





Abstract
The kinetics and structure of cell-free fetal DNA in maternal plasma is currently under investigation. Plasma fetal DNA seems quite stable albeit cleared rapidly following birth, suggesting continuous fetal DNA release into the maternal circulation during pregnancy. However, to understand better the kinetics of circulating DNA, studies to determine the biological (structural) form in which fetal and maternal DNA exist and the mechanisms underlying variation in plasma are warranted to ensure quantitative diagnostic reliability. It is likely that circulating fetal DNA is released from fetal and/or placental cells undergoing apoptosis. Thus, the majority of fetal DNA is proposed to circulate in membrane-bound vesicles (apoptotic bodies). This review summarizes the latest reports in this field.
Introduction
Prenatal genetic diagnosis has traditionally required invasive procedures such as amniocentesis or chorionic villous sampling (CVS); however, each carries a small but finite risk of fetal loss and injury. Maternal serum analyte screening and ultrasound can identify individuals at risk for fetal aneuploidy (predominantly trisomy 21), but like other non-invasive screening methods, they are hampered by non-optimal sensitivities and high false-positive (procedure) rates. For several years, we and others have focused on the isolation of intact fetal cells from maternal blood, a non-invasive method that can yield definitive results. Universal presence of fetal cells in maternal blood is now accepted, but their occurrence is rare and requires complex enrichment and identification strategies. There exist one to six fetal cells per millilitre of blood from normal pregnant women (Hamada et al., 1993; Krabchi et al., 2001). The largest study concerning efficacy is the multicentre National Institute of Child Health and Development (NICHD) fetal cell study group in which we and other groups collaborated. Of 2744 maternal samples (Bianchi et al., 2002), fetal male cells were correctly identified in 41.4% when the fetus was euploid (n=1292). Among confirmed aneuploid cases, the detection rate was higher: 74.4%. Although further improvement of existing enrichment and isolation protocols is warranted, progress remains hampered by both rarity of fetal cells and the lack of fetal-specific cell markers. More recently, we and other researchers have also focused on non-cellular fetal DNA in the plasma fraction of maternal blood for quantification and analysis of locus-specific sequences. In this review we emphasize advancements related to evaluation of the dynamic changes and biological nature of circulating nucleic acids in maternal plasma.
Existence of circulating nucleic acids in plasma of cancer patients
Nucleic acids (DNA and RNA) in plasma were first observed >50 years ago. In the early 1970s increased quantities of DNA were verified in the plasma of cancer patients (Leon et al., 1977). In the late 1980s and 1990s several groups demonstrated that plasma DNA derived from cancer patients displayed tumour-specific characteristics, including decreased strand stability, Ras and p53 mutations, mircrosatellite alterations, abnormal promoter hypermethylation of selected genes, mitochondrial DNA mutations and tumour-related viral DNA (Stroun et al., 1989; Sorenson et al., 1994; Vasioukhin et al., 1994; Chen et al., 1996; Nawroz et al., 1996; Anker et al., 1999; Chan et al., 2002). Tumour-specific DNA for a wide range of malignancies has been found: haematological, colorectal, pancreatic, skin, head-and-neck, lung, breast, kidney, ovarian, nasopharyngeal, liver, bladder, gastric, prostate and cervix. In aggregate, the above data show that tumour-derived DNA in plasma is ubiquitous in affected patients, and likely the result of a common biological process such as apoptosis. Investigations into the size of these plasma DNA fragments from cancer patients has revealed that the majority show lengths in multiples of nucleosomal DNA, a characteristic of apoptotic DNA fragmentation (Giacona et al., 1998; Jahr et al., 2001).
If a cancer shows specific viral DNA sequences or tumour suppressor and/or oncogene mutant sequences, PCR-specific strategies can be developed. However, for most cancers (and most Mendelian disorders), clinical application awaits optimization of methods to isolate, quantify and characterize the tumour-specific DNA compared to the patient's normal DNA, which is also present in plasma. Therefore, understanding the molecular structure and dynamics of DNA in plasma of normal individuals will be necessary to achieve further advancement in this field.
Presence of cell-free fetal nuceic acids in maternal blood
Circulating cell-free fetal DNA in maternal plasma and serum
As studies of tumour-derived DNA detection in plasma of cancer patients were being pursued, Lo et al. (1997) demonstrated fetal DNA in plasma and serum from healthy pregnant women. Using quantitative real-time PCR, surprisingly high mean concentrations (6.2% of total plasma DNA) of fetal DNA were found in maternal plasma in early and late pregnancy (Lo et al., 1998b). In plasma, fetal DNA reached a mean of 25.4 genome equivalents (GEq)/ml (range 3.3–69.4) in early pregnancy and 292.2 GEq/ml (range 76.9–769) in late pregnancy. Mean concentration was less (3.4% of total serum DNA) but still substantive in maternal serum. As assessed by the numbers of copies of SRY (a single-copy Y chromosome-specific sequence), the ratio of fetal to maternal DNA was 775–970-fold greater in the plasma than amount of DNA derived from intact fetal cells would indicate.
Interestingly, Jimenez et al. (2003) demonstrated detection of fetal DNA in the maternal serum of rhesus monkeys with apparently similar kinetics related to gestational age and postnatal clearance characteristics observed in humans. The availability of such an animal model system should prove very useful in addressing further questions relating to origin, mechanism and nature of fetal DNA in maternal circulation.
Fetal/placental-derived RNA in maternal plasma
In plasma from cancer patients, RNA is also shown to be present (Kopreski et al., 1999; Lo et al., 1999a; Chen et al., 2000; Silva et al., 2001). These molecules are likely packaged in apoptotic bodies and, hence, rendered more stable compared to ‘free RNA’ (Hasselmann et al., 2001; Anker et al., 2002; Tsui et al., 2002; Ng et al., 2003). Thus, it is not surprising that stable fetal RNA is also present in maternal plasma (Poon et al., 2000b). Ng et al. (2003a) detected maternal plasma mRNA transcripts exclusively expressed from the placenta. Two placenta-expressed genes (human placental lactogen, hPL; β subunit of hCG) were detected in each of 10 maternal samples. This study provides direct evidence that RNA is stable in whole blood prior to processing and clearance following delivery. Investigations involving another plasma-expressed gene, corticotrophin-releasing hormone (CRH), have shown increased CRH mRNA in plasma of women with pre-eclampsia (n=12; mean 1070 copies/ml) compared with normal gestational age-matched controls (n=10; 102 copies/ml) (Ng et al., 2003).
Temporal changes in circulating cell-free DNA
Total DNA
Given establishment of fetal DNA in maternal plasma, attention turned to kinetics of the phenomenon. The concentration of total DNA in plasma of healthy (non-pregnant) adults is in the range of 10–100 ng or 103–104 GEq/ml Wu et al. (2002). Total plasma DNA (fetal and maternal) levels are significantly higher during pregnancy (Lo et al., 1998b; Lo, 2000) as well as cancer (Wu et al., 2002; Taback et al., 2004). The explanation of increased concentration of total DNA during pregnancy is unclear. DNA from fetal sources comprises only a small portion (5–7%) of total circulating DNA. Moreover, fetal DNA in maternal plasma seems quite stable (Angert et al., 2003) albeit cleared at an extremely rapid rate following birth (Lo et al., 1999d). Thus, a continuous supply of fetal DNA is emitted into the maternal circulation. Such findings suggest that during pregnancy, cellular turnover of maternal cells is enhanced. Although such quantitative changes may serve as a means for distinguishing between euploid and abnormal pregnancies, clinical application will require a better understanding of the cause associated with variability among normal specimens.
Fetal DNA
As expected, detection of relatively low levels of fetal DNA sequences (as compared to maternal DNA levels) is dependent on the sensitivity of the assay as well as the amount of target fetal sequences. Several reports have confirmed that gestational age correlates positively with amount of fetal DNA in plasma; thus, higher detection rates are reported with increased gestation. Lo et al. (1998b) reported fetal concentrations to be low in the first trimester, rising in the second and third trimester. Ariga et al. (2001) combined real-time kinetic PCR with liquid oligomer hybridization with 32P-labelled probes to quantify Y chromosome-specific sequences throughout pregnancy. In 20 women confirmed to have a male fetus and followed from the first to third trimester, fetal DNA concentrations increased from 10.1 to 130.5 copies per 0.5 ml maternal plasma. Rijnders et al. (2003) studied pregnant women after assisted reproduction and reported detection of fetal DNA as early as 5 weeks and 2 days gestation in one of two patients; however, detection reached 100% by 9 weeks gestation. It is likely that earlier studies (i.e. Thomas et al., 1995) demonstrating 100% Y-sequence detection using maternal whole blood between 4 and 7 weeks were actually measuring cell-free fetal DNA and not intact cells. Although there appears to be considerable variability among subjects in the quantity and timing of fetal DNA's initial presence in maternal circulation, the overall trend of increased quantity of this DNA with increasing gestational age is consistent.
During the last 8 weeks of pregnancy there is a sharp increase of fetal DNA in maternal plasma (Lo et al., 1998). This might be related to gradual breakdown of the maternal–fetal interface/placental barrier (Bianchi, 2000). To address this, Chan et al. (2003) performed serial analysis of fetal DNA concentrations in late pregnancy, showing a positive correlation with gestational age in the third trimester. During the late third trimester, they observed a mean increase of 29.3% of fetal DNA each week. Thus, they provide normative values for comparative studies involving pregnancy related pathological conditions such as preterm labour and pre-eclampsia.
Ohashi et al. (2002) report a correlation of fetal DNA and hCG concentrations using second trimester maternal serum samples. Farina et al. (2002) demonstrated a significant correlation between early gestational age (10–12 weeks) and total fetal DNA concentrations among 63 euploid pregnancies that was normally distributed. In addition, several studies to assess the sensitivity and specificity of fetal DNA in first and second trimester maternal plasma have also been reported, with relatively large sample sizes (Costa et al., 2001; Sekizawa et al., 2001; Zhong et al., 2001a; Honda et al., 2002). Real-time quantitative PCR was employed based on Y-specific sequences in pregnancies carrying male fetuses. Overall, 95–100% sensitivity with 100% specificity was observed. Sensitivity of detection in first trimester (7–12 weeks) euploid samples was less efficient (70–95%), likely reflecting lower amounts of fetal DNA.
In what form does fetal DNA exist? Apoptotic bodies as likely vehicle
Apoptosis as ubiquitous source of circulating DNA
Three sources of circulating DNA can be plausibly hypothesized: (i) dying cells (necrotic or apoptotic); (ii) active secretion of DNA; (iii) terminal differentiation. Apoptosis (programmed cell death) is the most common form of cell death, continuing through life from early stages of embryogenesis to death. Because 1011–1012 cells divide daily and the same amount should be lost to maintain tissue homeostasis, ∼1–10 g of DNA can be expected to be degraded each day in the human (Rudin et al., 1997). Given the high turnover, it is not surprising that some DNA escapes final cleavage/degradation and thus appears in the plasma. The concentration of DNA in plasma is in the range of 10–100 ng or 103–104 GE/ml (Jen et al., 2000a; Wu et al., 2002). During pregnancy this DNA could be fetal and/or placental in origin.
Apoptosis giving rise to fetal DNA: distinguishable size fragments
The biochemical hallmark of apoptosis is fragmentation of genomic DNA, an irreversible event that commits the cell to die. This occurs before changes in plasma membrane permeability (prelytic DNA fragmentation) (Barrett et al., 2001; Martelli et al., 2001). In many systems, this DNA fragmentation has been shown to result from activation of endogenous Ca2+ - and Mg2+ -dependent nuclear endonuclease. This enzyme selectively cleaves DNA at sites located between nucleosomal units (linker DNA) generating mono- and oligonucleosomal DNA fragments. The process of DNA cleavage is not random but likely under regulatory control. Thus, certain DNA sequences are relatively more susceptible to cleavage, generating predictable size fragments. Given that the normal mechanisms of apoptosis may be saturated during pregnancy or that an entirely different mechanism of apoptosis is involved, fetal and/or placental cells undergoing apoptosis may contain DNA that is distinguishable from maternal DNA in plasma based on molecular characteristics. Indeed, a recent report by Chan et al. (2004) investigated the size distribution of plasma DNA among pregnant and non-pregnant women. They have shown that plasma DNA (based on amplicon sizes of the leptin gene) in maternal samples is significantly longer than DNA in plasma of non-pregnant women. In addition, among maternal samples, maternal-derived DNA (based on leptin gene sequences) was longer than fetal-derived (based on SRY) DNA. In their study of 31 pregnant women, median percentage of plasma DNA with size >201 bp was 57% compared to 14% among non-pregnant women (n=34). Of the fetal-derived DNA, 20% displayed sizes >193 bp but not exceeding 313 bp. These studies support the hypothesis that fetal DNA may be distinguishable from maternal DNA, enabling development of fetal DNA enrichment strategies.
Fetal DNA likely exists in the form of apoptotic bodies
Irrespective of the source, various forms of circulating DNA in plasma can be suggested: shed cells, apoptotic bodies, nucleosomes, other nucleoproteins, and free DNA. Halicka et al. (2000) described formation of apoptotic bodies during apoptosis of different cell types induced by various treatments. These authors reported that apoptosis is not a random or chaotic process but under regulatory control. Thus, formation of apoptotic bodies is a specific stage of programmed cell death. Apoptotic bodies usually contain either DNA or RNA, but not both (Halicka et al., 2000). Evidence that apoptotic bodies are the vehicle for cell-free DNA in plasma has been provided by detection of stabilized circulating RNA in patients with malignant melanoma (Kopreski et al., 1999) and breast cancer (Chen et al., 2000). The same holds for fetal DNA in normal pregnant women (Tsui et al., 2002). Given that plasma is rich in RNase activity and that RNA is highly sensitive to nuclease attack, detection of stable RNA further suggests that this nucleic acid is protected likely in vesicles or apoptotic bodies.
Evidence that apoptotic bodies contain fetal DNA: Baylor results
Although nucleosomes have been observed and quantified in plasma of cancer patients, little is known of the basic properties of apoptotic bodies which are known to contain nucleosomes. It is unclear how apoptotic bodies are distributed during blood fractionation into cellular and plasma fractions by centrifugation or ultrafiltration. Because we have such compelling evidence for the presence and utility of fetal DNA in maternal circulation for clinical diagnostic application, a better understanding of the nature of this potential source of fetal genetic material is warranted. To address some of these issues, we performed studies that enable more efficient recovery and concentration of circulating DNA from maternal plasma.
Transmission electron microscopy
Most isolation protocols target a particular form of DNA; thus, standard approaches will inevitably result in loss of non-targeted DNA (fetal and/or maternal). Thus, to maximize recovery of all possible forms of fetal DNA, we have used the Microcon centrifugal filter device (Millipore, USA). These filter units employ a low-binding, anisotropic, hydrophilic regenerated cellulose membrane that allows high yield recovery rates (>95% of the sample) with concentration factors as high as 100-fold. Using these filter units enables collection of all nucleic acids >10 bp in size in a plasma sample. By concentrating the plasma DNA content, suitable size plasma pellets were prepared and subjected to electron microscopic analysis that demonstrated for the first time presence of substantial quantities of nucleosomes in plasma of pregnant women. In Figure 1, the images display transmission electron micrographs of maternal plasma pellets at two magnifications. In Figure 1A, arrows identify the presence of small spherical bodies that appear quite symmetrical among various other structures that are likely ruptured vesicles, such as apoptotic bodies. These structures are nucleosomes (DNA bound to histones), which had likely been engulfed by apoptotic bodies. Because the initial step in our plasma concentration protocol involves a 16 000 g centrifugation to force the suspension through the filter, it is not surprising that such membrane-bound components would rupture and release their components in the plasma pellet. At higher magnification (Figure 1B), these structures clearly contain and are attached to chromatin (arrow). This chromatin is abundant, with varying size and level of condensation, consistent with an apoptotic pathway. This finding supports the hypothesis that fetal DNA may exist in distinct forms and thus be subject to enrichment.
Real-time quantitative PCR
In a preliminary study, maternal plasma (n=28; 15 male, 13 female) was separated by centrifugation (800 g) from 12–16 week gestations. Acridine Orange (AO), a nucleic acid stain, was used to label recovered plasma, followed by flow cytometric separation of the AO-positive non-cellular fraction. Sorted plasma was analysed by real-time PCR to quantify DYS1 (fetal) and GAPDH sequences. Microscopic analysis revealed the presence of apoptotic bodies and nucleosomes in plasma following separation. Real-time PCR quantification demonstrated significant enrichment with mean detection of 28% fetal sequences in sorted compared to 2.8% in non-sorted specimens. Thus, based on these observations, fetal DNA enrichment is feasible and can enhance fetal sequence detection.
Technical advances in assaying cell-free fetal DNA
Recovery of plasma DNA
More efficient or selective methods of plasma fetal DNA isolation should improve fetal DNA sequence detection in first trimester cases. The amount of DNA isolated from plasma is dependent on the specific method of DNA isolation employed (Houfflin-Debarge et al., 2000; Jen et al., 2000b). Centrifugation speed is critical for blood separation and recovery of fetal DNA from plasma (Lui et al., 2002). Chiu et al. (2001) showed that inefficient processing could lead to residual cells in the plasma and interfere with accuracy in quantification of fetal sequences. This likely explains the discrepancies between groups reporting on whether intact fetal cells are actually present in maternal plasma (Poon et al., 2000a; Bayrak-Toydemir et al., 2003; Bischoff et al., 2003b). Because intact fetal cells in maternal plasma are likely apoptotic (Kolialexi et al., 2001), their localization to the plasma fraction after centrifugation is likely the indirect result of these cells floating from the mononuclear cell layer due to reduced cellular density.
To address some of the obvious variables associated with plasma DNA isolation and utilization, the five participating centres of the NICHD fetal cell study group developed a standard protocol for inter-laboratory comparison of fetal DNA detection (Johnson et al., 2004). This collaborative effort examined variables impacting data quality, and determined sensitivity and specificity for detection of the Y chromosome. These studies conclusively showed not only that fetal DNA, as judged by the Y chromosome, is consistently present in maternal plasma, but defined the range of fetal DNA during the weeks of gestation relevant for prenatal diagnosis. Overall results among the five centres were based on matched maternal plasma specimens (total of 35 known male and 28 known female fetuses). Efficiency of amplification of known quantities of standard DNA was consistent between all centers. Surprisingly, we found that detecting the fetal allele, and thus sensitivity and negative predictive power, was disproportionately impacted by total DNA recovery. The quantity of male DNA (SRY sequences) amplified from maternal plasma when the fetus was male ranged from 51 to 228 GEq/ml, whereas control DNA (GAPDH sequences) ranged from 5939 to 12 397 GEq/ml in these cases. Sensitivity among centres varied from 31.4 to 97.1% with specificity of 92.8 to 100%. Given that the amount of DNA recovered correlates positively with sensitivity, variable sensitivities are predictable. This result puts a premium on efficient DNA purification.
Sampling of isolated plasma DNA
The quantity of background (circulating maternal) DNA can influence the detection rate of low copy number sequences (Stenman et al., 2001). When fetal DNA is present in very low copy numbers (e.g. 1–3 GEq/ml), real-time PCR is likely at the limit of its assay sensitivity. Thus, vicissitudes of sampling affect detection rate and quantitative measurements. Detection of the target DNA may therefore be influenced not only by the amount of material used per test but by the number of fractions tested. As a result, an increased number of replicates is likely necessary to ensure reliable results (Hromadnikova et al., 2003).
Stabilization and transportability of specimens prior to DNA isolation
It is not surprising that intact cell lysis due to time delay prior to blood processing or treatment conditions (i.e. centrifugation speed) would result in increased concentrations of maternal background DNA, thus causing dilution of relatively low levels of circulating fetal DNA (Dukes et al., 2004). Dhallan et al. (2004) propose treatment of blood samples with formaldehyde. This seems plausible in preventing lysis of intact cells (maternal and fetal). Data support this hypothesis with increased percentage of fetal DNA detected in seven of 10 matched formaldehyde-treated (mean 20.2% fetal) versus untreated (mean 7.7% fetal) cases. If fetal DNA exists in the form of apoptotic bodies, then treatment of maternal blood samples with formaldehyde is also likely to assist in the stabilization of this DNA. Formaldehyde-treated plasma DNA may be more resilient to the adverse affects caused by delay in processing time, centrifugation speeds, and storage conditions (i.e. −80 versus −20°C temperature) prior to DNA extraction. Lam et al. (2004) also report improved recovery of plasma DNA and analysis when blood is collected in EDTA rather than heparin or citrate if processing is to be delayed by >6 h after blood draw.
Transportability of specimens soon after venipuncture has been considered. This potential problem is likely to be obviated by technical advances as well. For example, our group reported success in detection of fetal sequences using dried maternal blood spots and real-time PCR (Bischoff et al., 2003a). Fetal Y-specific (DYS1) sequences were detected in all 19 (100%; 4.20–24.68 GEq/ml of blood) maternal blood specimens from women carrying male fetuses. Feasibility of detecting fetal sequences in maternal blood spots dried onto filter paper allows for more efficient collection and transport of specimens, thus enabling cell-free DNA to be incorporated into non-invasive screening regimes on a wide scale.
Cell-free fetal DNA for detecting aneuploidy
Trisomies 13, 18 and 21
Cell-free fetal DNA can play a role in non-invasive prenatal genetic diagnosis of aneuploidy by helping to identify pregnancies at sufficient risk for trisomy 21 or 18 such that an invasive diagnostic procedure should be offered for definitive diagnosis. The fetal DNA is likely derived from genes located throughout the fetal genome, not just fetal chromosome 21. Using real-time PCR to quantify Y-specific sequences, Lo et al. (1999b) demonstrated a 2-fold increase in fetal DNA levels for trisomy 21, compared to euploid cases. Subsequent studies have supported these observations on trisomy 21, although increase is not observed in trisomy 18 (Zhong et al., 2000a). This suggests that different fetal growth and placental pathologies may result in different levels of fetal DNA.
Interestingly, Hromadnikova et al. (2002) were not able to detect increased fetal DNA levels or differences in the fetal:maternal DNA ratio in maternal plasma of patients with affected trisomy 21 fetuses compared to normal controls. Although Spencer et al. (2003) could not detect increased levels of fetal cell-free DNA in serum of ten trisomy 21-affected women, total DNA (fetal and maternal) was observed to be increased. Based on real-time quantification of ubiquitous albumin gene sequences, the median level of total DNA was significantly greater in women with trisomy 21 (36 152 GEq/ml) compared to controls (5832 GEq/ml). Given that maternal cell lysis increases the overall quantity of DNA in serum, analysis using maternal serum rather than plasma in this study likely explains the inability to detect increased fetal DNA levels in trisomy 21.
Wataganara et al. (2003) found maternal serum fetal DNA levels to be elevated in cases of trisomy 13 (29.2–187.0 GEq/ml) but not trisomy 18 (18.6–77.6 GEq/ml). The level of fetal DNA detected was quite variable in the two groups as well as in controls (3.7–127.4 GEq/ml). Archival maternal serum samples displayed a 1.8-fold increase in the amount of fetal DNA levels in trisomy positive pregnancies compared to gestational age-matched controls.
Although methods to improve consistency of fetal DNA quantities are likely to be achieved with more efficient plasma/serum DNA isolation, these findings point to the possible use of either fetal DNA or total DNA (fetal and maternal) as an additional aneuploidy screening analyte. Farina et al. (2003) evaluated the use of circulating fetal DNA as a second trimester maternal serum marker of Down's syndrome. The median fetal DNA concentration was 1.7-fold greater in Down's syndrome cases than in controls. Used singularly as a non-invasive marker for Down's syndrome, fetal DNA gives a 21% detection rate with a set 5% false-positive rate. If combined with the quadruple marker screen, non-invasive aneuploidy detection would improve to 86% at a fixed 5% false-positive rate.
Cell-free fetal DNA to identify pregnancy complications
Increased fetal DNA concentrations
Circulating fetal DNA has also been targeted as a marker for assessing feto-maternal well-being. Increased fetal DNA concentrations have been reported for several pregnancy-related complications (Table I). The best studied is pre-eclampsia, a multisystem disorder characterized by hypertension (high blood pressure) and proteinuria (the presence of protein in urine). An example of a pedestrian complication of pregnancy, pre-eclampsia, has long been a leading cause of maternal mortality in the USA and UK. Although well-established diagnostic criteria exist (Roberts et al., 1993; Redman et al., 1999), these are specific to onset of clinical symptoms relatively late in pregnancy. In pre-eclampsia, placentas show failure of trophoblast cells to invade and remodel the maternal environment (Redman et al., 2001). Failure to remodel the maternal spiral arteries restricts the flow of blood to the fetus and thus contributes to onset of pre-eclampsia. Among possible explanations for the failure of trophoblasts to function properly, the most commonly speculated is an increased rate of apoptosis. Alternatively, motility and/or invasiveness of trophoblasts may be altered. Irrespective, there is growing evidence to support increased placental apoptosis, although the underlying biological basis remains unclear.
In pre-eclampsia, a significant increase in the number of copies of fetal DNA (Y-sequences) was observed at the time the disease was manifested (Lo et al., 1999c; Leung et al., 2001; Zhong et al., 2001b, 2002b). Elevated concentrations of fetal DNA in plasma of pre-eclamptic women has been shown to appear prior to the onset of clinical symptoms as well as correlate with severity of this pathological condition (Leung et al., 2001; Swinkels et al., 2002; Zhong et al., 2002b). Interestingly, Byrne et al. (2003) reported that fetal DNA concentrations were not correlated with severity of pre-eclampsia when whole blood analysis was performed. This further suggests that cell-free fetal DNA in plasma of these patients is derived from placental or fetal cells undergoing apoptosis rather than from intact circulating fetal cells. Such findings are consistent with previous reports demonstrating a lack of correlation between circulating intact fetal cells and cell-free fetal DNA (Bischoff et al., 2002; Zhong et al., 2002a).
Impaired clearance of fetal DNA
Fetal DNA clearance is also impaired in pre-clampsia (Lau et al., 2002). That abnormal clearance has not been observed in trisomy 21 suggests that the underlying mechanism and/or form of fetal DNA in maternal plasma may differ between the two conditions. Some investigators hypothesize that the increase of circulating fetal DNA in pre-eclampsia may be due to some form of cell injury and placental breakdown with release of cellular ‘micro-debris’ into the circulation (Holdenrieder et al., 2001; Reister et al., 2001Stroun et al., 2000). We speculate whether these micro-debris particles are likely apoptotic bodies or nucleosomes. More recently, Levine et al. (2004) reported that fetal DNA in cases of pre-eclampsia is elevated in two stages, at 17–28 weeks (36 GEq/ml) and then 29–41 weeks (176 GEq/ml) as compared to normal controls (16 and 75 GEq/ml respectively), reflecting placental necrosis and apoptosis in the first stage and impaired DNA clearance in the second.
Detection of paternally inherited alleles using maternal plasma
Single gene sequences
Initial studies characterizing fetal DNA in maternal circulation used PCR to detect Y-chromosome sequences corresponding to male DNA, presumably derived from the fetus. Because of high amounts of maternal (background) DNA, approaches have focused on cases where the allele of interest would not be present in the maternal genome (e.g. paternal inheritance of a dominant disease). PCR-based assays for fetal DNA have been developed for a number of single gene disorders. In particular, Rh determination during pregnancies in which a rhesus-negative mother is at risk for a rhesus-positive child has been validated (Faas et al., 1998; Bischoff et al., 1999). In our retrospective study of 20 frozen serum samples from sensitized RhD-negative pregnant women confirmed to have RhD-positive fetuses, we demonstrated positive fetal RhD detection in 70% of cases using conventional fluorescent PCR for simultaneous amplification of the RhD and RhCE (control) genes (Bischoff et al., 1999). Failure to detect RhD sequences in 100% of cases was most likely due to either analysis using serum samples (lysis of maternal cells further dilutes fetal DNA), DNA degradation as a result of freezing and thawing serum specimens or inefficient DNA isolation. Using improved methods of plasma DNA isolation and more sensitive sequence detection assays, namely real-time PCR, highly accurate (98–100%) fetal RhD genotyping can be achieved (Lo et al., 1998a; Zhong et al., 2000b; Finning et al., 2002). Indeed, the high detection and accuracy rates have resulted in the implementation of this non-invasive test at the International Blood Group Reference Laboratory (IBGRL) (Finning et al., 2002). Several other single gene disorders have also been tested for feasibility in detection or exclusion of an affected fetus based on DNA sequences isolated from maternal plasma. Table II summarizes these disorders; however, each reflects analysis of only one or very few cases.
Epigenetic modifications
Inability of PCR to distinguish readily between maternally inherited fetal DNA and native maternal DNA is clearly a diagnostic impediment. A comparable equivalent to Y-specific DNA that could serve as a facile internal control to verify presence of fetal DNA in the sample being assessed is lacking. However, studies of epigenetic changes resulting in distinguishable patterns of DNA methylation between fetal and maternal DNA provide promise in the development of unique gene or global fetal-specific sequence detection assays (Poon et al., 2002). Similarly, placental-specific transcripts in maternal plasma provide an alternative approach for quantification assays for detection of fetal trisomies or pregnancy complications.
Conclusion
Overall, apoptosis seems to be the most plausible source of fetal DNA in maternal circulation, accounting for quantitative increase of this DNA with gestational age and rapid clearance following delivery. Irrespective of the source, various forms of circulating DNA in plasma can be suggested: shed cells, apoptotic bodies, nucleosomes, other nucleoproteins, and free DNA. Thus, an important goal in this field should be to identify the predominant form of fetal DNA in maternal plasma among normal pregnancies. Identification of the forms of circulating fetal DNA can lead to selection or creation of more effective methods of enrichment and DNA isolation from plasma for improved clinical applications.
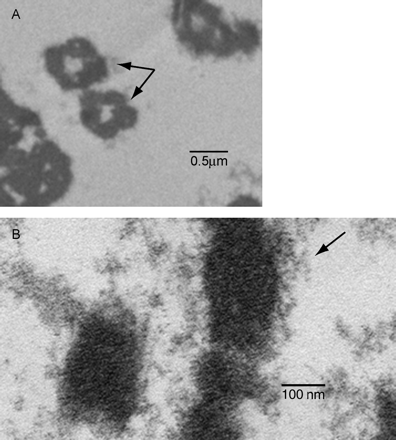
Transmission electron microscopic analysis of maternal plasma pellets. Images display electron micrographs of maternal plasma pellets at two magnifications. (A) Arrows identify the presence of nucleosomes among various structures that are likely ruptured vesicles (apoptotic bodies). (B) Higher magnification illustrates presence of chromatin (arrow).
Pregnancy-related disorders shown to be associated with increased fetal DNA concentrations in maternal plasma
| Pregnancy-related disorders | References |
|---|---|
| Pre-eclampsia | Lo et al., 1999; Zhong et al., 2001 |
| Preterm labour | Leung et al., 1998 |
| Invasive placentation | Sekizawa et al., 2002 |
| Hyperemesis gravidarum | Sekizawa et al., 2001; Sugito et al., 2003 |
| Intrauterine growth restriction (IUGR) | Caramelli et al., 2003 |
| Feto-maternal haemorrhage | Lau et al., 2000 |
| Polyhydramnious | Zhong et al., 2000 |
| Pregnancy-related disorders | References |
|---|---|
| Pre-eclampsia | Lo et al., 1999; Zhong et al., 2001 |
| Preterm labour | Leung et al., 1998 |
| Invasive placentation | Sekizawa et al., 2002 |
| Hyperemesis gravidarum | Sekizawa et al., 2001; Sugito et al., 2003 |
| Intrauterine growth restriction (IUGR) | Caramelli et al., 2003 |
| Feto-maternal haemorrhage | Lau et al., 2000 |
| Polyhydramnious | Zhong et al., 2000 |
Pregnancy-related disorders shown to be associated with increased fetal DNA concentrations in maternal plasma
| Pregnancy-related disorders | References |
|---|---|
| Pre-eclampsia | Lo et al., 1999; Zhong et al., 2001 |
| Preterm labour | Leung et al., 1998 |
| Invasive placentation | Sekizawa et al., 2002 |
| Hyperemesis gravidarum | Sekizawa et al., 2001; Sugito et al., 2003 |
| Intrauterine growth restriction (IUGR) | Caramelli et al., 2003 |
| Feto-maternal haemorrhage | Lau et al., 2000 |
| Polyhydramnious | Zhong et al., 2000 |
| Pregnancy-related disorders | References |
|---|---|
| Pre-eclampsia | Lo et al., 1999; Zhong et al., 2001 |
| Preterm labour | Leung et al., 1998 |
| Invasive placentation | Sekizawa et al., 2002 |
| Hyperemesis gravidarum | Sekizawa et al., 2001; Sugito et al., 2003 |
| Intrauterine growth restriction (IUGR) | Caramelli et al., 2003 |
| Feto-maternal haemorrhage | Lau et al., 2000 |
| Polyhydramnious | Zhong et al., 2000 |
Single gene mutations detected using cell-free fetal DNA in maternal plasma
| Single gene disorder | References |
|---|---|
| Achondroplasia | Saito et al., 2000 |
| Myotonic dystrophy | Amicucci et al., 2000 |
| Congenital adrenal hyperplasia | Chiu et al., 2002b |
| Beta-thalassaemia | Chiu et al., 2002a,c |
| Cystic fibrosis | Gonzalez-Gonzalez et al., 2002 |
| Huntington disease | Gonzalez-Gonzalez et al., 2003a,b |
| Single gene disorder | References |
|---|---|
| Achondroplasia | Saito et al., 2000 |
| Myotonic dystrophy | Amicucci et al., 2000 |
| Congenital adrenal hyperplasia | Chiu et al., 2002b |
| Beta-thalassaemia | Chiu et al., 2002a,c |
| Cystic fibrosis | Gonzalez-Gonzalez et al., 2002 |
| Huntington disease | Gonzalez-Gonzalez et al., 2003a,b |
Single gene mutations detected using cell-free fetal DNA in maternal plasma
| Single gene disorder | References |
|---|---|
| Achondroplasia | Saito et al., 2000 |
| Myotonic dystrophy | Amicucci et al., 2000 |
| Congenital adrenal hyperplasia | Chiu et al., 2002b |
| Beta-thalassaemia | Chiu et al., 2002a,c |
| Cystic fibrosis | Gonzalez-Gonzalez et al., 2002 |
| Huntington disease | Gonzalez-Gonzalez et al., 2003a,b |
| Single gene disorder | References |
|---|---|
| Achondroplasia | Saito et al., 2000 |
| Myotonic dystrophy | Amicucci et al., 2000 |
| Congenital adrenal hyperplasia | Chiu et al., 2002b |
| Beta-thalassaemia | Chiu et al., 2002a,c |
| Cystic fibrosis | Gonzalez-Gonzalez et al., 2002 |
| Huntington disease | Gonzalez-Gonzalez et al., 2003a,b |
We thank Dr William Brinkley for expertise in transmission election microscopy. We also thank Dianne Dang and Belinda Felder for assistance in manuscript preparation. This work was supported in part by NIH/NICHD contract N01-HD-43203.
References
Amicucci P, Gennarelli M, Novelli G and Dallapiccola B (
Angert RM, LeShane ES, Lo YM, Chan LY, Delli-Bovi LC and Bianchi DW (
Anker P and Stroun M (
Anker P, Mulcahy H, Chen XQ and Stroun M (
Ariga H, Ohto H, Busch MP, Imamura S, Watson R, Reed W and Lee TH (
Barrett KL, Willingham JM, Garvin AJ and Willingham MC (
Bayrak-Toydemir P, Pergament E and Fiddler M (
Bianchi DW (
Bianchi DW, Simpson JL, Jackson LG, Elias S, Holzgreve W, Evans MI, Dukes KA, Sullivan LM, Klinger KW, Bischoff FZ et al. (
Bischoff FZ, Nguyen DD, Marquez-Do D, Moise KJ, Jr, Simpson JL and Elias S (
Bischoff FZ, Sinacori MK, Dang DD, Marquez-Do D, Horne C, Lewis DE and Simpson JL (
Bischoff FZ, Dang DX, Marquez-Do D, Martinez D, Horne C, Lewis DE and Simpson JL (
Bischoff FZ, Hahn S, Johnson KL, Simpson JL, Bianchi DW, Lewis DE, Weber WD, Klinger K, Elias S, Jackson LG, Evans MI, Holzgreve W and de la CF (
Byrne BM, Crowley A, Taulo F, Anthony J, O'Leary JJ and O'Herlihy C (
Caramelli E, Rizzo N, Councu M, Simonazzi G, Carinci P, Bondavalli C, Bovicelli L and Farina A. (
Chan KC and Lo YM (
Chan KC, Zhang J, Hui AB, Wong N, Lau TK, Leung TN, Lo KW, Huang DW and Lo YM (
Chan LY, Leung TN, Chan KC, Tai HL, Lau TK, Wong EM and Lo YM (
Chen XQ, Stroun M, Magnenat JL, Nicod LP, Kurt AM, Lyautey J, Lederrey C and Anker P (
Chen XQ, Bonnefoi H, Pelte MF, Lyautey J, Lederrey C, Movarekhi S, Schaeffer P, Mulcahy HE, Meyer P, Stroun M and Anker P (
Chiu RW, Poon LL, Lau TK, Leung TN, Wong EM and Lo YM (
Chiu RW, Lau TK, Cheung PT, Gong ZQ, Leung TN and Lo YM (
Chiu RW, Lau TK, Cheung PT, Gong ZQ, Leung TN and Lo YM (
Chiu RW, Lau TK, Leung TN, Chow KC, Chui DH and Lo YM (
Costa JM, Benachi A, Gautier E, Jouannic JM, Ernault P and Dumez Y (
Dhallan R, Au WC, Mattagajasingh S, Emche S, Bayliss P, Damewood M, Cronin M, Chou V and Mohr M (
Dukes KA, Sullivan LM, Lewis D, Johnson KL, Bianchi DW, Simpson JL, Holzgreve W, Hahn S, Bischoff FZ and Jackson LG (
Faas BH, Beuling EA, Christiaens GC, dem Borne AE and Van der Schoot CE (
Farina A, Caramelli E, Concu M, Sekizawa A, Ruggeri R, Bovicelli L, Rizzo N and Carinci P (
Farina A, LeShane ES, Lambert-Messerlian GM, Canick JA, Lee T, Neveux LM, Palomaki GE and Bianchi DW (
Finning KM, Martin PG, Soothill PW and Avent ND (
Giacona MB, Ruben GC, Iczkowski KA, Roos TB, Porter DM and Sorenson GD (
Gonzalez-Gonzalez MC, Garcia-Hoyos M, Trujillo MJ, Rodriguez dA, Lorda-Sanchez I, Diaz-Recasens J, Gallardo E, Ayuso C and Ramos C (
Gonzalez-Gonzalez MC, Trujillo MJ, Rodriguez dA, Garcia-Hoyos M, Lorda-Sanchez I, Diaz-Recasens J, Ayuso C and Ramos C (
Gonzalez-Gonzalez MC, Trujillo MJ, Rodriguez dA and Ramos C (
Halicka HD, Bedner E and Darzynkiewicz Z (
Hamada H, Arinami T, Kubo T, Hamaguchi H and Iwasaki H (
Hasselmann DO, Rappl G, Tilgen W and Reinhold U (
Holdenrieder S, Stieber P, Bodenmuller H, Busch M, von Pawel J, Schalhorn A, Nagel D and Seidel D (
Honda H, Miharu N, Ohashi Y, Samura O, Kinutani M, Hara T and Ohama K (
Houfflin-Debarge V, O'Donnell H, Overton T, Bennett PR and Fisk NM (
Hromadnikova I, Houbova B, Hridelova D, Voslarova S, Calda P, Nekolarova K, Kofer J, Stejskal D, Doucha J, Cinek O and Vavrirec J (
Hromadnikova I, Houbova B, Hridelova D, Voslarova S, Kofer J, Komrska V and Habart D (
Jahr S, Hentze H, Englisch S, Hardt D, Fackelmayer FO, Hesch RD and Knippers R (
Jen J, Wu L and Sidransky D (
Jen J, Wu L and Sidransky D (
Jimenez DF and Tarantal AF (
Johnson KL, Dukes KA, Vidaver J, LeShane ES, Ramirez I, Weber WD, Bischoff FZ, Hahn S, Sharma A, Dang DX, Hire LM et al. (
Kolialexi A, Tsangaris GT, Mavrou A, Antsaklis A, Tzortzatou F, Touliatou V and Metaxotou C (
Kopreski MS, Benko FA, Kwak LW and Gocke CD (
Krabchi K, Gros-Louis F, Yan J, Bronsard M, Masse J, Forest JC and Drouin R (
Lam NY, Rainer TH, Chiu RW and Lo YM (
Lau TK, Lo KW, Chan LY, Leung TY and Lo YM. (
Lau TW, Leung TN, Chan LY, Lau TK, Chan KC, Tam WH and Lo YM (
Leon SA, Shapiro B, Sklaroff DM and Yaros MJ (
Leung TN, Zhang J, Lau TK, Hjelm NM and Lo YM (
Leung TN, Zhang J, Lau TK, Chan LY and Lo YM (
Levine RJ, Qian C, Leshane ES, Yu KF, England LJ, Schisterman EF, Wataganara T, Romero R and Bianchi DW (
Lo YM, Leung TN, Tein MS, Sargent IL, Zhang J, Lau TK, Haines CJ and Redman CW (
Lo YM, Tein MS, Lau TK, Haines CJ, Leung TN, Poon PM, Wainscoat JS, Johnson PJ, Chang AM and Hjelm NM (
Lo YM, Corbetta N, Chamberlain PF, Rai V, Sargent IL, Redman CW and Wainscoat JS (
Lo YM, Hjelm NM, Fidler C, Sargent IL, Murphy MF, Chamberlain PF, Poon PM, Redman CW and Wainscoat JS (
Lo YM, Tein MS, Lau TK, Haines CJ, Leung TN, Poon PM, Wainscoat JS, Johnson PJ, Chang AM and Hjelm NM (
Lo KW, Lo YM, Leung SF, Tsang YS, Chan LY, Johnson PJ, Hjelm NM, Lee JC and Huang DP (
Lo YM, Lau TK, Zhang J, Leung TN, Chang AM, Hjelm NM, Elmes RS and Bianchi DW (
Lo YM, Leung TN, Tein MS, Sargent IL, Zhang J, Lau TK, Haines CJ and Redman CW (
Lo YM, Zhang J, Leung TN, Lau TK, Chang AM and Hjelm NM (
Lui YY and Dennis YM (
Martelli AM, Zweyer M, Ochs RL, Tazzari PL, Tabellini G, Narducci P and Bortul R (
Nawroz H, Koch W, Anker P, Stroun M and Sidransky D (
Ng EK, Tsui NB, Lau TK, Leung TN, Chiu RW, Panesar NS, Lit LC, Chan KW and Lo YM (
Ohashi Y, Miharu N, Honda H, Samura O and Ohama K (
Poon LL, Leung TN, Lau TK and Lo YM (
Poon LL, Leung TN, Lau TK and Lo YM (
Poon LL, Leung TN, Lau TK, Chow KC and Lo YM (
Redman CW and Sargent IL (
Redman CW, Sacks GP and Sargent IL (
Reister F, Frank HG, Kingdom JC, Heyl W, Kaufmann P, Rath W and Huppertz B (
Rijnders RJ, Van der Luijt RB, Peters ED, Goeree JK, Van der Schoot CE, Ploos Van Amstel JK and Christiaens GC (
Roberts JM and Redman CW (
Rudin CM and Thompson CB (
Saito H, Sekizawa A, Morimoto T, Suzuki M and Yanaihara T (
Sekizawa A, Kondo T, Iwasaki M, Watanabe A, Jimbo M, Saito H and Okai T (
Sekizawa A, Jimbo M, Saito H, Iwasaki M, Sugito Y, Yukimoto Y, Otsuka J and Okai T (
Silva NH, Pimenta G, Pulcheri WA, Fournier MV, Spector N and Costa Carvalho MG (
Sorenson GD, Pribish DM, Valone FH, Memoli VA, Bzik DJ and Yao SL (
Spencer K, de Kok JB and Swinkels DW (
Stenman J and Orpana A (
Stroun M, Anker P, Maurice P, Lyautey J, Lederrey C and Beljanski M (
Stroun M, Maurice P, Vasioukhin V, Lyautey J, Lederrey C, Lefort F, Rossier A, Chen XQ and Anker P (
Sugito Y, Sekizawa A, Farina A, Yukimoto Y, Saito H, Iwasaki M, Rizzo N and Okai T (
Swinkels DW, de Kok JB, Hendriks JC, Wiegerinck E, Zusterzeel PL and Steegers EA (
Taback B, O'Day SJ and Hoon DS (
Thomas MR, Tutschek B, Frost A, Rodeck CH, Yazdani N, Craft I and Williamson R (
Tsui NB, Ng EK and Lo YM (
Vasioukhin V, Anker P, Maurice P, Lyautey J, Lederrey C and Stroun M (
Wataganara T, LeShane ES, Farina A, Messerlian GM, Lee T, Canick JA and Bianchi DW (
Wu TL, Zhang D, Chia JH, Tsao KH, Sun CF and Wu JT (
Zhong XY, Burk MR, Troeger C, Jackson LR, Holzgreve W and Hahn S (
Zhong XY, Holzgreve W and Hahn S (
Zhong XY, Holzgreve W and Hahn S (
Zhong XY, Holzgreve W, Li JC, Aydinli K and Hahn S (
Zhong XY, Hahn S and Holzgreve W (
Zhong XY, Laivuori H, Livingston JC, Ylikorkala O, Sibai BM, Holzgreve W and Hahn S (
Zhong XY, Holzgreve W and Hahn S (
Zhong XY, Holzgreve W and Hahn S (
Author notes
Departments of 1Obstetrics and Gynecology,2 Immunology and3 Human and Molecular Genetics, Baylor College of Medicine, Houston, TX 77030, USA

{kind=link}