





Abstract
NK cell transcript 4 (NK4), now denoted as IL-32, was originally identified as a transcript whose expression was increased in activated NK cells. It has been very recently demonstrated that NK4 is secreted from several cells upon the stimulation of some inflammatory cytokines such as IL-18, IL-1β, IFN-γ and IL-12. Furthermore, NK4 induces production of tumor necrosis factor, macrophage inflammatory protein (MIP)-2 and IL-8 in monocytic cell lines, indicating that this factor would be involved in the inflammatory responses. Based on these findings, NK4 was renamed IL-32. However, the biological activities of IL-32 on other cell types remained undetermined. Furthermore, it was still argued whether IL-32 acts on cells from outside or inside the cells. In this article, we first report that expression of IL-32 was up-regulated in activated T cells and NK cells, and that IL-32β was the predominantly expressed isoform in activated T cells. IL-32 was specifically expressed in T cells undergoing apoptosis and enforced expression of IL-32-induced apoptosis, whereas its down-regulation rescued the cells from apoptosis in HeLa cells. IL-32 existing in the supernatant would be derived from the cytoplasm of apoptotic cells. These results strongly indicated that IL-32 would be involved in activation-induced cell death in T cells, probably via its intracellular actions. Our present findings expand our understanding of the biological function of IL-32 and argue that IL-32 may act on cells, not only from the outside but also from the inside.
Introduction
NK cell transcript 4 (NK4), now denoted IL-32, was originally identified as a transcript whose expression was increased in a cDNA library derived from IL-2-activated NK cells (1). This transcript was also up-regulated in phorbol myristate acetate (PMA)-activated PBMCs as well as activated NK cells. Based on the deduced amino acid sequence, the NK4 product was assumed to be a secreted protein because it has a signal sequence, but not a transmembrane region. Furthermore, it was shown that this signal sequence was functional, cleaved in the in vitro translational system (1). Since the transcript of NK4 had been identified, several reports demonstrated that expression of NK4 was augmented at the mRNA level in some conditions such as memory T cells compared with naive T cells (2), EBV-infected B lymphocytes (3), hematopoietic progenitor cells (4) and neuroblastoma cells activated by an anti-neoplastic agent (5). However, the NK4 product had not been functionally characterized, and the physiological role of the NK4 product had not been defined.
Very recently, Kim et al. (6) identified NK4 as an IL-18-responsive gene in a human lung carcinoma cell line, A549. NK4 was secreted from A549 cells, Wish cells and NK cell lines upon the stimulation of some inflammatory cytokines such as IL-18, IL-1β, IFN-γ and IL-12. Furthermore, NK4 induced production of tumor necrosis factor (TNF), macrophage inflammatory protein (MIP)-2 and IL-8 in monocytic cell lines, indicating that this factor would be involved in the inflammatory responses. Based on these findings, NK4 was renamed IL-32. However, the biological activities of IL-32 on other cell types remained undetermined. Furthermore, it was still controversial whether IL-32 acts on cells from outside or inside, because the newly identified isoforms of IL-32 lacked the putative signal peptide existing in the original NK4 (IL-32γ), and the amounts of IL-32 secreted from the cells were small, compared with those in the cytoplasm (6).
In this article, we found up-regulation of IL-32 expression in activated T cells and NK cells, and identified isoforms of IL-32 expressed in T cells. Furthermore, we confirmed specific expression of IL-32 in T cells undergoing apoptosis and showed IL-32-dependent apoptosis in HeLa cells. IL-32 existing in the supernatant would be derived from the cytoplasm of apoptotic cells. These findings indicated that IL-32 would be involved in activation-induced cell death (AICD) in T cells, probably via its intracellular actions.
Methods
Plasmids
IL-32β and IL-32γ cDNAs were prepared by reverse transcription (RT)–PCR using a cDNA library derived from human lung fibroblasts purchased from Sanko Junyaku (Tokyo, Japan), and then those were incorporated into pME18S (7). The three tandem-lined FLAG and myc-His sequences derived from p3xFLAG-CMV™-7.1 (Sigma–Aldrich, St Louis, MO, USA) and pcDNA3.1-myc-HisB (Invitrogen, Carlsbad, CA, USA), respectively, were attached by PCR to the N-terminus and C-terminus, respectively, of the coding region of both IL-32β and IL-32γ cDNAs. These fragments were denoted as (3xFLAG)-IL-32γ-(myc-His), which was incorporated into pTRE2-hyg (BD Biosciences Clontech, Palo Alto, CA, USA).
Cells
DND-39 cells were maintained in RPMI 1640 medium supplemented with 10% of FCS, 100 μg ml−1 of streptomycin and 100 units ml−1 of penicillin G. HEK293T and HeLa cells were maintained in DMEM supplemented with 10% of FCS, 100 μg ml−1 of streptomycin and 100 units ml−1 of penicillin G. For HeLa Tet-OffTMM cells (BD Biosciences Clontech), 800 μg ml−1 of G418 (Sigma–Aldrich) and 200 ng ml−1 of tetracycline hydrochloride (Wako, Osaka, Japan) were added to the medium.
PBMCs were separated from healthy human volunteers using Lymphoprep™ (Axis-Shield, Oslo, Norway), and then B cells were isolated from PBMCs using Dynabeads™ M-450 CD19 (Dynal, Oslo, Norway). NK cells and T cells were selected from whole-blood cells using RosetteSep™ antibody cocktail (StemCell Technologies Inc., Vancouver, British Columbia, Canada) or Dynal CD2 CELLectin™ kit (Dynal).
B cells were stimulated with 10 ng ml−1 of IL-4 (Peprotech, Rocky Hill, NJ, USA) or 10 μg ml−1 of anti-IgM antibody (Cappel, Aurora, OH, USA) and/or 0.5 μg ml−1 of anti-CD40 antibody (Immunotech, Marseille, France) for 24 h. T cells were stimulated with 10 ng ml−1 of IL-4 or 10 ng ml−1 of IL-2 (Genzyme, Cambridge, MA, USA) or 2 μg ml−1 of coated anti-CD3 antibody (Immunotech) or 5 ng ml−1 of PMA (Sigma–Aldrich) and 1 μM of ionomycin (Sigma–Aldrich) for 24 or 48 h. NK cells were stimulated with 10 ng ml−1 of IL-2 for 24 h.
Transfection
Transient expression of IL-32β/γ alone into HEK293T was performed by TransFast™ transfection reagent (Promega, Madison, WI, USA). HeLa Tet-Off™ cells stably expressing (3xFLAG)-IL-32γ-(myc-His) (HeLa Tet-Off/NK4) were selected with 200 μg ml−1 of hygromycin (Sigma–Aldrich) after transfection.
Quantitative RT–PCR analysis of IL-32
Quantitative analysis of mRNA expression for IL-32 was performed using the ABI PRISM™ 7000 sequence detection system (Perkin-Elmer Japan, Urayasu, Japan), as described before (8). To calculate the copy numbers for IL-32, standard curves were generated using a plasmid encoding that gene whose copy numbers were known. The PCR primers used were 5′-CGTGGACAGGTGATGTCGAG-3′ and 5′-CCGCCACTGTCTCCAGGTAG-3′, and the TaqMan™ probe used was 5′-FAM-TGGCAGAGCTGGAGGACGACTTCA-TAMRA-3′.
RT–PCR
Total RNA was extracted by ISOGEN (Nippongene, Tokyo, Japan). The RT reaction primed with random hexamer was performed using GeneAmp RNA PCR Kit (Applied Biosystems Japan, Tokyo, Japan). The PCR reaction was performed with cDNA as a template using the primers below after an initial 1-min denaturation at 96°C, followed by the indicated cycles of 96°C for 1 min, 55 or 57°C for 1 min and 72°C for 1 min. The PCR primers used were 5′-GCCTTGGCTCCTTGAACTTTTG-3′ or 5′-CCGAAGGTCCTCTCTGATGA-3′ for the upper primer of IL-32, and 5′-CCGCCACTGTCTCCAGGTAG-3′ for the lower primer of IL-32, 5′-GGCTCCAGGCGGTGCTTGTTC-3′ and 5′-AGACGGCGATGCGGCTGATG-3′ for TNF and 5′-GAAGGTGAAGGTCGGAGT-3′ and 5′-CTTCTACCACTACCCTAAAG-3′ for glyceraldehyde-3-phosphate dehydrogenase (GAPDH), respectively.
Generation of anti-IL-32 serum
His-tagged exon 7/intron 7/exon 8 product of the IL-32 protein was expressed in an Escherichia coli strain, Rosetta™ (DE3; Novagen, Madison, WI, USA), and isolated by using Ni-NTA™ agarose (Qiagen, Hilden, Germany). Purified His-tagged exon 7/intron 7/exon 8 product was used together with CFA (Sigma–Aldrich) for immunization of New Zealand white rabbits. IgG was purified from the serum by ImmunoPure (G) Immobilized Protein G (Pierce, Rockford, IL, USA) and the purified IgG was biotinylated with EZ-Link™ Sulfo-NHS-LC-Biotin (Pierce).
Western blotting
The samples were applied to SDS-PAGE, and then electrophoretically transferred to polyvinylidene difluoride membranes (Amersham Biosciences). The membranes were blotted by anti-IL-32 serum or anti-histone deacetylase class 1 (HDAC1) antibody (Santa Cruz Biotechnology, Santa Cruz, CA, USA). The proteins were visualized by enhanced chemiluminescence (Amersham Biosciences). In some experiments, proteins in the supernatant were precipitated by trichloroacetic acid (Wako) and cytosolic proteins were treated with N-glycosidase (PNGase F, New England BioLabs, Beverly, MA, USA).
RNA interferance (RNAi) method
The short-hairpin RNA (shRNA)-expressing plasmid was generated in pSilencer™ 2.0-U6 (Ambion, Austin, TX, USA) according to the manufacturer's instructions. Briefly, the shRNA sequence contained a 19-mer sense sequence of IL-32 cDNA, separated by a 9-mer spacer, TTCAAGAGA, from the 19-mer anti-sense sequence complementary to the sense sequence, followed by five consecutive T's as the transcriptional terminator. Target shRNA sequence was inserted into BamHI and HindIII sites in the vector. The target sequence used was 5′-AGAGGGCTACCTGGAGACA-3′. The pSilencer vector was used as the negative control. The shRNA expressed by this vector had limited homology to any known sequences in the human, mouse or rat genomes.
Apoptosis assay
Quantitation of apoptotic HeLa cells was performed using DeadEnd™ Fluorometric TUNEL System (Promega) or an annexin V–FITC apoptosis detection kit I (BD PharMingen, San Diego, CA, USA). Tetracycline was removed either 5 or 4 days before harvest for detection of TUNEL or annexin, respectively, and the concentration of FCS was decreased to 0.1% 1 day after the removal of tetracycline. For the RNAi experiments, the shRNA-expressing plasmid was transfected 1 day after the removal of tetracycline.
Quantitation of apoptotic primary T cells was performed as follows. After activated T cells were fixed with 4% PFA, the cells were permeabilized with 70% ethanol for 72 h. For the analysis of double staining of IL-32 and TUNEL, TUNEL was first performed, and then the cells were labeled with biotinylated anti-IL-32 antibody, followed by PE–streptavidin (BD PharMingen). For the analysis of double staining of IL-32 and active caspase-3, the cells were first labeled with PE-conjugated anti-active caspase-3 antibody (BD PharMingen) and then with biotinylated anti-IL-32 antibody, followed by FITC–streptavidin. Quantitation of the staining was performed using FACSCalibur, and the data were analyzed using Cell Quest software (BD Biosciences, San Jose, CA, USA).
Results
Induction of IL-32 in T cells and NK cells
To identify IL-4-inducible genes in a human Burkitt's lymphoma cell line, DND-39, we subjected cRNA derived from IL-4-stimulated DND-39 cells to microarray analysis. One nucleotide sequence among the inducible genes, corresponding to the coding region of the IL-32 gene, was consistently and highly induced by IL-4 (9). To explore the possibility that expression of IL-32 would be up-regulated by IL-4 in B cells, we stimulated human primary B cells by IL-4 and analyzed expression of IL-32. We chose the primers for the quantitative RT–PCR analysis to detect all four isoforms of IL-32 (α, β, γ and δ; 6). IL-32 tended to be expressed in B cells by IL-4, anti-IgM antibody and anti-CD40 antibody using quantitative RT–PCR analysis; however, the copy number was extremely low, and RT–PCR did not show the band corresponding to IL-32 (data not shown). These results suggested that expression of IL-32 was negligible in primary B cells at the examined conditions. In contrast, the stimulus of either anti-CD3 antibody or PMA and ionomycin significantly induced expression of IL-32 by almost 6- and 7-fold, respectively, in human primary T cells, by quantitative RT–PCR analysis (Fig. 1). IL-2 and IL-4 slightly augmented the expression of IL-32 but the enhancement was negligible (Fig. 1, data not shown). In addition to T cells, we confirmed that IL-32 was induced in NK cells activated by IL-2 by almost 4-fold by quantitative RT–PCR analysis, as previously described (1; Fig. 1). These results demonstrated that activation of T cells and NK cells induce expression of IL-32 in these cells.
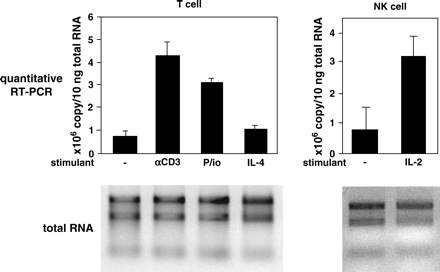
Induction of IL-32 in T cells and NK cells. Purified T cells or NK cells were incubated with either 2 μg ml−1 of coated anti-CD3 antibody or 5 ng ml−1 of PMA and 1 μM of ionomycin (P/io) or 10 ng ml−1 of IL-4 or 10 ng ml−1 of IL-2 for 24 h, and expression of the IL-32 gene was analyzed by quantitative RT–PCR analysis. The copy number was calculated using a standard plasmid. The applied RNA of each sample is depicted. The mean values of three independent experiments are shown.
Identification of isoforms of IL-32 expressed in activated T cells
Kim et al. (6) demonstrated that there exist at least four isoforms of IL-32 generated by alternative splicing. To examine which isoform of IL-32 is predominantly expressed in T cells, we sequenced the full length of 21 clones derived from activated T cells. Fourteen clones among the 21 were identical with the sequence of IL-32β (Fig. 2A). Only one clone of IL-32δ was detected, and no IL-32α or IL-32γ was observed. Interestingly, we found two novel isoforms, IL-32ε and IL-32ζ (Fig. 2A). IL-32ε lacked exon 4 and IL-32ζ lacked exons 2 and 3, respectively. Existence of IL-32ε has been very recently reported (accession no.: NM_001012634 and NM_001012635). There existed five clones of IL-32ε and one clone of IL-32ζ among 21 clones. These results indicated that although several isoforms of IL-32 were generated by alternative splicing in activated T cells, IL-32β is the predominantly expressed transcript in these cells.
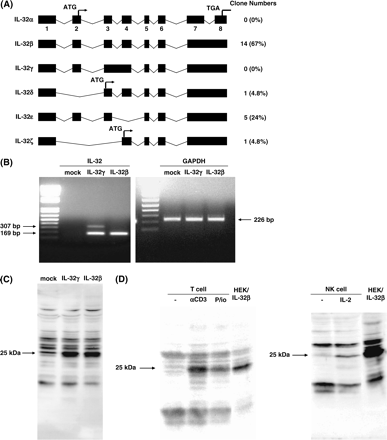
IL-32 isoforms expressed in activated T cells. The structure of IL-32 isoforms and the number of each clone in activated T cells are depicted. The figures under the boxes represent the exon numbers. The nomenclature of IL-32α, β, γ and δ is based on the literature of Kim et al. (6). IL-32ε and ζ are novel isoforms. (B) Total RNA was extracted from mock- or IL-32γ- or IL-32β-transfected HEK293T cells, and then RT–PCR for IL-32 or GAPDH was performed. The PCR reactions were performed for 25 cycles. (C) Cell lysates were prepared from mock- or IL-32γ- or IL-32β-transfected HEK293T cells, and then western blot analysis was performed using anti-IL-32 serum. (D) Cell lysates were prepared from T cells activated by anti-CD3 antibody or PMA/ionomycin (P/io) or NK cells activated by IL-2, as described in Fig. 1, or IL-32β-transfected HEK293T cells. Twenty-five or 10 μg of cell lysates for T cell or for NK cell, respectively, per lane, were submitted to western blot analysis using anti-IL-32 serum.
We next generated the plasmids encoding IL-32γ, originally identified as NK4, and IL-32β, and analyzed expression of both types at the mRNA and protein levels in HEK293T cells. The transcript of IL-32β appeared as a single band, whereas IL-32γ was transcribed as two bands, of which the shorter one, predominant compared with the longer one, had the same length as IL-32β (Fig. 2B). We confirmed that the nucleotide sequences of all of these bands corresponded to that of either IL-32β or IL-32γ (data not shown). When either IL-32β or IL-32γ was expressed in HEK293T cells, only the same mobility band of 25 kDa, which was almost the same as the predicted size of IL-32β, 22 kDa, was recognized by anti-IL-32 serum (Fig. 2C). These results indicated that the cDNA encoding IL-32γ could be transcribed as both IL-32γ and IL-32β, and that the IL-32β type would be translated more than the IL-32γ type, probably because of different efficiencies of translation or a difference in mRNA stability.
When T cells were stimulated with anti-CD3 antibody or PMA and ionomycin, or NK cells were stimulated with IL-2, only the 23-kDa band recognized by anti-IL-32 serum appeared (Fig. 2D). The mobility of this band was the same as that of IL-32β expressed in HEK293T cells. Taking these results together, although IL-32 was induced in activated T cells and NK cells as previously described (1), IL-32β was the major product in activated T cells.
Specific expression of IL-32 in AICD-caused T cells
It is well known that activated T cells undergo AICD, the mechanism to eliminate activated T cells (10–13). In fact, upon stimulation of anti-CD3 antibody for 48 h, T cells undergoing apoptosis were increased compared with unstimulated cells (32 versus 8.7%, evaluated from annexin V-positive, propidium iodide-negative cells, data not shown). Considering our findings that when T cells were stimulated with anti-CD3 antibody, expression of IL-32 was induced, we assumed that IL-32 would be correlated with AICD in T cells. To explore this possibility, we examined whether IL-32 was expressed only in activated T cells that are undergoing apoptosis. When T cells were unstimulated, no cells showed apoptosis as estimated by TUNEL assay (Fig. 3A) and expression of active caspase-3 (Fig. 3B), and no expression of IL-32 was detected. In contrast, when T cells were stimulated with anti-CD3 antibody, the number of T cells showing double staining of IL-32 and either TUNEL (Fig. 3A) or active caspase-3 (Fig. 3B) was augmented. We confirmed that addition of recombinant IL-32 blocked the signal of IL-32 expressed in T cells (data not shown). These results demonstrated that expression of IL-32 was specific in AICD-caused T cells.
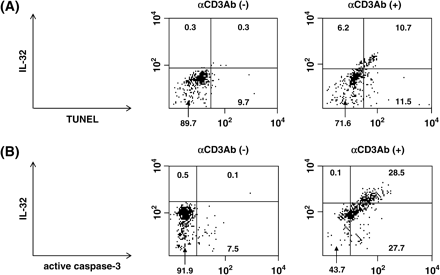
Induction of apoptosis by IL-32 in T cells. (A and B) Purified T cells were incubated with 2 μg ml−1 of coated anti-CD3 antibody for 48 h. Co-expression of either TUNEL and IL-32 (A) and active caspase-3 and IL-32 (B) in unstimulated and stimulated T cells are shown. The inserted figures represent the population of the cells in each region (%).
Induction of apoptosis by IL-32 in HeLa cells
We tried to establish cells stably expressing IL-32 or to transfect transiently IL-32 into primary T cells or Jurkat cells to examine the function of IL-32 in vitro; however, we found it difficult. One reason was that over-expression of IL-32 in cells might cause cell death and/or cell cycle arrest. To explore this possibility, we established HeLa cells in which IL-32 was induced by removal of tetracycline (HeLa Tet-Off/IL-32 cells), and analyzed the induction of apoptosis. In this cell system, only a small amount of IL-32 was expressed in the presence of tetracycline, whereas removal of tetracycline from the culture medium evoked expression of IL-32, predominantly IL-32β, in the cells (Fig. 4A).
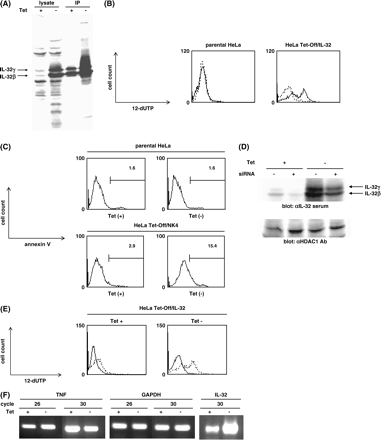
Induction of apoptosis by IL-32 in HeLa cells. (A) HeLa Tet-Off/IL-32 cells were cultured with or without tetracycline for 5 days, and then cell lysates or immunoprecipitates with anti-IL-32 serum were blotted with anti-IL-32 serum. (B and C) HeLa Tet-Off/IL-32 cells or parental HeLa cells were cultured with or without tetracycline for 5 days in the presence of 0.1% FCS, and then TUNEL assay (B) or annexin V assay (C) was analyzed by flow cytometry. Each experiment was done two times, and representative data are shown. The solid and dashed lines represent the absence or presence of tetracycline, respectively, in panel B. (D) HeLa Tet-Off/IL-32 cells were cultured with or without tetracycline for 1 day, and either mock- or IL-32-targeted shRNA-expressing plasmid was transfected into parental Hela cells or HeLa Tet-Off/IL-32 cells. After 4 days, cell lysates were blotted with either anti-IL-32 serum or anti-HDAC1 antibody. (E) IL-32-targeted shRNA-expressing plasmid was transfected into HeLa Tet-Off/IL-32 cells as (D), and then after 4 days, TUNEL assay was analyzed by flow cytometry. Each experiment was done three times, and representative data are shown. The solid and dashed lines represent the transfection of IL-32-targeted and mock shRNA-expressing plasmid, respectively. (F) HeLa Tet-Off/IL-32 cells were cultured with or without tetracycline for 5 days, and then RT–PCR for TNF or IL-32 or GAPDH was performed. The PCR reactions were performed for the indicated cycles.
We analyzed induction of apoptosis using these cells by TUNEL assay or annexin V assay. When parental HeLa cells were incubated without tetracycline, almost no cells showed positive staining for TUNEL assay in the presence of 0.1% FCS (Fig. 4B). In contrast, removal of tetracycline caused appearance of positive staining in HeLa Tet-Off/IL-32 cells (Fig. 4B). HeLa Tet-Off/IL-32 cells showed slightly positive staining even in the existence of tetracycline compared with parental HeLa cells, which would be due to leaky expression of IL-32. We next analyzed expression of annexin V that has a strong affinity for phosphatidylserine, a marker of apoptosis, in either the presence or absence of tetracycline. Removal of tetracycline caused a significant increase in annexin V-positive cells in HeLa Tet-Off/IL-32 cells (15.4 versus 2.9%), whereas the same treatment did not affect the ratio of positive cells for annexin V in parental HeLa cells (1.6 versus 1.6%) as well as TUNEL assay (Fig. 4C).
We next tested the effects of RNAi on IL-32-inducible apoptosis in HeLa cells. When the plasmid expressing an shRNA targeting IL-32 was transfected in HeLa Tet-Off/IL-32 cells, expression of IL-32 was decreased by almost half at the protein level, whereas there was no effect on expression of HDAC1 (Fig. 4D). When the shRNA-expressing plasmid was incorporated into HeLa Tet-Off/IL-32 cells, the population of positive cells for TUNEL assay was significantly decreased in either the presence or absence of tetracycline (Fig. 4E). These results clearly demonstrated that IL-32 was capable of causing apoptosis in HeLa cells.
Kim et al. (6) reported that IL-32 induces production of TNF in monocytic cell lines. We explored the possibility that IL-32-inducible apoptosis in HeLa cells would be due to induction of TNF by IL-32. However, removal of tetracycline did not affect the expression of TNF, contradicting the involvement of TNF in induction of apoptosis by IL-32 in HeLa cells (Fig. 4F).
Intracellular actions of IL-32 on cells
It was assumed that the IL-32γ, originally isolated as NK4, would be a secreted protein because its amino acid sequence had a signal sequence, but not a transmembrane region, and that the potential signal sequence was cleaved in the in vitro translation system (1). Kim et al. (6) demonstrated that enforced expression of either IL-32α or IL-32β led to secretion of these proteins and that activated PBMCs, NK cell lines, A549 cells and Wish cells, secreted IL-32, although both IL-32α and IL-32β lack the putative signal sequence. We next investigated whether secretion of IL-32 was involved in AICD in T cells. When T cells were activated by anti-CD3 antibody, small amounts of IL-32 were accumulated in the supernatant in a time-dependent manner (Fig. 5). However, the amounts of IL-32 in the supernatant were parallel with those of GAPDH, a cytosolic protein. These results strongly supported the notion that IL-32 in the supernatant was derived from the cytoplasm of apoptotic cells, rather than it being secreted from the cells.
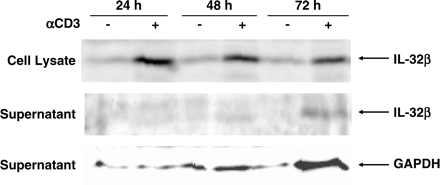
Expression of IL-32 in the supernatant and the lysate. Purified T cells were incubated with 2 μg ml−1 of coated anti-CD3 antibody for the indicated times. Twenty micrograms of cell lysates or precipitates of 300 μl supernatant by trichloroacetic acid, per lane, were submitted to western blot analysis using anti-IL-32 serum.
Discussion
In this article, we demonstrated that expression of IL-32 was specific in AICD-caused T cells (Fig. 3) and that induction of IL-32 caused apoptosis, whereas its down-regulation rescued the cells from apoptosis in HeLa cells (Fig. 4). These results strongly suggested that IL-32 would be involved in AICD in T cells. Previously, it had been reported that IL-32 induces production of TNF, MIP-2 and IL-8 in monocytic cell lines, indicating that IL-32 would play a role in the inflammatory responses (6). This had been the only known biological activity of IL-32. Our present findings have added a novel aspect of the biological function of IL-32.
AICD, the mechanism to eliminate activated T cells, is an important process for T cell homeostasis in that after the immune response peak, the majority of antigen-specific T cells is eliminated (10–13). Furthermore, this process may be also involved in peripheral tolerance. It is known that AICD in T cells occurred mainly via the CD95 signal pathway, by augmentation of CD95 ligand expression and the susceptibility to CD95 ligand by formation of death-inducing signaling complex, and down-regulation of c-FLIP and Bcl-xL, followed by activation of the caspase cascade (10–13). In addition to the CD95 signal pathway, the signal pathways of IL-2 (14, 15), TNF (16–18) and CD2 (19) would also be involved in AICD. The precise mechanism of how IL-32 is involved in AICD remains obscure at this moment because IL-32 has no known functional domain. It has been reported that IL-32 induces production of TNF in monocytic cell lines (6). However, expression of TNF was not influenced by induction of IL-32 in HeLa cells (Fig. 4E). IL-32 may accelerate the CD95 signal by modifying expression or activation of some protein involved in this pathway, or alternatively IL-32 may modify the signal pathways of IL-2 or CD2 that are known to be involved in AICD different from TNF, in addition to the CD95 signal pathway. Although we showed that IL-32 was induced by IL-2 in NK cells, the physiological role of IL-32 in NK cells are unclear at this moment. Because IL-2 is a critical factor for development and proliferation of NK cells (20, 21), the role of IL-32 in NK cells would be different in T cells.
We next demonstrated that IL-32 would cause apoptosis in T cells from inside the cells, rather than from outside. The amounts of accumulated IL-32 in the supernatant were parallel with those of GAPDH, a cytosolic protein (Fig. 5). Furthermore, the mobilities on SDS-PAGE of IL-32 in the cell lysate and supernatant were the same, and treatment of N-glycosidase of the cell lysate did not change the mobilities of IL-32 (data not shown), indicating that in our system there is no evidence that IL-32 is secreted via the conventional (classical) endoplasmic reticulum (ER)/Golgi-dependent pathway. These findings may argue against the results of Kim et al. (6). However, they also did not exclude the possibility that IL-32 exerts its actions from the inside of the cells because IL-32α, β and δ lack the putative signal peptide and the amounts of IL-32 secreted from the cells were very low. Both cytosolic and secreted types of IL-32 may exist. Alternatively, IL-32 may be secreted via the non-conventional (non-classical) pathway, independent from the functional ER/Golgi system (22).
In the previous report, it was shown that four isoforms of IL-32 (α, β, γ and δ) are generated by alternative splicing (6). In the present article, we have demonstrated that there exist two more novel isoforms of IL-32 (ε and ζ) and that IL-32β is the major product in activated T cells (Fig. 2). It is a matter to be resolved in the future to elucidate the expression pattern of each isoform in certain cells or tissues and the functional differences of each isoform. Particularly, it is of note that IL-32γ, originally identified as NK4, involving intron 3 in the transcript, contains a potential signal sequence consisting of 35 amino acids predicted by SignalP ver. 3.0, hidden Markov models (23, 24), whereas other isoforms lack its C-terminal half, which might affect localization of these isoforms. Furthermore, IL-32δ and ζ, lacking exon 2 and exons 2 and 3, respectively, should change the translation start site, which may influence the function of these molecules, although thus far no known functional domain has been found in the sequence of IL-32.
In conclusion, we found that expression of IL-32β was up-regulated in activated T cells. IL-32 would be involved in AICD in T cells, probably from inside the cells. These findings expand our understanding of the biological function of IL-32.
Transmitting editor: H. Karasuyama
We thank Dovie R. Wylie for critical review of this manuscript. This work is supported by a Research Grant for Immunology, Allergy and Organ Transplant from the Ministry of Health, Welfare, and Labor of Japan, a Grant-in-Aid for Scientific Research from Japan Society for the Promotion of Science.
References
Dahl, C. A., Schall, R. P., He, H. L. and Cairns, J. S.
Liu, K., Li, Y., Prabhu, V. et al.
Carter, K. L., Cahir-McFarland, E. and Kieff, E.
Brown, J., Matutes, E., Singleton, A. et al.
Park, G. H., Choe, J., Choo, H. J., Park, Y. G., Sohn, J. and Kim, M. K.
Kim, S. H., Han, S. Y., Azam, T., Yoon, D. Y. and Dinarello, C. A.
Umeshita-Suyama, R., Sugimoto, R., Akaiwa, M. et al.
Yuyama, N., Davies, D. E., Akaiwa, M. et al.
Tanaka, G., Kanaji, S., Hirano, A. et al.
Krueger, A., Fas, S. C., Baumann, S. and Krammer, P. H.
Hildeman, D. A., Zhu, Y., Mitchell, T. C., Kappler, J. and Marrack, P.
Janssen, O., Sanzenbacher, R. and Kabelitz, D.
Refaeli, Y., Van Parijs, L., London, C. A., Tschopp, J. and Abbas, A. K.
Van Parijs, L., Refaeli, Y., Lord, J. D., Nelson, B. H., Abbas, A. K. and Baltimore, D.
Zheng, L., Fisher, G., Miller, R. E., Peschon, J., Lynch, D. H. and Lenardo, M. J.
Speiser, D. E., Sebzda, E., Ohteki, T. et al.
Sytwu, H. K., Liblau, R. S. and McDevitt, H. O.
Mollereau, B., Deckert, M., Deas, O. et al.
Bonavida, B., Lebow, L. T. and Jewett, A.
Williams, N. S., Klem, J., Puzanov, I. J. et al.
Nickel, W.
Nielsen, H., Engelbrecht, J., Brunak, S. and von Heijne, G.
Nielsen, H. and Krogh, A.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}