M. (
Massa, O., Iafusco, D., D'Amato, E., Gloyn, A.L., Hattersley, A.T., Pasquino, B., Tonini, G., Dammacco, F., Zanette, G., Meschi, F. et al. (
Flanagan, S.E., Edghill, E.L., Gloyn, A.L., Ellard, S. and Hattersley, A.T. (
Gloyn, A.L., Reimann, F., Girard, C., Edghill, E.L., Proks, P., Pearson, E.R., Temple, I.K., Mackay, D.J., Shield, J.P., Freedenberg, D. et al. (
Temple, I.K., Gardner, R.J., Mackay, D.J., Barber, J.C., Robinson, D.O. and Shield, J.P. (
Hattersley, A.T. and Ashcroft, F.M. (
Shyng, S.L. and Nichols, C.G. (
Miki, T. and Seino, S. (
Tucker, S.J., Gribble, F.M., Zhao, C., Trapp, S. and Ashcroft, F.M. (
Ashcroft, F.M. (
Gribble, F.M. and Reimann, F. (
Zung, A., Glaser, B., Nimri, R. and Zadik, Z. (
Codner, E., Flanagan, S., Ellard, S., Garcia, H. and Hattersley, A.T. (
Klupa, T., Edghill, E.L., Nazim, J., Sieradzki, J., Ellard, S., Hattersley, A.T. and Malecki, M.T. (
Nahi-Buisson, N., Bellanne-Chantelot, C., Eisermann, M., Nabbout, R., Bach, N., Nivot, S., Plouin, P., Robert, J.J. and de Lonlay, P. (
Proks, P., Girard, C. and Ashcroft, F.M. (
Proks, P., Antcliff, J.F., Lippiat, J., Gloyn, A.L., Hattersley, A.T. and Ashcroft, F.M. (
Tammaro, P., Girard, C., Molnes, J., Njolstad, P.R. and Ashcroft, F.M. (
Proks, P., Girard, C., Haider, S., Gloyn, A.L., Hattersley, A.T., Sansom, M.S. and Ashcroft, F.M. (
Higgins, C.F. and Linton, K.J. (
Babenko, A.P., Gonzalez, G. and Bryan, J. (
Chan, K.W., Zhang, H. and Logothetis, D.E. (
Bryan, J., Vila-Carriles, W.H., Zhao, G., Babenko, A.P. and Aguilar-Bryan, L. (
Nichols, C.G., Shyng, S.L., Nestorowicz, A., Glaser, B., Clement, J.P., Gonzalez, G., Aguilar-Bryan, L., Permutt, M.A. and Bryan, J. (
Gribble, F.M., Tucker, S.J. and Ashcroft, F.M. (
Gribble, F.M., Tucker, S.J. and Aschroft, F.M. (
Gloyn, A.L., Siddiqui, J. and Ellard, S. (
Mikhailov, M.V., Campbell, J.D., de Wet, H., Shimomura, K., Zadek, B., Collins, R.F., Sansom, M.S., Ford, R.C. and Ashcroft, F.M. (
Reimann, F., Huopio, H., Dabrowski, M., Proks, P., Gribble, F.M., Laakso, M., Otonkoski, T. and Ashcroft, F.M. (
Deist, M., Repp, H. and Dreyer, F. (
Liss, B., Bruns, R. and Roeper, J. (
Tammaro, P., Proks, P. and Ashcroft, F.M. (
Bienengraeber, M., Olson, T.M., Selivanov, V.A., Kathmann, E.C., O'Cochlain, F., Gao, F., Karger, A.B., Ballew, J.D., Hodgson, D.M., Zingman, L.V. et al. (

{kind=link}
{kind=link}
{kind=link}