





Abstract
β2-Glycoprotein I (β2-GPI) is a major antigen for anti-cardiolipin antibodies and their epitopes are cryptic. Conformation of each domain of β2-GPI was optimized from its crystal structure by energy minimization and by molecular dynamics simulation. Three electrostatic interactions, i.e. D193–K246, D222–K317 and E228–K308, were observed between domains IV and V in the optimized structure that was constructed based on the consensus sequences obtained by the phage-displayed random peptide library. Antigenic structures determined by the epitope mapping mainly consisted of hydrophobic amino acids located on two discontinuous sequences in domain IV. These amino acid clusters, as an epitope, were covered by domain V and were of a hidden nature. A similar but incomplete counterpart to the epitopic clusters was found in domain I but was not in domains II or III. Binding of anti-β2-GPI auto-antibodies to solid-phase β2-GPI was significantly reduced either by L replacement for W235, a common amino acid component for the epitopes, or by V replacement for all of D193, D222 and E228. Structural analysis indicated a hypothesis that these electrostatic interactions between domains IV and V retained exposure to W235 and that epitope spreading occurred in the region surrounding W235. Thus, epitopic structures recognized by anti-β2-GPI auto-antibodies are cryptic and inter-domain electrostatic interactions are involved in their in exposure.
Introduction
The anti-phospholipid syndrome (APS) is a multi-system disorder characterized by arterial/venous thrombosis and pregnancy complication (1–4). Anti-phospholipid antibodies, in particular, anti-cardiolipin antibodies (aCLs) and lupus anticoagulant (5), are of considerable clinical importance in APS. In 1990, three individual research groups reported that a 50 kD plasma co-factor is required for the binding of such aCL to cardiolipin (CL) (6–8)and β2-glycoprotein I (β2-GPI), which binds to anionic phospholipids (PLs), is currently thought to be the major antigen for aCL in APS patients.
We reported that aCL-recognized cryptic epitopes appear on the β2-GPI molecule only when β2-GPI interacts with lipid membranes containing anionic PLs, such as CL, or also with a polyoxygenated polystyrene surface (9, 10). We also reported that domain IV of β2-GPI was dominantly involved in expression of antigenicity for anti-β2-GPI auto-antibodies (11). Other research groups reported the presence of anti-β2-GPI auto-antibodies directed to domain V (12, 13) or to domain I (13). It has also been proposed that binding of β2-GPI to anionic PLs increased the local concentration of β2-GPI, thus leading to an increase in intrinsic affinity and binding of the antibody to β2-GPI, an event which argues against the interpretation that epitopes for aCL are cryptic (14, 15).
β2-GPI, a 50-kD protein with a carbohydrate content of 17%, is present in normal human plasma at ∼200 μg ml−1 and was apparently first described by Schultze et al. (16). β2-GPI is composed of five homologous motifs of ∼60 amino acids which contain highly conserved half-cysteine residues related to the formation of two internal disulfide bridges. These repeating motifs were designated as short consensus repeats (SCR)/complement control protein (CCP) repeats or sushi domains (17–19). The fifth domain is a modified form that contains 82 amino acid residues and 6 half cysteines. The primary amino acid sequence is highly conserved (>80%) among human, bovine and mouse counterparts (20). The crystal structure of human β2-GPI was recently reported (21, 22).
We characterized epitopic structures for anti-β2-GPI auto-antibodies derived from APS by making use of epitope mapping based on the crystal structure of β2-GPI. Our results show that cryptic epitopes for monoclonal anti-β2-GPI auto-antibodies located on a particular region in its domain IV and that the region is naturally hidden by domain V. Three electrostatic interactions between domains IV and V were involved in both crypt and exposure of the amino acid clusters, as an epitope.
Methods
Microtiter plates
Plain plates (Sumilon S-type) and polyoxygenated plates (Sumilon C-type) were obtained from Sumitomo Bakelite Co., Ltd (Tokyo, Japan). Densities of oxygen atoms covalently introduced onto the polystyrene surfaces were analyzed by X-ray photoelectron spectroscopy and the results were as follows: plain plate, C–O (1.3 mol%); polyoxygenated plate, C–O (10.5 mol%); C=O (5.6 mol%); O–C=O (3.7 mol%).
Sera and mAbs
Anti-β2-GPI antibody-positive sera were obtained from systemic lupus erythematosus patients who fulfilled the criteria of the American Rheumatism Association revised in 1982. All patients had significantly high levels of β2-GPI-dependent aCL, as compared with healthy subjects, and had one or more symptoms of APS, i.e. thromboembolic complications (venous and/or arterial), recurrent spontaneous abortion and/or thrombocytopenia. Monoclonal anti-β2-GPI auto-antibodies, EY1C8, EY2C9 and TM1G2, were derived from the APS patients (23). Patients EY and TM had recurrent fetal losses and recurrent deep vein thrombosis accompanied by multiple pulmonary embolism, respectively. EY1C8 and EY2C9 competitively inhibit 50–80% of the IgG aCL activity of sera from the patient from whom these monoclonal aCLs were established. A mouse monoclonal anti-β2-GPI auto-antibody, WB-CAL-1, was established from splenocytes of a (NZW × BXSB) F1 male mouse (24). Pathogenic roles of these mAbs have been known in vitro and in vivo. Five mouse monoclonal anti-human β2-GPI antibodies, Cof-18, Cof-20, Cof-21, Cof-22 and Cof-23, were established from the splenocytes of BALB/c mice immunized with human β2-GPI in CFA (11, 25).
Biopanning and selection of clones from random peptide libraries
Consensus peptide sequences recognized by anti-β2-GPI antibodies were obtained with combinatorial libraries of a 7-mer and/or 12-mer random peptide fused to a minor coat protein (pIII) of the filamentous coilphage, M13 (PH.D-7 and/or PH.D-12 Phage Display Peptide Library Kit, New England Biolabs Inc., Beverly, MA, USA). These libraries consist theoretically of 2.0 × 109 or 1.9 × 109 electroporated sequences, respectively. A plain plate was coated by incubating with 150 μl of anti-β2-GPI mAbs (100 μg ml−1) in 0.1 M NaHCO3 overnight at 4°C, followed by blocking with 10 mM Tris–HCl, 150 mM NaCl, pH 7.4 [Tris-buffered saline (TBS)] containing 5 mg ml−1 of BSA for 1 h. The plate was washed six times with 200 μl of TBS containing 0.05% Tween 20 (TBS–Tween) and 100 μl of phages (2 × 1012 ml−1 in TBS–Tween), then incubated for 1 h. Following 10 washings with TBS–Tween, bound phages were eluted with 0.2 M glycine–HCl (pH 2.2) containing 1 mg ml−1 of BSA. After three rounds of selection and amplification with the mAbs, the clones were isolated and sequenced.
Optimization of the structure of human β2-GPI
A conformation of each domain was first constructed from the crystal structure (implemented in Protein Data Bank: 1C1Z) and was then optimized by 2000 cycles of energy minimization by the CHARMm program (26), with hydrophilic hydrogen atoms and TIP3 water molecules (27). Molecular dynamics simulation (5 ps) was then done with a 0.002 ps time step. Cutoff distance for non-bonded interactions was set to 15+ and the dielectric constant was 1.0. A non-bonded pair list was updated every 10 steps. The most stable structure of each domain in the dynamics iterations was then optimized by 2000 cycles of energy minimization. The final structures of domains I, II, III, VI and V consisted of 1867, 1722, 2097, 1828 and 2602 atoms, including 443, 408, 515, 435 and 598 TIP3 water molecules, respectively.
A structure of complex domains IV and V was further constructed by considering the location of the oligosaccharide attachment site in domain IV, location of epitopic regions of the Cof-18 and Cof-20 mAbs, junction between domains IV and V and the molecular surface charges of both domains. This model was again optimized by molecular dynamics simulation and by energy minimization. The final structure consisting of 3775 atoms, including hydrophilic hydrogen atoms and 807 TIP3 water molecules had a total energy of −2.43 × 104 kcal mol−1 with a root mean square (r.m.s.) force of 0.985 kcal mol−1. A model of mutant domain IV in which D193, D222 and E228 were replaced by valines (V) or of cleaved domain V at a K317–T318 was constructed. After optimization, the final structures consisted of 3774 and 3839 atoms, including hydrophilic hydrogen atoms and 808 and 827 TIP3 water molecules and had a total energy of −2.42 × 104 and −2.02 × 104 kcal mol−1 with a r.m.s. force of 0.892 and 0.979 kcal mol−1, respectively.
Production of β2-GPI mutant proteins
By site-directed mutagenesis, we designed four kinds of β2-GPI mutant proteins of which three particular amino acids, i.e. D193, D222 and/or E228 were replaced by V. Another mutant, W235 replaced by L, was also designed. The isolation and sequencing of a full-length cDNA for human β2-GPI was as described (20). The cDNA was digested with EcoR1 and BglII and ligated to the corresponding site of the pVL1393 transfer vector. To assess the mutation, we used the GeneEditor in vitro site-directed mutagenesis system (Promega Corp., Madison, WI, USA) and the sequence of primers used is as follows: 5′-TGCCCATTCCCATCAAGACCAGTCAATG-3′ for a mutant D193 → V (GAC → GTC), 5′-GGATATTCTCTGGTTGGCCCGGAAGAAATAG-3′ for a mutant D222 → V (GAT → GTT), 5′-GGCCCGGAAGAAATAGTATGTACCAAACTG-3′ for a mutant E228 → V (GAA → GTA), 5′-TGCCCATTCCCATCAAGACCAGTCAATG-3′ and 5′-CTCTGGTTGGCCCGGAAGAAATAGTATGTACC-3′ for a mutant ‘triple’, D193 → V (GAC → GTC), D222 → V (GAT → GTT) and E228 → V (GAA → GTA) and 5′-TGTACCAAACTGGGAAACTTGTCTGCCATGCC-3′ for a mutant W235 → L (TGG → TTG). A DNA sequence of the mutants was verified by analysis using Prism Model 310 (PE Applied Biosystems, Foster City, CA, USA). Recombinant β2-GPI and its mutant proteins were produced in the baculovirus Spodoptera frugiperda (Sf-9) expression system (11) using linearized virus cDNA (Baculogold, Pharmingen, San Diego, CA, USA). β2-GPI proteins were purified from serum-free culture media (Sf-900 II, Life Technologies, Grand Island, NY, USA) using anti-β2-GPI mAb affinity column chromatography. Apparent molecular mass of these mutant proteins in SDS-PAGE was 43 kD and there were no significant changes (data not shown).
ELISA for anti-β2-GPI antibodies
Anti-β2-GPI ELISA was done as described (9) but with slight modification. A polyoxygenated plate was coated by incubation with 50 μl per well of recombinant β2-GPI or its mutant proteins, dissolved at a concentration of 2.5 μg ml−1 in PBS, pH 7.4, overnight at 4°C. The plates were blocked with 200 μl of PBS containing 3% gelatin (Difco Laboratories, MI, USA) in PBS. Human or mouse anti-β2-GPI mAbs diluted with PBS containing 0.5% gelatin were applied for 1 h. Antibody binding to β2-GPI was probed by HRP-conjugated anti-mouse IgG or anti-human IgG/IgM and o-phenylenediamine solution (0.2 mg ml−1) containing 0.01% H2O2 was applied for 10 min. Color reaction was halted by adding 100 μl of 2 N H2SO4 and optical density was measured at 492 nm. Between each step, we used 200 μl of PBS containing 0.05% Tween 20 (PBS–Tween) for extensive washings.
Inhibition ELISA for the binding between β2-GPI and anti-β2-GPI mAb
β2-GPI at the concentration of 10 μg ml−1 was coated on the polyoxygenated plates and was blocked in the same way as described above. Solutions of WB-CAL-1 (1 μg ml−1) together with different concentrations (up to 1 μM) of β2-GPI or triple mutant of β2-GPI, in the presence or absence of CL–liposome (10 μg ml−1), were distributed to the wells in triplicate. HRP-conjugated anti-mouse mAb was utilized for detection.
Results
Epitopic structures of anti-β2-GPI antibodies
Peptide libraries displayed on filamentous phages were used to define the antigenic structure specific for anti-β2-GPI mAbs. In all screenings, we carried out at least three rounds of biopanning with coated antibodies on a plain plate. After the latest biopanning, 20- to 10,000-fold increase in the number of eluted phages and 6 to 10 clones were randomly picked up and nucleotide sequences coding fused peptides were analyzed (Table 1). The conformation of each domain of β2-GPI was constructed from the crystal structure's coordinates of human β2-GPI implemented in the Brookhaven Protein Data Bank as 1C1Z. We had earlier characterized the fine specificities of Cof-18, Cof-20 and Cof-21 mAbs, established from human β2-GPI immunized BALB/c mice (11). All these antibodies recognize the native structure of human β2-GPI and their epitopes are located on the surface of domains V, III and IV of β2-GPI in solution, respectively. As shown in Fig. 1, all antigenic structures for these mAbs were identified on the outer surface in individual domains. Antigenic structures for human anti-β2-GPI auto-mAbs, namely EY1C8, EY2C9 and TM1G2, and for a mouse monoclonal auto-antibody, WB-CAL-1, were identified in domain IV (Fig. 2). In case of EY1C8, two possible candidates for the antigenic structure have to be considered (Table 2, Fig. 2). All these epitopes were located on two individual loops, i.e. the N- and C-terminal loops of the domain.
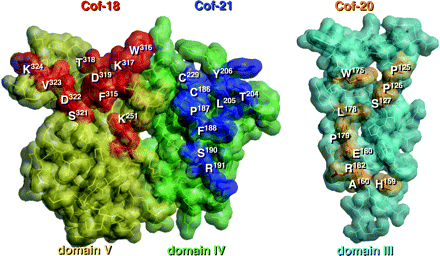
Epitopic structures recognized by mouse anti-β2-GPI mAbs, Cof-18, Cof-20 and Cof-21. All antigenic regions for Cof-18, Cof-21 and Cof-20 were present on the outer surface of domains V, IV and III, respectively. Each amino acid residue composed of these epitopes is displayed by a bold stick and space-filling model with a residue number and the others by a light stick and space-filling model. The correspondences of the deduced amino acid sequences from the phage-displayed peptide libraries and epitopic regions on the β2-GPI molecule are summarized in Table 2.
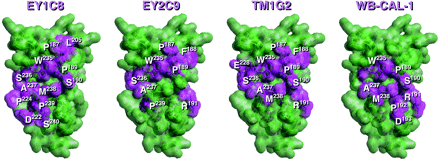
Antigenic regions for auto-antibodies, i.e. EY1C8, EY2C9, TM1G2 and WB-CAL-1 mAbs were present in domain IV. Each amino acid residue composed of these epitopes is colored violet with a residue number and the others by green. Correspondences of the deduced amino acid sequences from the phage-displayed peptide libraries and epitopic regions on the β2-GPI molecule are summarized in Table 1.
Sequences of synthetic oligonucleotides used for mutagenesis
Mutant | Synthetic primer | Codon change |
|---|---|---|
| D193 → V | 5′-TGCCCATTCCCATCAAGACCAGTCAATG-3′ | GAC → GTC |
| D222 → V | 5′-GGATATTCTCTGGTTGGCCCGGAAGAAATAG-3′ | GAT → GTT |
| E228 → V | 5′-GGCCCGGAAGAAATAGTATGTACCAAACTG-3′ | GAA → GTA |
| Triple | ||
| (D193 → V | 5′-TGCCCATTCCCATCAAGACCAGTCAATG-3′ | GAC → GTC |
| D22 2 → V, E228 → V) | 5′-CTCTGGTTGGCCCGGAAGAAATAGTATGTACC-3′ | GAT → GTT, GAA → GTA |
| W235 → L | 5′-TGTACCAAACTGGGAAACTTGTCTGCCATGCC-3′ | TGG → TTG |
Mutant | Synthetic primer | Codon change |
|---|---|---|
| D193 → V | 5′-TGCCCATTCCCATCAAGACCAGTCAATG-3′ | GAC → GTC |
| D222 → V | 5′-GGATATTCTCTGGTTGGCCCGGAAGAAATAG-3′ | GAT → GTT |
| E228 → V | 5′-GGCCCGGAAGAAATAGTATGTACCAAACTG-3′ | GAA → GTA |
| Triple | ||
| (D193 → V | 5′-TGCCCATTCCCATCAAGACCAGTCAATG-3′ | GAC → GTC |
| D22 2 → V, E228 → V) | 5′-CTCTGGTTGGCCCGGAAGAAATAGTATGTACC-3′ | GAT → GTT, GAA → GTA |
| W235 → L | 5′-TGTACCAAACTGGGAAACTTGTCTGCCATGCC-3′ | TGG → TTG |
Sequences of synthetic oligonucleotides used for mutagenesis
Mutant | Synthetic primer | Codon change |
|---|---|---|
| D193 → V | 5′-TGCCCATTCCCATCAAGACCAGTCAATG-3′ | GAC → GTC |
| D222 → V | 5′-GGATATTCTCTGGTTGGCCCGGAAGAAATAG-3′ | GAT → GTT |
| E228 → V | 5′-GGCCCGGAAGAAATAGTATGTACCAAACTG-3′ | GAA → GTA |
| Triple | ||
| (D193 → V | 5′-TGCCCATTCCCATCAAGACCAGTCAATG-3′ | GAC → GTC |
| D22 2 → V, E228 → V) | 5′-CTCTGGTTGGCCCGGAAGAAATAGTATGTACC-3′ | GAT → GTT, GAA → GTA |
| W235 → L | 5′-TGTACCAAACTGGGAAACTTGTCTGCCATGCC-3′ | TGG → TTG |
Mutant | Synthetic primer | Codon change |
|---|---|---|
| D193 → V | 5′-TGCCCATTCCCATCAAGACCAGTCAATG-3′ | GAC → GTC |
| D222 → V | 5′-GGATATTCTCTGGTTGGCCCGGAAGAAATAG-3′ | GAT → GTT |
| E228 → V | 5′-GGCCCGGAAGAAATAGTATGTACCAAACTG-3′ | GAA → GTA |
| Triple | ||
| (D193 → V | 5′-TGCCCATTCCCATCAAGACCAGTCAATG-3′ | GAC → GTC |
| D22 2 → V, E228 → V) | 5′-CTCTGGTTGGCCCGGAAGAAATAGTATGTACC-3′ | GAT → GTT, GAA → GTA |
| W235 → L | 5′-TGTACCAAACTGGGAAACTTGTCTGCCATGCC-3′ | TGG → TTG |
Consensus amino acid sequences deduced from phage libraries and epitopes for anti-β2-GPI antibodies
Antibody (location of epitope) | Phage no. | Consensus sequences from phage libraries | Frequency | Corresponding amino acid residues on the β2-GPI molecule |
|---|---|---|---|---|
| Cof-18 (domain V) | Cof-18-1 | GMKLTSEQKAML | 4/9 | * *K324V323*S321D322*K251**F315D319T318W316*317 |
| Cof-18-2 | SDWRLMFESWIR | 3/9 | ||
| Cof-20 (domain III) | Cof-20 | AHKDHVVSTWVP | 5/10 | A160H159R182E180* P179 L178S127* W175P126P125 |
| Cof-21 (domain IV) | Cof-21 | NFCASCLLPVSR | 7/10 | *Y206C229L205T204C186**P187F188 S190 R191 |
| EY1C8 (domain IV) | EY1C8-1 | DLFVTAW | 4/9 | aD222S240P 239M238*S190P189W235P187L205*** |
| EY1C8-2 | SLMVSPWPLHGV | 4/9 | aD222S240P 239M238A237S236*W235P187L205*** | |
| EY2C9 (domain IV) | EY2C9 | HNIWSAPRLIFN | 4/10 | **P187W235S236A237P239R191 * P189F188 * |
| TM1G2 (domain IV) | TM1G2-1 | DLTIMPWSLYPP | 4/10 | E228*S236A237M238*W235*P187S190F188P189R191 |
| TM1G2-2 | TMMQTMSFLRGF | 2/10 | ||
| WB-CAL-1 (domain IV) | WB-CAL-1 | SLWQDRLNAMQS | 5/6 | S190I189W235*D193R191P192*A237M238** |
Antibody (location of epitope) | Phage no. | Consensus sequences from phage libraries | Frequency | Corresponding amino acid residues on the β2-GPI molecule |
|---|---|---|---|---|
| Cof-18 (domain V) | Cof-18-1 | GMKLTSEQKAML | 4/9 | * *K324V323*S321D322*K251**F315D319T318W316*317 |
| Cof-18-2 | SDWRLMFESWIR | 3/9 | ||
| Cof-20 (domain III) | Cof-20 | AHKDHVVSTWVP | 5/10 | A160H159R182E180* P179 L178S127* W175P126P125 |
| Cof-21 (domain IV) | Cof-21 | NFCASCLLPVSR | 7/10 | *Y206C229L205T204C186**P187F188 S190 R191 |
| EY1C8 (domain IV) | EY1C8-1 | DLFVTAW | 4/9 | aD222S240P 239M238*S190P189W235P187L205*** |
| EY1C8-2 | SLMVSPWPLHGV | 4/9 | aD222S240P 239M238A237S236*W235P187L205*** | |
| EY2C9 (domain IV) | EY2C9 | HNIWSAPRLIFN | 4/10 | **P187W235S236A237P239R191 * P189F188 * |
| TM1G2 (domain IV) | TM1G2-1 | DLTIMPWSLYPP | 4/10 | E228*S236A237M238*W235*P187S190F188P189R191 |
| TM1G2-2 | TMMQTMSFLRGF | 2/10 | ||
| WB-CAL-1 (domain IV) | WB-CAL-1 | SLWQDRLNAMQS | 5/6 | S190I189W235*D193R191P192*A237M238** |
Two possible correspondences are considered with EY1C8 libraries.
Consensus amino acid sequences deduced from phage libraries and epitopes for anti-β2-GPI antibodies
Antibody (location of epitope) | Phage no. | Consensus sequences from phage libraries | Frequency | Corresponding amino acid residues on the β2-GPI molecule |
|---|---|---|---|---|
| Cof-18 (domain V) | Cof-18-1 | GMKLTSEQKAML | 4/9 | * *K324V323*S321D322*K251**F315D319T318W316*317 |
| Cof-18-2 | SDWRLMFESWIR | 3/9 | ||
| Cof-20 (domain III) | Cof-20 | AHKDHVVSTWVP | 5/10 | A160H159R182E180* P179 L178S127* W175P126P125 |
| Cof-21 (domain IV) | Cof-21 | NFCASCLLPVSR | 7/10 | *Y206C229L205T204C186**P187F188 S190 R191 |
| EY1C8 (domain IV) | EY1C8-1 | DLFVTAW | 4/9 | aD222S240P 239M238*S190P189W235P187L205*** |
| EY1C8-2 | SLMVSPWPLHGV | 4/9 | aD222S240P 239M238A237S236*W235P187L205*** | |
| EY2C9 (domain IV) | EY2C9 | HNIWSAPRLIFN | 4/10 | **P187W235S236A237P239R191 * P189F188 * |
| TM1G2 (domain IV) | TM1G2-1 | DLTIMPWSLYPP | 4/10 | E228*S236A237M238*W235*P187S190F188P189R191 |
| TM1G2-2 | TMMQTMSFLRGF | 2/10 | ||
| WB-CAL-1 (domain IV) | WB-CAL-1 | SLWQDRLNAMQS | 5/6 | S190I189W235*D193R191P192*A237M238** |
Antibody (location of epitope) | Phage no. | Consensus sequences from phage libraries | Frequency | Corresponding amino acid residues on the β2-GPI molecule |
|---|---|---|---|---|
| Cof-18 (domain V) | Cof-18-1 | GMKLTSEQKAML | 4/9 | * *K324V323*S321D322*K251**F315D319T318W316*317 |
| Cof-18-2 | SDWRLMFESWIR | 3/9 | ||
| Cof-20 (domain III) | Cof-20 | AHKDHVVSTWVP | 5/10 | A160H159R182E180* P179 L178S127* W175P126P125 |
| Cof-21 (domain IV) | Cof-21 | NFCASCLLPVSR | 7/10 | *Y206C229L205T204C186**P187F188 S190 R191 |
| EY1C8 (domain IV) | EY1C8-1 | DLFVTAW | 4/9 | aD222S240P 239M238*S190P189W235P187L205*** |
| EY1C8-2 | SLMVSPWPLHGV | 4/9 | aD222S240P 239M238A237S236*W235P187L205*** | |
| EY2C9 (domain IV) | EY2C9 | HNIWSAPRLIFN | 4/10 | **P187W235S236A237P239R191 * P189F188 * |
| TM1G2 (domain IV) | TM1G2-1 | DLTIMPWSLYPP | 4/10 | E228*S236A237M238*W235*P187S190F188P189R191 |
| TM1G2-2 | TMMQTMSFLRGF | 2/10 | ||
| WB-CAL-1 (domain IV) | WB-CAL-1 | SLWQDRLNAMQS | 5/6 | S190I189W235*D193R191P192*A237M238** |
Two possible correspondences are considered with EY1C8 libraries.
Structural model of the domain IV–V complex
To investigate interactions between domains IV and V, a structural model of the domain IV–V complex of β2-GPI in solution was constructed by considering crypticity of the epitope for the monoclonal auto-antibodies (i.e. EY1C8, EY2C9, TM1G2 and WB-CAL-1) and exposing of epitopic structures for Cof-18 and Cof-21. The r.m.s. deviation of the main-chain structure of each domain in the complex model and the original X-ray structure was <1.52 Å with super-impositions of only each domain, and the secondary structure of the model was not significantly altered by optimization. Three electrostatic interactions (D193–K246/K250, D222–K305 and E228–K308) between domains IV and V, and the interaction of K242–E302 in the IV–V junction were the only significant differences observed (Fig. 3). The model was more stable than the unbent structure shown in the X-ray structure. Although W235, exposed in the X-ray structure, was naturally covered by domain V in the model, N234, an oligosaccharide attachment site, was exposed in both structures. All the above antigenic structures for EY1C8, EY2C9, TM1G2 and WB-CAL-1 on domain IV were partly hidden by the overlying of domain V.
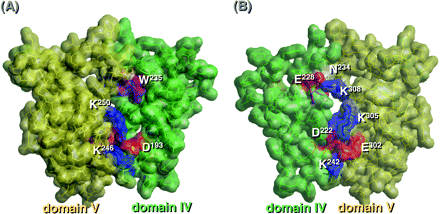
Interaction of domains IV and V. (A) A space-filling model of domains IV–V is shown from one direction (left) and from another one (right) and the structure of each domain is displayed in different colors: domain IV, blue; domain V, yellow.
Alteration on antigenicity for anti-β2-GPI antibodies by the mutations
Binding of anti-β2-GPI mAbs to the solid-phase β2-GPI mutant proteins was then determined (Fig. 4). Binding of the mouse anti-human β2-GPI mAbs Cof-21 which recognize a native structure in domain IV (Fig. 1) was not affected by any V replacements for D193, D222 and E228. In contrast, binding of human and mouse anti-β2-GPI auto-mAbs, i.e. EY2C9 and WB-CAL-1 (recognized a cryptic epitope in domain IV), was affected considerably by all four types of mutations, and almost completely disappeared by the triple one. It was supported by our model that the reduction of antigenicity might be caused by turning of W235, which might have the major role for epitopic construction, into the inner side of domain IV, due to the interaction with domain V via these three hydrogen bonds. The side chain of the W residue appeared outside of the VI–V model. In contrast, the side chain directed to the inner space of the IV domain was displayed in the X-ray structure and in our other model with the triple mutation (Fig. 5).
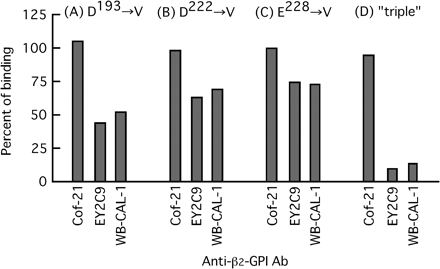
Binding of anti-β2-GPI mAbs to solid-phase mutant proteins. Antibody binding to the mutant proteins is indicated (as a percent of control) and as compared with that to control β2-GPI. mAbs (1 μg ml−1) were used for the assay.
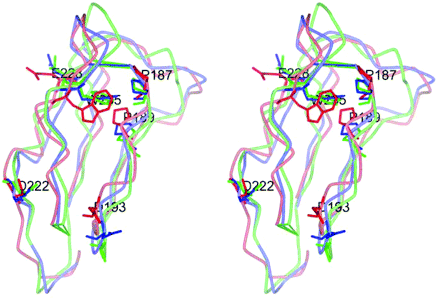
Superimposed stereo view of domain IV structures of X-ray, IV–V interacted and ‘triple’ mutated models. Three domain IVs of the X-ray structure, the IV–V model and its triple mutation were superimposed using their main-chain atoms, and backbone smooth lines were colored blue, red and green, respectively. Each W235, P187, P189, 193rd, 222nd and 228th residue was also described by the stick model in the same color scheme, with their residue names and numbers (only IV–V model) written in white. The side chains of P187 and P189 were close to the side chain of W235 by hydrophobic interaction.
When we tested for 30 anti-β2-GPI antibody-positive APS serum samples, antibody binding in 24 serum samples (80%) was significantly attenuated by the triple mutation (Fig. 6A). The ‘type A’ group was defined as having higher binding to native β2-GPI compared with the mutant. In contrast, six samples (20%) showed increased binding to the mutant protein (defined as ‘type B’). Two typical binding profiles of anti-β2-GPI antibodies in sera of patients with APS are shown in Fig. 6(B). The above-described major antibodies showed a binding profile similar to that of the type A antibody and a significantly reduced binding to the triple mutant protein was likely the result of the sum of weak reductions caused by each mutation for D193, D222 or E228. In contrast, the latter minor group of antibodies revealed a profile similar to that of the type B antibody.
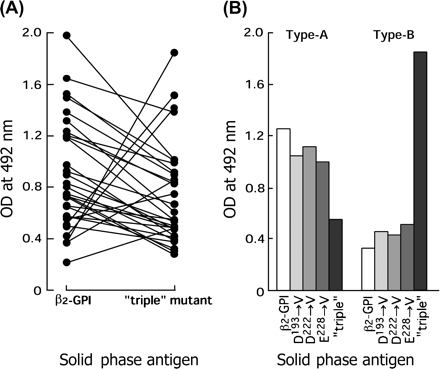
Binding of antibodies to solid-phase mutant proteins. (A) Binding of antibodies in 30 anti-β2-GPI-positive APS serum samples to the ‘triple’ mutant protein was determined, as compared with that to control β2-GPI in anti-β2-GPI ELISA. (B) Binding of two typical antibodies (type A and type B) to mutant proteins was determined. β2-GPI and its mutant proteins were coated on a polyoxygenated plate and 100-fold diluted anti-β2-GPI antibody-positive sera were used.
The importance of the domain IV–V interaction was also investigated using an inhibition assay. Binding between WB-CAL-1 and β2-GPI coated on polyoxygenated plates was disrupted by the addition of β2-GPI in the presence of CL–liposome in a dose-dependent manner. This inhibition was not observed by the addition of β2-GPI alone or the triple mutant of β2-GPI with the CL–liposome (Fig. 7). In the absence of the CL–liposome, neither native β2-GPI nor the triple mutant had any inhibition for the binding of WB-CAL-1.
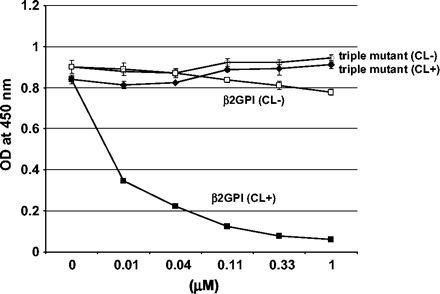
Inhibition of the binding between WB-CAL-1 and β2-GPI coated on a polyoxygenated surface. β2-GPI was coated on polyoxygenated plates at a concentration of 10 μg ml−1 and the binding of WB-CAL-1 (1 μg ml−1) was tested in the presence of inhibitors. The whole recombinant β2-GPI or the ‘triple’ mutant of β2-GPI was added as an inhibitor in the presence or absence of CL–liposome. Binding of WB-CAL-1 to the solid-phase β2-GPI was detected using HRP-conjugated anti-mouse IgG. Closed squares indicate β2-GPI with CL–liposome and open squares indicate β2-GPI without CL. Closed diamonds indicate triple mutant of β2-GPI with CL–liposome and open diamonds indicate the triple mutant of β2-GPI without CL.
As shown in Table 1 and Fig. 3, amino acid W235 was always used to form the antigenic structures recognized by anti-β2-GPI auto-antibodies so that we prepared another mutant protein in which W235 was replaced by L. With this replacement, β2-GPI binding of auto-antibodies, i.e. EY2C9, WB-CAL-1, and of antibodies in 30 of the anti-β2-GPI antibody-positive APS serum samples was significantly diminished (Fig. 8). In contrast, the mutation did not affect antibody binding of mouse anti-human β2-GPI mAb Cof-21 (Fig. 8A).
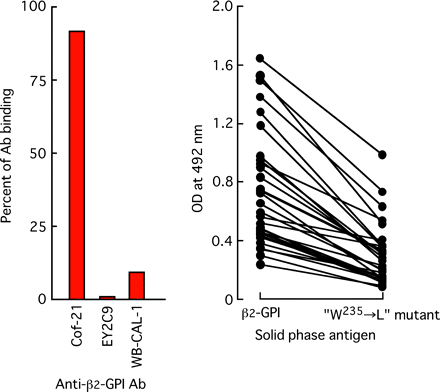
Binding of anti-β2-GPI antibodies to solid-phase ‘W235 → L’ mutant protein. β2-GPI and its mutant proteins were coated on a polyoxygenated plate. Binding of mouse or human mAb (1 μg ml−1) (A) and of antibodies in 30 anti-β2-GPI-positive APS sera to W235 → L mutant protein was determined. A 100-fold diluted anti-β2-GPI antibody-positive sera were used.
Exposure of W235 by the inter-domain interaction between IV and V
Location of W235, which was commonly found in all epitopic structures, was analyzed by making use of molecular modeling. As shown in Fig. 9, W235 partly located in an inner region in domain IV was exposed on the domain's outer surface by electrostatic interaction between domains IV and V, however, the residue was still hidden by domain V. Locations of W235 and P189 in the model of nicked β2-GPI and in the triple-mutant model were similar to that in the X-ray structure. Nicked β2-GPI is a proteolytically cleaved form of β2-GPI that is generated mainly by plasmin in vitro and in vivo (28). This form of β2-GPI does not bind to PL surface and loses its function as a self-antigen (29). The analysis also showed that a structural strain can occur by structural obstacles from the interactions of domain IV and V and from the location of the oligosaccharide attachment site (N234) and it may result in the appearance of configurational changes in domain IV to expose the clusters to the epitopes.
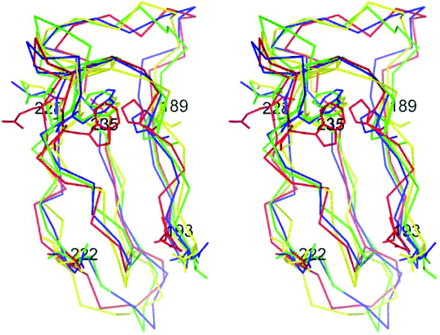
Location of W235 predicted by molecular modeling. A stereoscopic superimposed view of Cα trace of domain IV in the X-ray structure (blue), the predicted domain IV–V model (red), the V-mutated model (green), or the nicked model (yellow) was shown with each structure of D (V)193, D (V)222, E (V)228, W235 and P189.
Structural similarity to the region surrounding W235 residue in domain IV
Corresponding structures surrounding a tryptophan residue, i.e. W53, W111, W175 and W235 in domains I, II, III and IV, respectively, were compared (Fig. 10). The cryptic structure in domain IV was located on two discontinuous loops and a counterpart similar to the epitopic cluster was partly observed in domain I but amino acid composition was not fully complete. In domains II and III, some amino acid residues with different features were present in these counterparts for the epitopic cluster and the locations of the two loops were relatively different.
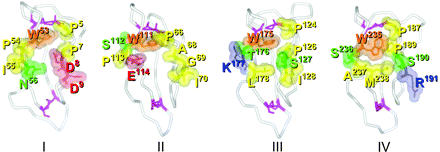
Structural similarity of the corresponding region surrounding a tryptophan (W) residue in each domain. Cα traces of domains I, II, III and IV (of the domain IV–V model) in solvent phase are shown as white curves and with disulfide bridges (violet). Amino acid residues which commonly appeared on at least three out of four epitopic structures (for E1YC8, E1Y2C9, TM1G2 and WB-CAL-1) were superimposed on domain IV. Residues on domains I, II and III corresponding with the epitopic residues are also indicated in each domain, respectively. Positive-charged, negative-charged, non-charged hydrophilic and hydrophobic residues are in blue, red, yellow and green, respectively.
Discussion
β2-GPI was first described in 1961 to be a plasma protein (16) and complete amino acid sequences of human and bovine β2-GPI were determined by peptide sequencing (18, 19). Later, nucleotide sequence and deduced amino acid sequence of human β2-GPI were defined by cDNA cloning from human liver cells and by sequencing (30, 31). The secondary structure of β2-GPI was found to be composed of five homologous motifs, i.e. SCR/CCP repeats or sushi domains, which contain highly conserved half-cysteine residues, related to the formation of two internal disulfide bridges. The crystal structure of human β2-GPI has been reported (20, 21). In the present study, a model of the domain IV–V complex was constructed by considering the crypticity of epitopic structures recognized by the auto-antibodies, as based on the elongated crystal structure of β2-GPI, and three inter-domain electrostatic interactions were observed in the optimized model (Fig. 3).
aCL bound to the solid-phase CL only in the presence of a plasma co-factor, i.e. β2-GPI, and the binding was not prevented in the excessive presence of fluid phase β2-GPI (8, 9). We also reported that such anti-β2-GPI auto-antibodies could bind to β2-GPI adsorbed on a polyoxygenated but not on a plain plate (10). Use of deletion mutant proteins indicated that domain IV was dominantly involved in expression of such epitopes and exposure of the epitopes was attributed to lack of domain V (11). All these data led to the hypothesis that such epitopes might be cryptic and locate on a hidden side of domain IV. We identified the structure of the cryptic epitopes recognized by human and mouse anti-β2-GPI auto-antibodies by making use of epitope mapping and phage libraries in the domain IV structure optimized from its crystal structure. In our β2-GPI model, all the antigenic amino acid clusters for the auto-antibodies mainly consisted of hydrophobic amino acids and were located on two discontinuous sequences in domain IV (Fig. 2). We also noted that it was able to construct a stable conformation of the domain IV–V complex, in which these regions of domain IV were partly on the inner side of the complex and were structurally hidden by overlying domain V (Fig. 3). In contrast, all epitopes specific for non-pathogenic mouse anti-human β2-GPI mAbs, such as Cof-18, Cof-20 and Cof-21, are located on the outer side of individual domains (Fig. 1).
The amino acid W235 was non-exceptionally utilized to form those antigenic structures for anti-β2-GPI auto-antibodies (Fig. 2). As shown in Fig. 7, L replacement of W235 significantly reduced antigenicity for either EY2C9, WB-CAL-1, and antibodies in 30 tested anti-β2-GPI antibody-positive serum samples. Thus, cryptic epitopes in domain IV might be recognized by the major population of anti-β2-GPI antibodies derived from APS and epitope spreading may occur around the W residue. This hypothesis is also supported by the data in the study that replacement of a single amino acid at position 247, which is important for the interaction between domain IV and V, can alter the antigenicity of β2-GPI for pathogenic auto-antibodies, and also can be a risk factor for APS (32, 33).
In contrast, other research groups reported the presence of anti-β2-GPI auto-antibodies directed to domain V (12) or domain I (13), indicating that anti-β2-GPI auto-antibodies still seem to be heterogeneous. In 1996, we first indicated another possible epitopic location of domain I as well as domain IV (11) and as shown in Fig. 10, a similar structure to epitopic clusters located in domain IV was also found in domain I, albeit not fully complete. So, such auto-antibodies from APS may cross-react with the mimic structure in domain I but exact characterization will need to be done. According to the study done by de Laat et al. (34), two of three mutant β2-GPI which bear single amino acid mutation in domain I had reduced reactivity to IgGs from APS patients, indicating the importance of domain I for the binding of anti-β2-GPI antibodies. According to our inhibition ELISA, the mutation of three amino acids which are predicted to be critical for the interaction between domain IV and V of β2-GPI completely lost the inhibitory function found in native β2-GPI in the presence of CL–liposome. This result implies that exposure of the epitopes induced by the domain IV–V interaction in the presence of a negatively charged surface is critical for the binding of WB-CAL-1 (Fig. 7). Discrepancy in the results from these two different studies may indicate the heterogeneity of anti-β2-GPI antibodies, or structural similarity between the candidative epitopes predicted in domain I and IV (Fig. 10).
Cryptic epitopes that appear in the structure of domain IV were conformationally or configurationally altered by PL binding via the particular binding patch in domain V. The key amino acid residue W235 at the epitopic center was located in the inner region in domain IV of the crystal structure of the nicked β2-GPI model, and the β2-GPI model of the three V-replaced the β2-GPI model; however, we considered that the residue was exposed to the outer surface of the domain IV molecule via the three electrostatic interactions. Further, a structural strain possibly occurs by PL binding to the patch which may result in configurational changes in domain IV via the structural obstacles from the domain IV–V interaction and from the oligosaccharide attachment site N234.
The artificial triple mutation on of three specific electrostatic interactions between domains IV and V resulted in reduced antibody binding to the antigenic protein β2-GPI (Figs 3, 6 and 7). Thus, the constructed β2-GPI model and results from binding experiments indicated that domain V interacts with domain IV via these three specific electrostatic interactions and that these interactions are essential for expression of a group of cryptic epitopes in domain IV. These three electrostatic interactions between domain IV and V could contribute to the encrypting by covering W235, the center residue of cryptic epitopes on domain IV, from the solvent phase and auto-antibody binding. Breaking of these interactions by mutations cause the de-encrypting by exposing the W235 residue to the solvent phase, and cryptic epitopes with W235 would be placed to the positions where they can bind to auto-antibodies. In contrast, our present study also indicated that there is another novel group of antigenic structures recognized by anti-β2-GPI auto-antibodies, which appeared only by the triple mutation in these in vitro systems (Fig. 6). Thus, cryptic epitopes which locate in domain IV would also be heterogeneous and at least two kinds of antibody populations would be present in APS patients. We entertained the notion that both populations of antibodies may be in close proximity with domain V yet be hidden. These electrostatic interactions linking domains IV and V may regulate the appearance of both types of cryptic epitopes closely located in domain IV. The core of the lysine-rich region of domain V seems to be constructed by K282, K284, K286, K287, K308 and K317 because of density of lysine on the molecular surface of the X-ray structure. By assuming that this core region could be strongly interacting with anionic PLs, K246, K250 and K305 would not be affected by the binding of PLs, and it is also suggested that domain V combines with domain IV by the contact surface different from PLs.
In addition, a solution structure of this protein was reported by small-angle X-ray scattering (35) that the total conformation would be S shaped and had no direct interaction between domains IV and V. It was reliable as a total shape in the experimental solution. However, some oligosaccharide chains had been employed to compensate the moieties which could not be constructed by protein atoms, although each chain length was not measured correctly in the report.
In contrast, our model had been considered no oligosaccharide chain. The attachment of an oligosaccharide chain to the N234 side chain would not disturb the overlying of domain IV by domain V because the N234 side chain could have the opposite direction to the core of the cryptic epitope, the W235 side chain, on the same β-sheet conformation. It is necessary to get more information on the oligosaccharide chains to obtain the accurate structure.
It is absolutely true that dimerization of the protein increases the avidity to antibodies but the possibility that the interaction between PL and the protein creates a neoepitope also remains. Critical observation to support the latter idea is that even the triple mutant had a similar PL-binding property, and the auto-antibody did not bind the mutant protein in the presence of PL. Thus, the interaction model between domains IV–V may be essential to expose the suitable structure for antibody binding. Antigenic structures which are recognized by different pathogenic anti-β2-GPI antibodies are not identical, even though they were found to be in a particular region on the inner side of domain IV of the β2-GPI molecule. Such responsiveness of antibodies to related multiple epitopes may indicate that intra-molecular epitope spreading may have occurred following initiation by a single epitope on the β2-GPI molecule. Although the majority of clinical manifestations associated with anti-β2-GPI antibodies is vascular occlusions by thrombosis either in vein or in arteries, symptomatic heterogeneity still exists in this syndrome. Establishment of clinical manifestations may depend on epitope spreading and specificity of anti-β2-GPI antibodies in each patient. The occurrence of epitope spreading and whether it contributes significantly to disease pathogenesis is potentially a key issue in designing antigen-directed therapeutics. Epitope mapping and the protein model we used may lead to identification of critical elements related to disease progression.
Transmitting editor: S. Izui
This work was supported in part by a grant for Scientific Research from the Ministry of Education, Science, Sports and Culture of Japan and by grants from the Ministry of Health and Welfare of Japan. We thank M. Ohara for pertinent comments.
References
Harris, E. N., Gharavi, A. E. and Hughes, G. R.
Hughes, G. R., Harris, E. N. and Gharavi, A. E.
McNeil, H. P., Chesterman, C. N. and Krilis, S. A.
Harris, E. N., Gharavi, A. E., Boey, M. L. et al.
Feinstein, D. I. and Rapaport, S. I.
McNeil, H. P., Simpson, R. J., Chesterman, C. N. and Krilis, S. A.
Galli, M., Comfurius, P., Maassen, C. et al.
Matsuura, E., Igarashi, Y., Fujimoto, M., Ichikawa, K. and Koike, T.
Matsuura, E., Igarashi, Y., Fujimoto, M. et al.
Matsuura, E., Igarashi, Y., Yasuda, T., Triplett, D. A. and Koike, T.
Igarashi, M., Matsuura, E., Igarashi, Y. et al.
Wang, M. X., Kandiah, D. A., Ichikawa, K. et al.
Iverson, G. M., Victoria, E. J. and Marquis, D. M.
Roubey, R. A.
Sheng, Y., Kandiah, D. A. and Krilis, S. A.
Schultze, H. E., Heide, K. and Haupt, H.
Bendixen, E., Halkier, T., Magnusson, S., Sottrup-Jensen, L. and Kristensen, T.
Lozier, J., Takahashi, N. and Putnam, F. W.
Kato, H. and Enjyoji, K.
Matsuura, E., Igarashi, M., Igarashi, Y. et al.
Bouma, B., de Groot, P. G., van den Elsen, J. M. et al.
Schwarzenbacher, R., Zeth, K., Diederichs, K. et al.
Ichikawa, K., Khamashta, M. A., Koike, T., Matsuura, E. and Hughes, G. R.
Hashimoto, Y., Kawamura, M., Ichikawa, K. et al.
Takeya, H., Mori, T., Gabazza, E. C. et al.
Brooks, B. R., Bruccoleri, R. E., Olafson, B. D. and States, D. J.
Carlson, W., Karplus, M. and Haber, E.
Hunt, J. E., Simpson, R. J. and Krilis, S. A.
Matsuura, E., Inagaki, J., Kasahara, H. et al.
Steinkasserer, A., Estaller, C., Weiss, E. H., Sim, R. B. and Day, A. J.
Mehdi, H., Nunn, M., Steel, D. M. et al.
Atsumi, T., Tsutsumi, A., Amengual, O. et al.
Yasuda, S., Atsumi, T., Matsuura, E. et al.
de Laat, B., Derksen, R. H., Urbanus, R.T., de Groot, P.G.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}