





Abstract
The membrane attack complex (MAC) of the complement system is causing membrane damage and cell death. For protection, cells have adopted several resistance mechanisms, including removal of the membrane-inserted MAC by vesiculation. To identify proteins involved in MAC vesiculation, extracellular proteins released from K562 cells in response to treatment with sub-lytic complement were separated by acrylamide gel electrophoresis and protein bands were extracted, digested into peptides and the peptides were analyzed by mass spectrometry. A 75-kDa protein that was abundant in the supernatant of complement-treated cells was identified as mortalin/GRP75. Analysis by western blotting demonstrated that as early as 5 min after exposure to sub-lytic doses of complement, mortalin was released from K562 cells. Mortalin was released after complete activation of the complement system and formation of C5b–8, and even more so when C5b–9 was formed. Other pore formers, such as streptolysin O and melittin, did not induce release of mortalin. As shown, mortalin can bind to complement C8 and C9 and is shed in vesicles containing C9 and complement MACs. Anti-mortalin antibodies reduced mortalin release from complement-treated cells and elevated the extent of cell death by complement. Inhibitors of protein kinase C and extracellular signal-regulated protein kinase also prevented mortalin release from complement-activated cells. These results suggest that mortalin/GRP75 promotes the shedding of membrane vesicles loaded with complement MAC and protects cells from complement-mediated lysis.
Introduction
The complement system consists of >20 blood plasma proteins that cooperate with other sections of the innate and acquired immune systems in the clearance of pathogenic organisms, immune complexes and apoptotic cells (1). The complement activation cascade culminates in the formation of the membrane attack complex (MAC), made of complement C5b, C6, C7, C8 and C9 proteins (C5b–9), and its insertion into the plasma membrane of target cells (2). Membrane insertion begins when C5b–7 forms, is enhanced in C5b–8 and is maximal upon binding and oligomerization of C9 and formation of a transmembrane, cylinder-shaped polyC9 complex attached to C5b–8. At a high dose, the MAC inflicts major damages to cells, culminating in rapid cell death by necrosis (3) or apoptosis (4). At a low, sub-lytic dose, the MAC acts as a potent stimulator of many cellular activities [for review see (5)]. Treatment with sub-lytic MAC was shown to transduce into various cells either anti-necrotic (6) or anti-apoptotic (7) signals.
As a means of protection from complement, cells can remove the MAC from their plasma membrane by endocytosis or vesiculation (8–10). Physical removal of MAC by vesiculation has been demonstrated in several cell types including neutrophils, oligodendrocytes and platelets, and in the tumor cell lines U937 and K562 (8, 11–13). The shed vesicles have a high content of cholesterol and diacylglycerol (14) and are loaded with MAC and C9, suggesting a selective sorting on the cell surface prior to shedding. To date, little is known about the molecular mechanism responsible for MAC vesiculation.
As shown here, mortalin is released, in association with the MAC, from K562 cells treated with sub-lytic doses of complement. Mortalin, also known as GRP75, PBP74, mot-2 and mitochondrial heat shock protein (mthsp) 70, was described in mitochondria, cytoplasm, endoplasmic reticulum and cytoplasmic vesicles (15, 16). It is constitutively expressed in cells, participates in stress response (17) and mitochondrial import (18) and is frequently up-regulated in tumors (19, 20). Mortalin is shown here to be located, together with C9, in extracellular vesicles. Its release is dependent on MAC formation and on the activation of protein kinase C (PKC) and extracellular signal-regulated protein kinase (ERK). Finally, our data show that anti-mortalin antibodies augment cell death by complement. It is suggested that mortalin is defending cells from complement by eliminating MAC from the cell surface.
Methods
Cells and cell lysates
K562, a human erythroleukemic cell line, was cultured in RPMI-1640 supplemented with 10% (v/v) heat-inactivated fetal bovine serum (GIBCO laboratories, Grand Island, NY, USA), 1% glutamine, 2% pyruvate and antibiotics mixture (Bio-Lab, Jerusalem, Israel) at 37°C and 5% CO2. To prepare cell lysates, 20 × 106 cells were mixed with 1 ml lysis buffer composed of 100 mM Tris, pH 7.5, 10 mM EDTA, protease inhibitor cocktail (Sigma, Rehovot, Israel) and 0.7% Triton-X100. After three cycles of freezing and thawing, the cell lysate was subjected to centrifugation for 15 min at 14 000 × g and the supernatant was collected and diluted with 1 ml HBSS (Sigma).
Sera, antisera and reagents
Normal human serum (NHS) was prepared from healthy individuals. Heat inactivation of NHS was performed by incubation at 56°C for 30 min. C7- or C8-deficient human sera were prepared from C7- or C8β-deficient patients, respectively, as described (21). C9-depleted human serum albumin and BSA were purchased from Sigma. Human sera were kept frozen at −70°C in small aliquots and thawed only once. Purified human C5b,6 complex, C7, C8 and C9 proteins were purchased from Advanced Research Technologies (San Diego, CA, USA). C3 was purified from human serum as described (6). A polyclonal antiserum directed to K562 cells was prepared in rabbits or mice. Rabbit anti-mortalin antibodies were kindly provided by Sunil Kaul (AIT, Tsukuba Ibaraki, Japan) (22) and Alex Merrick (NIEHS, Research Triangle, NC, USA) (23). Peroxidase-conjugated goat anti-rabbit IgG, streptolysin O (SLO), melittin and HEPES were purchased from Sigma. PD98059, GF109203X and polymyxin B (PMB) were from Calbiochem (San Diego, CA, USA).
Cell lysis and induced protection assays
Cytotoxicity and induced protection assays were performed as described before (24). Briefly, for a direct cell lysis assay, cells were incubated with diluted anti-K562 antiserum for 30 min at 4°C and then with complement [NHS, C7- or C8-deficient serum or heat-inactivated normal human serum (HI-NHS), 50%] for 60 min at 37°C. Cell lysis was determined by Trypan blue exclusion. For the complement-induced protection assays, the cells were first treated with sub-lytic doses of antibody (diluted 1 : 24 to 1 : 30, depending on antiserum batch; 30 min at 4°C) and complement (NHS or HI-NHS, 50%; 60 min at 37°C), washed and then incubated with lytic doses of antibody (usually 2-fold more concentrated than in the first stage; 30 min at 4°C) and NHS (diluted 1 : 4 or 1 : 6; 60 min at 37°C). Cell death was determined by Trypan blue exclusion. Statistical significance was analyzed by using the two-sided unpaired Student's t-test.
Collection of extracellular protein
Cells were treated with antibodies for 30 min at 4°C and then with NHS or HI-NHS (50%) for 10 min at 37°C. Then, they were extensively washed in HBSS and incubated at 37°C. At different times, cells were removed by centrifugation at 250 × g and supernatants were collected and kept frozen until examined. In some experiments, cells were first metabolically labeled for 2 h at 37°C with an L-[35S]methionine, L-[35S]cysteine mix (Amersham Pharmacia, Uppsala, Sweden) at 200 μCi ml−1 in RPMI (lacking methionine and cysteine) and 20 mM HEPES, pH 7.0. Cytotoxicity assay and collection of extracellular proteins were performed as described above. Radioactivity in the supernatants was quantified in a β-Scintillation Counter (Tri-Carb, Packard).
Protein analyses
Protein concentration was analyzed with the BCA Protein Assay Kit (Pierce, Rockford, IL, USA). Protein (20 μg) was subjected to SDS-PAGE under reducing conditions [50 mM dithiothreitol (DTT)], in a 10% acrylamide gel, and stained with Coomassie blue or silver. Selected protein band was cut off a Coomassie blue-stained gel and analyzed by mass spectrometry (MALDI-MS) at the Maiman Institute for Proteom Research (Tel Aviv University). Alternatively, the proteins were transferred onto a nitrocellulose membrane (Schleicher & Schuell, Dassel, Germany). The membrane was blocked with 5% skim milk (Tnuva, Rehovot, Israel) in Tris-buffered saline containing 0.05% Tween 20 (TBST; Sigma) for 1 h at room temperature. The membrane was treated with rabbit anti-mortalin antibodies and then with peroxidase-conjugated goat anti-rabbit IgG. Bands were developed with an enhanced chemiluminescence reagent (Pierce) and exposed to a SuperRX film (Fuji, Tokyo, Japan).
Binding of mortalin to complement C9
Binding of mortalin to the complement C9 protein was examined by using an ELISA and a modified western blotting technique. In brief, purified human C9, C8, C7, C5b,6, C3 and BSA (0.5 μg per well each) were bound overnight at 4°C to a 96-well EIA plate (Corning, NY, USA). Diluted cell lysate prepared from 40 × 106 K562 cells as described above was added to each well for 60 min at 37°C. After washes, monoclonal mouse anti-mortalin antibody (diluted 1 : 500) was added for 30 min at 37°C and then peroxidase-conjugated goat anti-mouse IgG was added for 30 min at room temperature. Next, o-phenylenediamine (Sigma) was added and the plate was examined in a Spectrafluor Plus plate reader (Tecan, Austria) at 450 nm. In addition, purified human C9 and BSA (1 μg each) were subjected to SDS-PAGE under reducing conditions (50 mM DTT), in a 10% acrylamide gel, and transferred onto a nitrocellulose membrane. The membrane was then incubated for 2 h at 37°C with K562 cell lysates supplemented or not with human C9 (2 μg). After two washes with TBST, western blotting analysis was performed, as described above, with anti-mortalin antibody.
Results
Release of mortalin after a sub-lytic complement attack
Proteins are spontaneously released from cells cultured at 37°C. Protein release is elevated after exposure of cells to a sub-lytic dose of antibody and NHS (data not shown). In order to find qualitative differences between protein profiles of supernatants collected from cells treated with complement (NHS) or control cells (HI-NHS), supernatants were analyzed by SDS-PAGE and stained by silver or Coomassie blue (Fig. 1A). An ∼75-kDa protein band was found in NHS-treated cells but was poorly visible in control cells. To identify this protein, it was excised after Coomassie blue staining from the gel and subjected to MALDI-MS analysis (Fig. 1B). Seven different peptides identified in the mass spectrometry analysis gave complete matching with mortalin (mot-2, GRP75) (gene: HSPA9B; gi:24234688). The kinetics of the release of mortalin was next studied. Supernatants were collected from K562 cells at different times after treatment with sub-lytic NHS or with HI-NHS, and were analyzed by western blotting (Fig. 2A and B). A significant level of extracellular mortalin was found in supernatants collected 15 min after treatment with complement and the amount of mortalin kept slightly rising up until 60 min. In contrast, the spontaneous release of mortalin in control cells was very low. Concomitantly, the intracellular level of mortalin gradually decreased from 15 to 60 min (Fig. 2C and D). In contrast, the concentration of mortalin within control cells was stable. To rule out the possibility that mortalin leaks out of dead cells, a correlation between the amount of released mortalin and the level of cell damage was searched for. Cells were treated with increasing concentrations of antibodies and 50% NHS. Percent cell lysis increased from 0 to 86% (Fig. 2E). Next, the quantity of extracellular mortalin was assessed on western blots. As shown in Fig. 2(E), the most significant increase in mortalin release occurred under sub-lytic conditions (14–21% lysis). At higher lytic conditions, there was no additional increase in the amount of extracellular mortalin. These findings indicate that mortalin is released from intact and not from damaged cells.
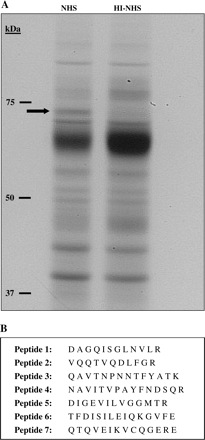
Identification of mortalin outside K562 cells attacked by sub-lytic complement. K562 cells were treated with a sub-lytic dose of anti-K562 antibody and NHS or HI-NHS. After 60 min at 37°C, the cells were sedimented and the supernatants were subjected to SDS-PAGE (5.5 μg per lane) and the gel was silver stained (A). The arrow points at a protein that appears stronger in NHS-treated cells than in HI-NHS-treated cells. (B) This protein band was excised from a Coomassie blue-stained gel and analyzed by MALDI-MS. Seven peptides were identified; all of them corresponded to mortalin.
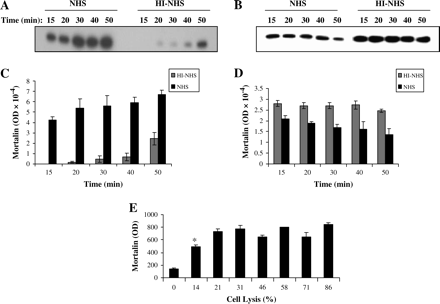
Kinetics and dose response of mortalin release. K562 cells were treated with a sub-lytic dose of antibody and NHS or HI-NHS. After 10 min at 37°C, they were washed and incubated at 37°C. At different times, the supernatants (A) and cells (B) were separated and analyzed each by SDS-PAGE and western blotting with rabbit anti-mortalin antibody. (C and D) Densitometric analysis of the bands shown in A and B, respectively. Results are representative of three independent experiments. (E) Mortalin is released by viable cells. Cells were subjected to different doses of anti-K562 antibody (dilutions: 1 : 3, 1 : 6, 1 : 9, 1 : 12, 1 : 15, 1 : 18 and 1 : 21) and NHS or HI-NHS. After 10 min at 37°C, supernatants released from the cells were analyzed as described above for Fig. 2(A). Cells were also kept for 60 min at 37°C and their percent lysis was determined. Results show the amount of extracellular mortalin (as determined by western blotting and densitometry) versus the level of cell death. Results are representative of three independent experiments. *P < 0.005, between 0 and 14% lysis.
Indications that mortalin is associated with membrane vesicles
Supernatants collected from K562 cells treated with sub-lytic NHS or HI-NHS were sedimented first at 5000 × g to remove cell debris. Then they were subjected to centrifugation at 100 000 × g, conditions known to spin down small membrane vesicles. The high-speed pellet and the supernatant were subjected to SDS-PAGE and western blotting. Analysis with anti-mortalin antibodies (Fig. 3A) or with anti-C9 antibodies (Fig. 3B) indicated that both mortalin and C9 released from K562 cells treated with sub-lytic complement could be spun down at 100 000 × g. Pre-treatment of the supernatants with 0.1% Triton-X100, prior to the high-speed centrifugation, resulted in translocation of both mortalin and C9 from the pellet to the supernatant (Fig. 3C and D), probably due to solubilization of the membrane vesicles bearing mortalin and C9.
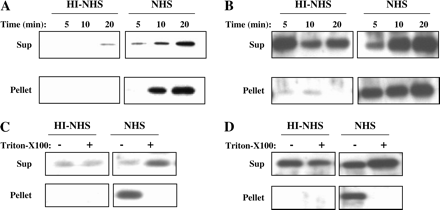
Mortalin and C9 are released in membrane vesicles. K562 cells treated with a sub-lytic dose of antibody and NHS or HI-NHS for 10 min at 37°C were washed in HBSS, incubated at 37°C and supernatants were collected after 5, 10 and 20 min. Released proteins were sedimented at 100 000 × g and supernatants (Sup.) and pellets were analyzed by SDS-PAGE and western blotting with anti-mortalin (A) or anti-C9 (B) antibodies. (B) Proteins released after 20 min incubation (as above) were treated with Triton-X100 (0.1%) and then sedimented at 100 000 × g. Sup and pellets were analyzed by SDS-PAGE and western blotting with anti-mortalin (C) or anti-C9 (D) antibodies. Results are each representative of three independent experiments.
Mortalin release depends on C5b–8 and C5b–9
In the absence of C8 or C9, the terminal complement complex is composed of C5b–7 or C5b–8, respectively (2). To determine whether only C5b–9 can cause release of mortalin, the effect of NHS was compared with that of C8-deficient and C9-depleted human sera. Supernatants were collected from K562 cells treated with antibody and C8-deficient or C9-depleted human sera or NHS and analyzed by western blotting for mortalin binding (Fig. 4A and B). Unlike NHS, only a small amount of mortalin was released from cells treated with C8-deficient serum or HI-NHS. C9-depleted serum caused a substantial release of mortalin, albeit only about half of the release induced by NHS (P < 0.01). Upon reconstitution of the C8-deficient serum with purified C8 (to 55 μg ml−1 final concentration) and of the C9-depleted human serum with C9 (to 60 μg ml−1 final concentration), they caused as much mortalin release as NHS.
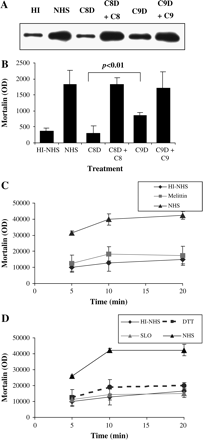
Mortalin secretion is induced by C5b–9 and not by melittin and SLO. (A and B) K562 cells were treated with anti-K562 antibody followed by NHS, HI-NHS (HI), C8-deficient serum (C8D) or C8D mixed with 55 μg ml−1 C8 (C8D + C8), C9-depleted serum (C9D) or C9D mixed with 60 μg ml−1 C9 (C9D + C9). After 10 min at 37°C, the cells were washed and incubated for 10 min at 37°C. Supernatants were collected and analyzed by SDS-PAGE and western blotting with anti-mortalin antibodies. Densitometric analysis of three independent experiments is shown in B. (C) K562 cells were treated with a sub-lytic dose of melittin (70 μM) or with PBS as control. After 10 min at 37°C, they were washed and incubated for different times at 37°C. Supernatants were collected and analyzed by SDS-PAGE and western blotting with anti-mortalin antibodies. Densitometric analysis of three independent experiments is shown. (D) Cells were treated with sub-lytic SLO (900 units ml−1) or with DTT as control. After 10 min at 37°C, they were washed and incubated for different times at 37°C. Supernatants were collected and analyzed by SDS-PAGE and western blotting with anti-mortalin antibodies. Densitometric analysis of three independent experiments is shown. Cells treated with NHS or HI-NHS were also included in C and D, as described for A. Statistical analyses indicated a significant (P < 0.01) difference between the NHS, C8D + C8 and C9D + C9 groups and the HI-NHS, C8D and C9D groups, respectively.
SLO and melittin are pore-forming proteins related to complement C9 (25–27). In order to find out whether the release of mortalin is selectively induced by C5b–9 or can also be induced by other pore formers, K562 cells were subjected to treatment with sub-lytic doses of SLO and melittin. Analysis of supernatants collected from these cells clearly demonstrated that mortalin was not released in response to SLO or melittin treatment (Fig. 4C and D).
Anti-mortalin antibodies sensitize cells to complement-mediated lysis
To address the biological relevance of mortalin release, we examined the effect of anti-mortalin antibodies on cell death induced by MAC. K562 cells were treated with antibody and NHS in the presence of anti-mortalin antibodies or pre-bleed serum as control. The anti-mortalin antibodies significantly increased complement-mediated lysis of K562 cells (Fig. 5A). In order to rule out the possibility that the anti-mortalin antibodies simply recruit more complement to the cell surface, the assay was performed in two stages. First, cells were treated with C8-deficient human serum (C8D) and washed, and then with anti-mortalin antibodies and C7-deficient-human serum (C7D) in presence of EDTA. In this case, the anti-mortalin antibodies were added to cells bearing C5b–7 complexes and could only block the formation of C5b–8 and C5b–9. Again, pre-bleed serum served as control. In this experiment too, anti-mortalin antibodies increased sensitivity of K562 cells to lysis by complement (Fig. 5B). As shown in Fig. 5(C), in the presence of anti-mortalin antibodies, release of mortalin and C9 from complement-attacked cells was blocked. Hence, apparently, mortalin emerges, during complement attack, from the cells and is involved in the C9 release process.
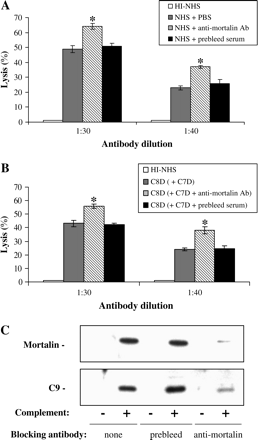
Anti-mortalin antibodies promote cell death by C5b–9. (A) K562 cells were treated with anti-mortalin antibodies or a pre-bleed serum (diluted 1 : 500) mixed with anti-K562 antibodies (diluted 1 : 30 or 1 : 40) and then with NHS or HI-NHS. After 60 min at 37°C, cell lysis was determined by Trypan blue exclusion. Results are representative of three independent experiments. (B) K562 cells were treated with anti-K562 antibodies (diluted 1 : 30 or 1 : 40) and C8D (or HI-NHS as negative control). After 15 min at 37°C, the cells were washed and mixed with C7-deficient human serum (C7D) containing 20 mM EDTA (or HI-NHS and EDTA). The C7D was pre-mixed with anti-mortalin antibodies (1 : 500) or pre-bleed serum (or buffer control). Cell lysis was determined as described above. *P < 0.01 between groups with anti-mortalin antibodies and pre-bleed serum. (C) K562 cells were treated or not (none) with anti-mortalin antibodies or a pre-bleed serum (diluted 1 : 500) mixed with anti-K562 antibodies and then with NHS (+complement) or HI-NHS (−complement). After 10 min at 37°C, the cells were washed and incubated for 10 min at 37°C. Supernatants were collected and analyzed by SDS-PAGE and western blotting with mortalin or C9 antibodies. Results are representative of three independent experiments.
Involvement of PKC and ERK in mortalin release
Both PKC and ERK are known to contribute to cell resistance to complement damage (28, 29). The effect of two PKC inhibitors, PMB and GF109203X, and a MEK inhibitor, PD98059, on mortalin release was examined. Both PMB (Fig. 6A and B) and PD98059 (Fig. 6C and D) significantly reduced the release of mortalin from complement-treated cells. A similar effect was observed with cells treated with another PKC inhibitor, GF109203X (data not shown). PMB and PD98059 treatments also blocked the complement-induced release of metabolically labeled proteins (Fig. 6E). To show a correlation between the effect of the PKC and MEK inhibitors on mortalin release and their effect on cell resistance to complement, the complement-induced protection assay was used (6). Cells were pre-treated with the inhibitors and then with a sub-lytic dose of antibody and complement. Next, the cells were treated with a lytic dose of the antibody and the complement. As shown in Fig. 6(F and G), cells pre-treated with sub-lytic complement and PBS or DMSO (solvent control to PD98059) acquired resistance to lytic complement doses (cell lysis after pre-treatment with NHS was lower than with HI-NHS). In contrast, cells pre-treated with sub-lytic complement and with PMB (Fig. 6F) or PD98059 (Fig. 6G) did not acquire the resistance to lytic complement.
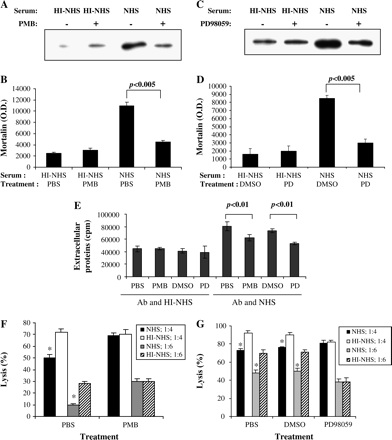
PKC and ERK are involved in mortalin release. K562 cells were pre-treated for 30 min at 37°C with 100 μg ml−1 PMB or PBS as control (A and B) or with 10 μM PD98059 (PD) or 0.1% dimethyl sulfoxide (DMSO) as control (C and D). Then, they were treated with a sub-lytic dose of anti-K562 antibody and NHS or HI-NHS. After 10 min at 37°C, they were washed and incubated for 10 min at 37°C. Supernatants were collected and analyzed by SDS-PAGE and western blotting with anti-mortalin antibodies. Densitometric analysis of three independent experiments is shown in B and D. (E) Metabolically labeled K562 cells were pre-treated for 30 min at 37°C with PBS, 100 μg ml−1 PMB, 0.1% DMSO or 10 μM PD. Then, they were treated with anti-K562 antibody and NHS or HI-NHS for 30 min at 37°C. Radioactivity in cell supernatants was determined. Results are representative of three independent experiments. (F and G) Cells pre-treated with PMB (F) or PD (G), as for A–D, were subjected to a complement-induced protection assay (see Methods). In the first step, the cells were treated with sub-lytic antibody and complement (legend: NHS or HI-NHS). Then, they were washed and treated with a lytic dose of antibody and NHS diluted 1 : 4 or 1 : 6 (legend: 1 : 4 or 1 : 6). Percent cell lysis was determined after 1 h incubation with NHS at 37°C. *P < 0.05 (between each pair of NI-NHS and NHS).
Mortalin binds to C9 and C8
The possibility that mortalin binds to the complement C9 protein was first examined by ELISA. As shown in Fig. 7(A), mortalin bound to human C9 stronger than to C8 and showed some poor binding also to C7. Mortalin did not bind to the C5b,6 complex or to C3 and BSA. In addition, a protein–protein-binding assay was performed, as described before (30) and briefly under Methods. Human C9 was subjected to SDS-PAGE and transferred to a nitrocellulose membrane. The membrane was incubated with cell lysate, as a source for mortalin, and then with anti-mortalin antibody. Alternatively, the membrane was incubated with cell lysate pre-mixed with C9. As shown in Fig. 7(B), mortalin bound to blotted C9 and not to BSA. This binding could be competed with soluble C9, indicating that mortalin can bind to native epitopes on C9.
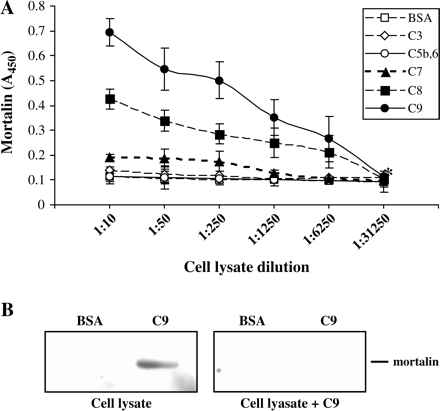
Mortalin binds to C9, C8 and C7. (A) C9, C8, C7, C5b,6, C3 or BSA bound to microtiter plate (0.5 μg per well) were incubated with K562 cell lysate (40 × 106 cells ml−1 lysis buffer) diluted 1 : 5 to 1 : 31250. The wells were then washed, treated with anti-mortalin antibodies and peroxidase-labeled second antibody, developed with o-phenylenediamine and analyzed in an ELISA reader (see Methods). Results are representative of five independent experiments. Differences between the C9 and the C8 wells and between them and the C7 wells were significant (P < 0.01) at lysate dilutions 1 : 5 to 1 : 6250. Differences between the C7 wells and the BSA wells were significant (P < 0.01) at lysate dilution 1 : 5 to 1 : 250. (B) C9 and BSA (5 μg each) were subjected to SDS-PAGE and transferred to a nitrocellulose membrane. The membrane was then incubated with K562 cell lysate pre-mixed with PBS (Cell lysate) or with 5 μg ml−1 C9 (Cell lysate + C9). Protease inhibitors cocktail was included to block proteases in cell lysate. Membranes were then washed and analyzed by western blotting with anti-mortalin antibodies. Results are representative of three independent experiments.
Discussion
The results of this study demonstrate that sub-lytic complement attack induces shedding of mortalin associated with membrane vesicles. This requires the complete assembly of the C5b–9 MAC, as heat inactivation of complement or the absence of complement C7 or C8 precludes the release of mortalin. Our data also show that shedding of mortalin in response to complement attack is PKC and ERK dependent and occurs from viable cells. In support to our claim that the extracellular mortalin originates from viable cells and not from damaged cells, we show that a similar level of sub-lytic damage caused by two other pore formers, SLO and melittin, does not induce the release of mortalin. That anti-mortalin antibodies reduce mortalin secretion and increase cell sensitivity to complement-mediated lysis suggests that shedding of mortalin is translated into resistance to complement, possibly due to its function in acceleration of complement MAC elimination by membrane vesiculation.
Mortalin, also known as GRP75, PBP74, mthsp75 and mot-2, is a member of the heat shock protein (hsp) 70 family (GeneCard no. GC05M137967). As reviewed in Wadhwa et al. (31), it was assigned multiple functions including stress response, glucose regulation, p53 inactivation, control of cell proliferation, differentiation, tumorigenesis and mitochondrial import. Mortalin is ubiquitously and constitutively expressed in normal tissues, and its expression level is up-regulated in some tumors (19, 20) and during infection and inflammation (32, 33). Furthermore, over-expression of mortalin in normal cells considerably extends their lifespan (34), while reduction of mortalin levels in immortalized cells causes growth arrest (35). Thus far, mortalin has been mainly described inside cells, in mitochondria and several other cytoplasmic locations (15). In unstimulated K562 cells, we saw a diffused distribution pattern of mortalin in the cytoplasm that partly shifted, after sub-lytic complement attack, to the cell periphery (data not shown). This occurred concomitantly with the shedding of mortalin from the cells. By flow cytometry analysis, mortalin was previously detected on the surface of mouse B cells and macrophages (36). In addition, profiling of cell-surface proteome of biotinylated cancer cells identified mortalin on neuroblastoma, lung adenocarcinoma, leukemia and ovarian cancer cells (37). Intriguingly, in our study, analysis by flow cytometry failed to detect mortalin on the surface of control and complement-triggered K562 cells (data not shown). This may appear contradictory to the fact that following complement attack we find mortalin outside K562 cells associated with membrane vesicles and that anti-mortalin antibodies block the shedding of mortalin and C9. Our data show that the shedding process is very fast and within 5 min after a sub-lytic MAC attack >50% of the total releasable mortalin is already in the supernatant. It is, therefore, conceivable that the amount of mortalin in transit on the cell surface is too small for detection by flow cytometry and that its identification on or within the plasma membrane will require a more sensitive technique.
MAC elimination from Ehrlich ascites tumor cells was shown to be dependent on Ca2+ influx and PKC activity (38). Both PKC and ERK contribute to cell protection from complement-mediated lysis (28, 29, 39) and, as shown here, also to mortalin release. It is likely that the activation of PKC and ERK, following MAC-induced Ca2+ influx, mobilizes mortalin to the cell surface. This is followed, somehow, by sorting of MAC complexes and membrane vesiculation. Hsc70, another member of the hsp70 family, also migrates, in response to sub-lytic MAC, from the cytoplasm to the plasma membrane (40). Both mortalin and hsc70 have an N-terminal ATPase domain and a C-terminal peptide-binding domain (41–43). Possibly, mortalin and hsc70 regulate or compose the machinery that uses ATP to force vesicle formation (budding) at the extracellular surface. Both mortalin and hsc70 bind to numerous other proteins, probably via their peptide-binding domain. For example, mortalin was shown to bind to GRP94 (44), p53 (45) and FGF-1 (46). An ATP-sensitive association between mortalin and IL-1R type 1, that was independent on IL-1 binding, was also described (47). This raises the possibility that mortalin functions both in internalization and externalization of proteins. Within the cell, mortalin plays a major role in import of proteins into mitochondria (18). Our finding of a possible mortalin–C9 and mortalin–-C8 binding (Fig. 7) requires further investigation. It suggests, however, that on the surface of MAC-attacked cells, mortalin recognizes the C5b–9 complexes via C9 and C8 and is involved in the activation or regulation of the MAC elimination process. This is supported by the observation that SLO and melittin do not induce the release of mortalin.
Spontaneous shedding of membrane vesicles has been observed with normal and malignant cells. These vesicles, named exosomes or prostasomes, are apparently secreted by exocytosis as a consequence of fusion of multivesicular late endosome/lysosome bodies (MVBs) with the plasma membrane (48–50). Exosomes and prostasomes are enriched in raft molecules, both are found intracellularly in MVBs and their release is sensitive to wortmannin (49). High cholesterol content was found in exosomes (51), prostasomes (52) and MAC-containing membrane vesicles (14). Nevertheless, at present, it seems improbable that the MAC–mortalin-containing membrane vesicles are exosomes-like. It seems unlikely that the large MAC complexes are endocytosed, transported to the late endosomal compartment, packed into MVBs and get exocytosed within 5–10 min. Transferrin receptors reach the recycling endosome only after ∼20 min (53, 54). In addition, extracellular application of anti-mortalin antibodies blocked the MAC-induced vesiculation process, suggesting that the process occurs on the cell surface. Theoretically, the simplest way to shed MAC–mortalin membrane vesicles is by a direct budding-off from the plasma membrane or by sloughing of membrane blebs. Electron microscopy studies performed on neutrophils activated with sub-lytic complement (9) or with fMLP (55) demonstrated direct budding of membrane vesicles (named ectosomes) from the surface of the neutrophils. Direct membrane vesiculation was also described with human erythrocytes under MAC attack (56). Alternatively, MAC–mortalin complexes may recycle via an early endosomal compartment. The reported t1/2 of recycling of transferrin receptors and lipids via the early endosomal compartment ranges between 1.4 and 6 min (53, 54), which fits well with the observed rate of MAC–mortalin release. Elucidation of the mechanism of MAC–mortalin release requires further investigation.
In conclusion, assembly of a sub-lytic dose of the complement MAC on K562 cells is followed by a rapid release of the MAC on membrane vesicles. Our data suggest that mortalin becomes integrated into these membrane vesicles and is somehow involved in the process of vesiculation. Elimination of the MAC is a protective mechanism from complement-mediated lysis. Cells subjected to sub-lytic MAC become, within 20–50 min, more resistant to lytic MAC doses (6). This could be, at least partly, due to translocation of mortalin to the cell cortex, thus permitting elimination of the second wave MAC at an accelerated rate. The physiological significance of this membrane vesiculation to disease remains to be further examined. Release of MAC-loaded vesicles from endothelial cells was proposed to contribute to fibrin deposition associated with immune endothelial injury (57).
Transmitting editor. I. Pecht
This work was supported by the Israel Science Foundation, the Israel Cancer Association and the Israel Cancer Research Foundation. We are grateful to Sunil Kaul (AIT) and Alex Merrick (NIEHS) for providing anti-mortalin antibodies. This work was performed in partial fulfillment of the requirements for a PhD degree by D.P., Sackler Faculty of Medicine, Tel Aviv University.
References
Koski, C. L., Ramm, L. E., Hammer, C. H., Mayer, M. M. and Shin, M. L.
Cragg, M. S., Howatt, W. J., Bloodworth, L., Anderson, V. A., Morgan, B. P. and Glennie, M. J.
Bohana-Kashtan, O., Ziporen, L., Donin, N., Kraus, S. and Fishelson, Z.
Reiter, Y., Ciobotariu, A. and Fishelson, Z.
Dashiell, S. M., Rus, H. and Koski, C. L.
Sims, P. J. and Wiedmer, T.
Morgan, B. P., Dankert, J. R. and Esser, A. F.
Carney, D. F., Koski, C. L. and Shin, M. L.
Scolding, N. J., Morgan, B. P., Houston, W. A., Linington, C., Campbell, A. K. and Compston, D. A.
Morgan, B. P., Imagawa, D. K., Dankert, J. R. and Ramm, L. E.
Morgan, B. P.
Stein, J. M. and Luzio, J. P.
Ran, Q., Wadhwa, R., Kawai, R. et al.
Wadhwa, R., Yaguchi, T., Hasan, M. K., Mitsui, Y., Reddel, R. R. and Kaul, S. C.
Carette, J., Lehnert, S. and Chow, T. Y.
Voisine, C., Craig, E. A., Zufall, N., von Ahsen, O., Pfanner, N. and Voos, W.
Takano, S., Wadhwa, R., Yoshii, Y., Nose, T., Kaul, S. C. and Mitsui, Y.
Dundas, S. R., Lawrie, L. C., Rooney, P. H. and Murray, G. I.
Schlesinger, M., Nave, Z., Levy, Y., Slater, P. E. and Fishelson, Z.
Wadhwa, R., Kaul, S. C., Ikawa, Y. and Sugimoto, Y.
Merrick, B. A., Walker, V. R., He, C., Patterson, R. M. and Selkirk, J. K.
Reiter, Y. and Fishelson, Z.
Laine, R. O., Morgan, B. P. and Esser, A. F.
Bhakdi, S., Tranum-Jensen, J. and Sziegoleit, A.
Reiter, Y., Ciobotariu, A., Jones, J., Morgan, B. P. and Fishelson, Z.
Kraus, S. and Fishelson, Z.
Kraus, S., Seger, R. and Fishelson, Z.
Deng, J., Gold, D., LoVerde, P. T. and Fishelson, Z.
Wadhwa, R., Taira, K. and Kaul, S. C.
Kirmanoglou, K., Hannekum, A. and Schafler, A. E.
Johannesen, J., Pie, A., Karlsen, A. E. et al.
Kaul, S. C., Yaguchi, T., Taira, K., Reddel, R. R. and Wadhwa, R.
Wadhwa, R., Takano, S., Taira, K. and Kaul, S. C.
VanBuskirk, A. M., DeNagel, D. C., Guagliardi, L. E., Brodsky, F. M. and Pierce, S. K.
Shin, B. K., Wang, H., Yim, A. M. et al.
Carney, D. F., Lang, T. J. and Shin, M. L.
Cybulsky, A. V., Bonventre, J. V., Quigg, R. J., Lieberthal, W. and Salant, D. J.
Fishelson, Z., Hochman, I., Greene, L. E. and Eisenberg, E.
Krimmer, T., Rassow, J., Kunau, W. H., Voos, W. and Pfanner, N.
Flaherty, K. M., DeLuca-Flaherty, C. and McKay, D. B.
Demand, J., Luders, J. and Hohfeld, J.
Takano, S., Wadhwa, R., Mitsui, Y. and Kaul, S. C.
Wadhwa, R., Takano, S., Robert, M. et al.
Mizukoshi, E., Suzuki, M., Loupatov, A. et al.
Sacht, G., Brigelius-Flohe, R., Kiess, M., Sztajer, H. and Flohe, L.
Fevrier, B. and Raposo, G.
Llorente, A., de Marco, M. C. and Alonso, M. A.
Ronquist, G. and Brody, I.
Wubbolts, R., Leckie, R. S., Veenhuizen, P. T. et al.
Arvidson, G., Ronquist, G., Wikander, G. and Ojteg, A. C.
Sheff, D., Pelletier, L., O'Connell, C. B., Warren, G. and Mellman, I.
Hao, M. and Maxfield, F. R.
Hess, C., Sadallah, S., Hefti, A., Landmann, R. and Schifferli, J. A.
Iida, K., Whitlow, M. B. and Nussenzweig, V.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}