




Abstract
Epimutations, such as the hypermethylation and epigenetic silencing of tumor suppressor genes, play a role in the etiology of human cancers. In contrast to DNA mutations, which are passively inherited through DNA replication, epimutations must be actively maintained because they are reversible. In fact, the reversibility of epimutations by small-molecule inhibitors provides the foundation for the use of such inhibitors in novel cancer therapy strategies. Among the compounds that inhibit epigenetic processes, the most extensively studied are DNA methyltransferase inhibitors. In this review, we examine the literature on DNA methyltransferase inhibitors and discuss the efficacy of such compounds as antitumor agents, as evaluated in phase I–III clinical trials. We also discuss future areas of research, including the development of nonnucleoside inhibitors, the application of novel bioanalytical tools for DNA methylation analysis (which will be important for the clinical application of these compounds by allowing rational approaches to trial design), the need to optimize treatment schedules for maximal biologic effectiveness, and the need to define molecular endpoints so that changes induced by demethylating drugs in patients can be monitored during treatment. Assays for genome-wide and tumor-specific DNA methylation also need to be further developed to establish the pharmacodynamic parameters of DNA methyltransferase inhibitors in patients and to provide rational approaches to maximizing the therapeutic efficacy of these compounds.
Epigenetic mechanisms regulate the expression of genetic information. Epigenetic modifications of DNA and histones are stable and heritable but are also reversible ( 1 ) . They include covalent modifications of bases in the DNA and of amino acid residues in the histones. DNA methyltransferases are a family of enzymes that methylate DNA at the carbon-5 position of cytosine residues ( 2 ) . Methylated DNA can then be bound by methyl-binding proteins ( 3 ) that function as adaptors between methylated DNA and chromatin-modifying enzymes (e.g., histone deacetylases and histone methyltransferases) by recruiting histone-modifying enzymes to stretches of methylated DNA ( Fig. 1 ). Histone-modifying enzymes then covalently modify the amino-terminal residues of histones to induce the formation of chromatin structures that repress gene transcription ( 4 ) . Examples of covalent histone modifications include the methylation of the lysine at position 9 in histone H3 and the deacetylation of the lysine at position 16 in histone H4, both of which are associated with gene silencing. The interplay between DNA methylation and histone modifications has a profound effect on the epigenetic regulation of gene expression patterns ( 5 ) and can thus become an important factor in the development of cancer.
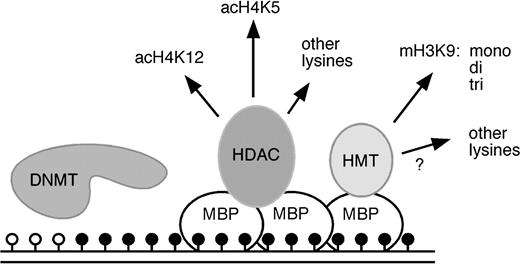
Epigenetic regulation by DNA methyltransferases methyl-binding proteins and histone modifying enzymes. DNA is methylated by DNA methyltransferases (DNMTs). Methylated cytosine residues ( solid circles ) are bound by methyl-binding proteins (MBPs) that subsequently recruit histone deacetylases (HDACs) and histone methyltransferases (HMTs). These enzymes mediate complex changes in the histone modification pattern of methylated genes that result in the establishment of repressive chromatin structures. acH4K12 = lysine 12–acetylated histone H4; acH4K5 = lysine 5–acetylated histone H4; mH3K9 = lysine 9–methylated histone H3; mono, di, tri = mono-, di-, tri-methylated; open circles = unmethylated cytosine residues.
DNA methylation is a crucial mechanism associated with epigenetic regulation. DNA methylation triggers chromatin reorganization that is mediated by methyl-binding proteins ( Fig. 1 ), it has a comparatively simple binary pattern (i.e., methylated versus nonmethylated bases) compared with the highly complex pattern of histone modifications ( Fig. 1 ), and it is particularly amenable to experimental analysis. Changes in the pattern of DNA methylation, either increased (hypermethylation) or decreased (hypomethylation), have been identified in all types of cancer cells examined so far. In addition, it has also been shown that genetic lesions in human cancer cells can promote epigenetic alterations. The leukemia-promoting PML–RAR fusion protein, for example, can recruit DNA methyltransferases to the target genes for the fusion protein and thereby induce epigenetic silencing ( 6 ) . These results provided an important paradigm for the cooperation of genetic and epigenetic lesions in promoting tumorigenesis.
Genomic tumor DNA is generally characterized by distinct methylation changes that have also been termed epimutations. At the global level, the DNA is often hypomethylated, particularly at centromeric repeat sequences, and this hypomethylation has been linked to genomic instability ( 7 ) . Another class of epimutations is characterized by the local hypermethylation of individual genes, which has been associated with aberrant gene silencing ( 8 ) . Epimutations have been described in many types of cancer and appear to play an important role in tumorigenesis. For instance, epigenetic silencing, rather than gene mutation, is the main mechanism of inactivation of the DNA mismatch repair gene hMLH1 in sporadic colon cancer ( 9 ) , and the methylation of E-cadherin has a central role in metastasis and invasion of breast cancers ( 10 ) . Such epimutations rarely appear in healthy tissue, indicating that epigenetic therapies may have high tumor specificity.
The reversibility of epigenetic modifications renders them attractive targets for therapeutic interventions ( 11 ) . In contrast to genetic mutations, which are inherited passively through DNA replication, epigenetic mutations must be actively maintained. Consequently, pharmacologic inhibition of certain epigenetic modifications could correct faulty modification patterns and thus directly change gene expression patterns and the corresponding cellular characteristics.
Progress in the development of pharmacologic inhibitors differs widely between individual enzyme families. The development of histone methyltransferase inhibitors is still in an early preclinical stage. Several histone deacetylase inhibitors are currently being tested in phase I and phase II clinical trials. However, many other cellular proteins are acetylated, including key regulators of tumor cell growth, and so it is unclear whether the growth inhibition induced by histone deacetylase inhibitors is the result of alterations in the histone acetylation patterns or alterations in signaling pathways that regulate cell proliferation. DNA methyltransferase inhibitors are at a more clinically advanced stage of development than inhibitors of histone deacetylases or histone methyltransferases, having been extensively tested in phase I–III clinical trials. In addition, the prototypical DNA methyltransferase inhibitor 5-azacytidine (i.e., Vidaza) has recently been approved by the Food and Drug Administration (FDA) as an antitumor agent for the treatment of myelodysplastic syndrome. DNA methyltransferase activity has an important role in tumor growth, and the activity of DNA methyltransferase inhibitors can be analyzed directly at the DNA level. Consequently, in this review, we will focus on the role of DNA methyltransferase inhibitors in the further development of epigenetic cancer therapies. We also discuss future areas of research, including the development of nonnucleoside inhibitors, the application of novel bioanalytical tools for DNA methylation analysis, and the need to optimize treatment schedules on the basis of defined molecular endpoints that allow changes induced by demethylating drugs in patients to be monitored.
Nucleoside DNA Methyltransferase Inhibitors
The archetypal DNA methyltransferase inhibitor 5-azacytidine, a simple derivative of the nucleoside cytidine ( Fig. 2 ), was first described more than 40 years ago ( 12 ) . Its demethylating activity was discovered later as the result of its ability to influence cellular differentiation ( 13 ) . 5-Azacytidine is a nucleoside inhibitor that is incorporated into DNA. DNA methyltransferases methylate both cytosine residues and 5-azacytosine residues in the DNA. However, 5-azacytosine prevents the resolution of a covalent reaction intermediate ( 14 ) , which leads to DNA methyltransferase being trapped and inactivated in the form of a covalent protein–DNA adduct ( Fig. 3 ). As a result, cellular DNA methyltransferase is rapidly depleted, and concomitantly genomic DNA is demethylated as a result of continued DNA replication. 5-Azacytidine is a ribose nucleoside and thus must be chemically modified to a deoxyribonucleoside triphosphate to be incorporated into DNA. However, before all 5-azacytidine is converted to a deoxyribonucleoside triphosphate, a portion of it is incorporated into RNA, which affects a variety of RNA functions including ribosome biogenesis ( 15 ) and, therefore, has cellular consequences independent of demethylation ( 16 ) .
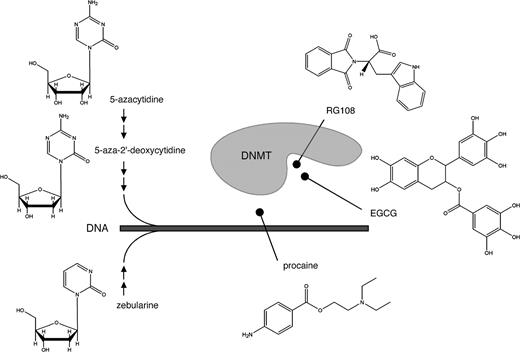
DNA methyltransferase (DNMT) inhibitors and their inhibitory mechanisms. The nucleoside inhibitors 5-azacytidine, 5-azadeoxycytidine, and zebularine are extensively metabolized by cellular pathways ( small arrows ) before being incorporated into DNA. After incorporation, they function as suicide substrates for DNMT enzymes. The nonnucleoside inhibitors procaine, epigallocatechin-3-gallate (EGCG), and RG108 have been proposed to inhibit DNA methyltransferases by masking DNMT target sequences (i.e., procaine) or by blocking the active site of the enzyme (i.e., EGCG and RG108).
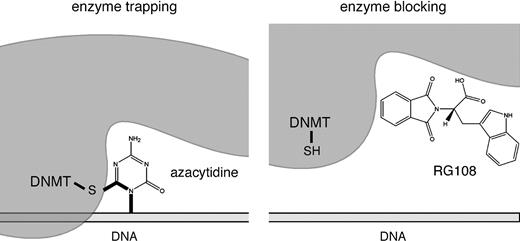
DNA methyltransferase (DNMT) inhibition by enzyme trapping or enzyme blocking. Left panel ) Aza-nucleotides can become incorporated into DNA during replication and then are recognized by DNMT enzymes. A stable reaction intermediate is formed via the sulfhydryl side chain of the catalytic cysteine residue. Thus, DNMT is trapped and concomitantly degraded. By this mechanism, cells are depleted of DNMT protein. Right panel ) Small molecules, such as RG108, can block the catalytic pocket of free DNMT proteins without the formation of covalent reaction intermediates.
Another DNA methyltransferase inhibitor is 5-aza-2′-deoxycytidine (i.e., decitabine), the deoxyribose analogue of 5-azacytidine. This compound does not need to be modified to a deoxy form and can be more directly incorporated into DNA ( Fig. 2 ). Therefore, decitabine may be more specific and less toxic than 5-azacytidine; indeed, the drug shows greater inhibition of DNA methylation and antitumor activity in experimental models ( 16 ) . Decitabine has single-agent activity in myeloid malignancies ( 17 , 18 ) , including myelodysplastic syndrome, acute myelogenous leukemia, and chronic myelogenous leukemia (for details, see below). However, decitabine also has substantial toxic effects, in particular myelosuppression with neutropenic fever ( 19 ) , that may be linked to the formation of covalent adducts between DNA and trapped DNA methyltransferase proteins ( 20 ) . These toxic effects highlight one of the central problems in interpreting much of the laboratory and clinical data for DNA methyltransferase inhibitors: It is often not clear whether effects on gene expression and cellular phenotype (or even on antitumor activity) associated with inhibitor treatment are due to cytotoxicity or to the demethylation of genomic DNA. Indeed, a recent phase II study of decitabine in patients with chronic myelogenous leukemia appeared to indicate that drug-induced hypomethylation of DNA in peripheral blood cells was less pronounced in responders than in nonresponders ( 19 ) .
The most recent addition to the group of DNA methyltransferase inhibitors is zebularine ( 21 ) , another derivative of 5-azacytidine ( Fig. 2 ). After several chemical modifications, zebularine is incorporated into DNA as a cytosine analogue. Zebularine is more stable than 5-azacytidine or decitabine and may also be less toxic. Orally ingested zebularine causes detectable demethylation and inhibits tumor growth in nude mice ( 22 ) . However, the oral bioavailability of zebularine in monkeys appears to be low ( 23 ) , and the drug has yet to be evaluated in clinical trials. Although zebularine appears to have some specificity toward cancer cells ( 24 ) , its mechanism of action is similar to that of the aza-nucleoside inhibitors. Thus, the demethylating activity of zebularine may also be difficult to separate from the toxic effects of DNA methyltransferase depletion that result from covalent enzyme trapping.
In fact, the inherent cytotoxicity of nucleoside DNA methyltransferase inhibitors poses a considerable limitation for their further development as therapeutic agents. However, their effectiveness in reversing epimutations warrants further investigations to optimize clinical treatment schedules, so that the risks associated with these drugs might be reduced and the benefits maximized.
Nonnucleoside DNA Methyltransferase Inhibitors
Some nonnucleoside compounds can also inhibit DNA methyltransferase activity. These substances directly block DNA methyltransferase activity and therefore do not appear to have the inherent toxicity caused by the covalent trapping of the enzyme ( Fig. 3 ). One nonnucleoside DNA methyltransferase inhibitor is (–)-epigallocatechin-3-gallate (EGCG), the main polyphenol compound in green tea. EGCG affects various biologic pathways ( 25 ) and inhibits DNA methyltransferase activity in protein extracts and in human cancer cell lines ( 26 ) . After examining the chemical structure of EGCG, Fang et al. ( 26 ) proposed that EGCG binds to and blocks the active site of human DNA methyltransferase 1 ( Fig. 2 ). However, degradation of EGCG generates a substantial amount of the strong oxidizing agent hydrogen peroxide ( 27 ) , and the oxidation of DNA methyltransferases and other proteins might contribute to the inhibition of DNA methylation by EGCG in vitro and to its cytotoxicity in human cell lines. Oxidation of DNA methyltransferases may also be involved in the mechanism of action of organoselenium compounds, such as benzyl selenocyanate ( 28 ) ; however, to our knowledge, no organoselenium compound has been shown to inhibit DNA methylation under in vivo conditions.
The discovery of most DNA methyltransferase inhibitors discussed above was fortuitous. The rational design of DNA methyltransferase inhibitors has been hampered by the lack of three-dimensional structures for the most relevant eukaryotic DNA methyltransferases, and it has been difficult to establish stringent DNA methyltransferase assays that are suitable for high-throughput screening. However, a three-dimensional homology model for the human DNA methyltransferase1 catalytic domain has been established ( 29 ) and used in an in silico screening assay to identify RG108, a small-molecule inhibitor of human DNA methyltransferases ( 30 ) . RG108 appears to block the active site of DNA methyltransferase ( Fig. 2 ), as indicated by the modeling data and its ability to inhibit the catalytic activity of purified recombinant DNA methyltransferase. The inhibitory mechanism of RG108 also appears to be direct and specific for DNA methyltransferases, in that RG108 has comparatively low toxicity in human cancer cell lines ( 30 ) . Thus, RG108 is an attractive candidate for further evaluation as a lead compound for new drug development.
The group of nonnucleoside DNA methyltransferase inhibitors also contains three additional classes of less well-characterized compounds. 1) 4-Aminobenzoic acid derivatives, such as the antiarrhythmic drug procainamide and the local anesthetic procaine, have shown demethylating activity in cellular assays and in mouse xenograft tumors ( 31 , 32 ) . Procaine appears to bind to CpG-rich sequences and thereby block the binding of DNA methyltransferases to DNA ( Fig. 2 ) ( 32 ) . However, procaine must be present in a high concentration (100–500 μ M ) to be an effective DNA methyltransferase inhibitor, and it has not been effective in all cell lines tested ( 33 ) . 2) The psammaplins inhibit DNA methyltransferase activity in cell-free assays, but their inhibitory mechanism has not been elucidated ( 34 ) . The psammaplins also inhibit histone deacetylase activity ( 34 ) and thus should be evaluated further as inhibitors of both histone deacetylases and DNA methyltransferases. 3) The third class of DNA methyltransferase inhibitors are oligonucleotides, including hairpin loops and specific antisense oligonucleotides, such as MG98. Hairpin loops have been used as competitor substrates for DNA methyltransferases in mouse erythroleukemia cells and have been able to induce the weak expression of the p16 tumor suppressor gene in human HT29 colon carcinoma cells ( 35 ) . In addition, transfection of human HCT116 and SW48 colon cancer cell lines with antisense oligonucleotides against DNA methyltransferase 1 resulted in the demethylation and reactivation of p16 ( 36 ) . Although the reproducibility of these results has been directly questioned by others ( 37 ) , the most advanced oligonucleotide (MG98) appears to have antitumor activity in preclinical models and is currently being tested in a phase II clinical trials (unpublished data; see http://www.methylgene.com ). Because only a limited response was observed in trials of single-agent MG98 therapy, current phase II trials in metastatic renal cell cancer are evaluating combination therapy of MG98 and interferon.
Pharmacodynamic Responses to DNA Methyltransferase Inhibitors
Alterations in the level or pattern of genomic DNA methylation may be an important endpoint for analyzing the effect of DNA methyltransferase inhibitor treatment. Changes in DNA methylation patterns can be analyzed with various methods, but most methods have substantial limitations, as outlined in Table 1 . Chromatographic and electrophoretic analysis of genomic DNA, for instance, allows for a straightforward determination of the methylation level of the whole genome but does not provide information about the distribution of methylated bases, i.e., the genomic methylation pattern. In contrast, bisulfite sequencing provides a detailed map of DNA methylation patterns, but the method is very time-consuming and limited to a few hundred base pairs of DNA per experiment. In addition, microarray analysis may allow a genome-wide analysis of DNA methylation patterns at a high resolution, but this technology is still challenging experimentally, and better microarray platforms need to be developed for such analyses.
Methods for the analysis of DNA methylation *
| Method | Parameter analyzed | Limitation |
|---|---|---|
| 5-Methylcytosine–specific immunohistochemistry | Presence/absence of methylated DNA | Not quantitative |
| HPLC | Whole-genome methylation level | Not locus specific, requires large amounts of DNA |
| Capillary electrophoresis | Whole-genome methylation level | Not locus specific |
| Methylation-sensitive Southern blot | Methylation status of selected restriction sites | Restricted to a few enzyme target sites, requires large amounts of DNA |
| Methylation-specific PCR | Methylation status of defined primer binding sites | Only semiquantitative, restricted to primer binding sites |
| COBRA | Methylation status of selected restriction sites | Restricted to a few enzyme target sites |
| Bisulfite sequencing | Methylation pattern of defined regions | Time-consuming, expensive |
| Bisulfite pyrosequencing | Methylation pattern of defined regions | Restricted to small regions, expensive |
| Methylation-sensitive microarray analysis | Methylation pattern of multiple regions | Not fully developed yet |
| Method | Parameter analyzed | Limitation |
|---|---|---|
| 5-Methylcytosine–specific immunohistochemistry | Presence/absence of methylated DNA | Not quantitative |
| HPLC | Whole-genome methylation level | Not locus specific, requires large amounts of DNA |
| Capillary electrophoresis | Whole-genome methylation level | Not locus specific |
| Methylation-sensitive Southern blot | Methylation status of selected restriction sites | Restricted to a few enzyme target sites, requires large amounts of DNA |
| Methylation-specific PCR | Methylation status of defined primer binding sites | Only semiquantitative, restricted to primer binding sites |
| COBRA | Methylation status of selected restriction sites | Restricted to a few enzyme target sites |
| Bisulfite sequencing | Methylation pattern of defined regions | Time-consuming, expensive |
| Bisulfite pyrosequencing | Methylation pattern of defined regions | Restricted to small regions, expensive |
| Methylation-sensitive microarray analysis | Methylation pattern of multiple regions | Not fully developed yet |
HPLC = high-performance liquid chromatography; PCR = polymerase chain reaction; COBRA = combined bisulfite restriction analysis.
Methods for the analysis of DNA methylation *
| Method | Parameter analyzed | Limitation |
|---|---|---|
| 5-Methylcytosine–specific immunohistochemistry | Presence/absence of methylated DNA | Not quantitative |
| HPLC | Whole-genome methylation level | Not locus specific, requires large amounts of DNA |
| Capillary electrophoresis | Whole-genome methylation level | Not locus specific |
| Methylation-sensitive Southern blot | Methylation status of selected restriction sites | Restricted to a few enzyme target sites, requires large amounts of DNA |
| Methylation-specific PCR | Methylation status of defined primer binding sites | Only semiquantitative, restricted to primer binding sites |
| COBRA | Methylation status of selected restriction sites | Restricted to a few enzyme target sites |
| Bisulfite sequencing | Methylation pattern of defined regions | Time-consuming, expensive |
| Bisulfite pyrosequencing | Methylation pattern of defined regions | Restricted to small regions, expensive |
| Methylation-sensitive microarray analysis | Methylation pattern of multiple regions | Not fully developed yet |
| Method | Parameter analyzed | Limitation |
|---|---|---|
| 5-Methylcytosine–specific immunohistochemistry | Presence/absence of methylated DNA | Not quantitative |
| HPLC | Whole-genome methylation level | Not locus specific, requires large amounts of DNA |
| Capillary electrophoresis | Whole-genome methylation level | Not locus specific |
| Methylation-sensitive Southern blot | Methylation status of selected restriction sites | Restricted to a few enzyme target sites, requires large amounts of DNA |
| Methylation-specific PCR | Methylation status of defined primer binding sites | Only semiquantitative, restricted to primer binding sites |
| COBRA | Methylation status of selected restriction sites | Restricted to a few enzyme target sites |
| Bisulfite sequencing | Methylation pattern of defined regions | Time-consuming, expensive |
| Bisulfite pyrosequencing | Methylation pattern of defined regions | Restricted to small regions, expensive |
| Methylation-sensitive microarray analysis | Methylation pattern of multiple regions | Not fully developed yet |
HPLC = high-performance liquid chromatography; PCR = polymerase chain reaction; COBRA = combined bisulfite restriction analysis.
The efficacy of a DNA methyltransferase inhibitor is generally determined by its ability to induce demethylation and reactivation of one or a few marker genes. Locus-specific DNA methylation patterns are usually determined by bisulfite modification procedures that deaminate unmethylated cytosine residues to uracil but do not affect methylated cytosines. The methylation-dependent polymorphism patterns are then determined after amplifying the bisulfite-treated DNA with a polymerase chain reaction, by use of discriminative primers, by restriction enzymes, or by DNA sequencing ( 38 – 40 ) . Although demethylation (and reactivation) of specific genes has been frequently observed after treatment with DNA methyltransferase inhibitors, these results need to be interpreted with caution because local changes in DNA methylation might also be a side effect of altered gene expression patterns in response to cytotoxic or other properties of DNA methyltransferase inhibitors.
To avoid misinterpretation of methylation data for a single gene, data from DNA methyltransferase inhibitor studies have been analyzed at the whole-genome level, by determining the level of genomic cytosine methylation—one of the most important and experimentally most straightforward parameters. Genomic cytosine methylation levels have traditionally been measured by high-performance liquid chromatography ( 41 ) . Although this method is fairly robust and quantitative, it requires comparatively large amounts of DNA. Consequently, novel analytical methods have been developed that use the capillary electrophoretic separation of nucleotides ( 42 , 43 ) ; these methods are both robust and sensitive and are now used to determine the pharmacodynamic parameters of DNA methyltransferase inhibitors.
To comprehensively characterize the molecular effects of reduced DNA methyltransferase activity, high-resolution methods need to be developed to analyze genome-wide DNA methylation patterns. To analyze DNA methylation, several laboratories are currently investigating the use of complex microarrays to detect bisulfite-induced, methylation-dependent polymorphisms in the DNA sequence or the use of array hybridization to detect resistance to methylation-sensitive restriction enzyme digestion ( 44 – 46 ) . In addition, such methylation-sensitive microarray assays may be able to classify tumors via the observed pattern of methylation ( 44 ) .
Methods that detect changes in methylation patterns can also be used to develop and refine epigenetic therapies for cancer. In clinical trials of demethylating agents, changes in patients' DNA methylation patterns should be monitored closely with appropriately validated and standardized methods to determine whether such changes can be used as endpoints in future clinical trials. If such methods can be established, they will allow the direct comparison of the biologic effectiveness of demethylating agents, as well as the optimization of schedules and the rational design of combination treatments with DNA methyltransferase inhibitors and other anticancer drugs. For example, in peripheral blood mononuclear cells from xenograft tumor–bearing mice treated with decitabine, decreased levels of genomic DNA methylation ( Fig. 4 ) are associated with the demethylation of the human MLH1 promoter in the xenograft tumors ( 47 ) . Thus, peripheral blood appears to be a surrogate tissue for determining pharmacodynamic characteristics of the effect of DNA methyltransferase inhibitors in tumors ( 48 ) .
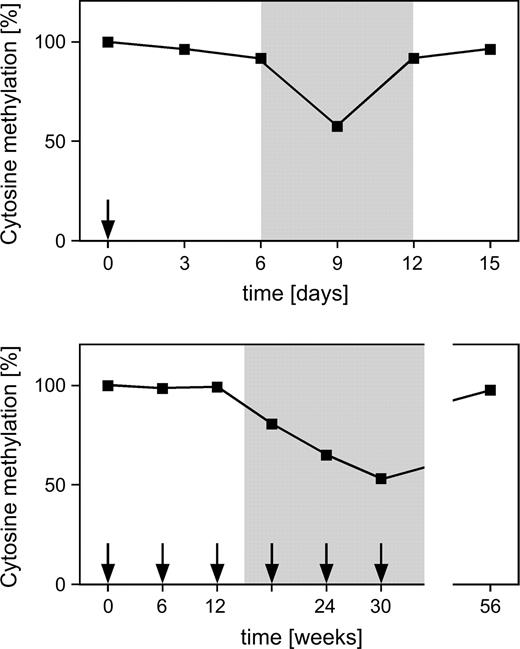
Pharmacodynamic response to demethylating drugs. Administration of 5-azadeoxycytidine ( arrows ) causes delayed and transient demethylation of genomic DNA. Upper panel ) Short-term demethylation dynamics after single treatment with decitabine ( 47 ) . Lower panel ) Long-term demethylation dynamics after successive pulses of decitabine ( 51 ) . The transient demethylation creates a window ( shaded background ) that can be used for epigenetic reprogramming and/or combinatorial therapies with cytotoxic drugs.
In other studies, the MAGE family of genes has been used to determine the biologic activity of DNA methyltransferase inhibitors. These genes are biallelically methylated in adult somatic tissues but demethylated in sperm ( 49 , 50 ) . Demethylation of the MAGE family genes in normal tissue can therefore be used to monitor drug-dependent methylation changes in response to treatment. In a phase I trial of decitabine in patients with refractory solid tumors ( 48 ) , the analysis of the methylation level of the whole genome and of the MAGE1 promoter in peripheral blood revealed substantial drug-induced decreases. Repeated sampling of tumor cells can occur in hematologic tumors; therefore, a more direct definition of molecular endpoints may be possible in such malignancies. Repeated sampling of bone marrow in decitabine-treated patients with myelodysplastic syndrome has revealed complex demethylation dynamics ( 51 ) ; demethylation of the p15 tumor suppressor gene appeared to coincide with the hematologic response in the same group of decitabine-treated patients ( 52 ) . However, this finding could not be confirmed in another study that also included patients with acute myelogenous leukemia ( 53 ) . Consequently, changes in the p15 methylation pattern may not be representative of the decitabine-induced changes in the global DNA methylation pattern, and the prognostic value of methylation changes at individual loci, such as p15 and p16 ( 54 ) , remains to be determined.
Ultimately, it may be the changes in gene transcription and growth suppression in the tumor that are monitored in patients as markers of a biologic response to treatment with DNA methyltransferase inhibitors. When reexpression of epigenetically silenced genes, such as fetal hemoglobin in blood cells, was evaluated in clinical trials of decitabine in patients with sickle-cell anemia, it was found to be associated with marked improvements in clinicopathological parameters related to red blood cell adhesion, endothelial damage, and coagulation pathway activity ( 55 ) . However, results of recent studies ( 50 ) suggest that demethylation of specific genes need not always result in their reexpression. For example, demethylation of the MAGE genes appears to lead to gene reexpression only when the appropriate tissue-specific transcription factors are also present ( 50 ) . Consequently, the unwanted gene reexpression induced by demethylating agents may be avoided in normal tissues if they lack such transcription factors, and there may be greater specificity of gene reexpression than has been predicted by examining methylation patterns. However, the broad alterations of gene expression patterns observed in cell lines treated with established DNA methyltransferase inhibitors do not indicate that such treatments have the ability to specifically reactivate epigenetically silenced genes. For example, decitabine induced both increased expression for some genes and decreased expression for other genes, as did the histone deacetylase inhibitor trichostatin A ( 56 ) . Thus, various factors, including DNA hypomethylation and cytotoxicity, may contribute to the complex effects observed after epigenetic drug treatment, and the characterization of these effects and development of compounds that specifically reverse epimutations will be required for future cancer therapies.
Cancers Targeted in Epigenetic Therapy
Abnormal DNA methylation patterns or epimutations have been documented for various cancers, and these epimutations may be used as biomarkers for tumor classification. Epimutations appear to accumulate over time at various sites in the genome and to promote tumorigenesis by increasing genomic instability or by silencing tumor suppressor genes. The silencing of tumor suppressor genes is closely associated with DNA hypermethylation and can be effectively reversed by DNA methyltransferase inhibitors. For these reasons, DNA methyltransferase inhibitors may be an attractive treatment option for most tumors. Although some mouse tumor models responded to a moderately reduced level of DNA methylation ( 57 ) , other models did not ( 58 ) . In addition, strong demethylation of the mouse genome that was induced by a mutant DNA methyltransferase allele appeared to increase genome instability and to cause concomitant tumorigenesis ( 59 ). These results indicate that the compounds and treatment schedules currently in use might not be sufficiently advanced to allow their broad therapeutic application.
The nucleoside inhibitors 5-azacytidine and decitabine have been tested in many phase I and II trials against many forms of cancer. The dose-limiting toxicity for both is myelosuppression, and the most commonly reported nonhematologic adverse effect was nausea and vomiting, which was grade 3–4 in approximately 10% of patients ( 60 ) . In lung cancer patients, toxicity was both dose and schedule dependent, with decitabine being less myelosuppressive when the same dose was administered over a shorter treatment period than over a longer period ( 60 ) . At cytotoxic doses, decitabine was active against leukemias and myelodysplastic syndromes ( 61 ) , but only limited activity with these schedules and doses was observed against solid tumors ( 62 ) . More recently, phase II and III clinical trials have been conducted for the treatment of myelodysplastic syndrome, a preleukemic bone marrow disorder that is usually diagnosed in older patients. For instance, a randomized trial of 5-azacytidine (75 mg/m 2 of body surface area subcutaneously for 7 days every 28 days) revealed substantially higher response rates (60% versus 5%) associated with 5-azacytidine treatment than with supportive care ( 63 ) . These results led to the recent FDA approval of 5-azacytidine (Vidaza, Pharmion) for the treatment of myelodysplastic syndrome and to the fast-track status of decitabine (Dacogen, MGI Pharma) for treatment of the same disorder. Because decitabine also showed promising response rates for other leukemias ( 19 , 53 ) , it might be effective for other tumor types. However, the activity of decitabine in solid tumors remains unclear, although prolonged disease stabilization has been reported in patients with lung cancer or prostate cancer ( 60 , 64 ) . Unfortunately, there is little evidence linking therapeutic efficacy to DNA demethylation, and drug-induced toxicity might play a major role in the patient responses observed.
Clinical Treatment Schedules
To allow effective clinical evaluation and a broader use of DNA methyltransferase inhibitors in clinical practice, doses and treatment schedules will need to be maximized for biologic effects and minimized for toxicity. The previously described pharmacodynamic markers could be used in phase I trials to determine the optimal dose for future clinical study. The initial phase I/II trials of 5-azacytidine or decitabine used doses near the maximum tolerated dose, rather than the optimal biologically effective dose ( 61 , 62 ) ; therapeutic responses were disappointing, and patients experienced many severe side effects that were probably related more to the inherent toxicity of aza-nucleoside inhibitors than to their demethylating activity. However, we now have a greater understanding of the mechanism of action of demethylating agents, and the broad use of decitabine in cell culture experiments indicates that demethylation of tumor suppressor genes can occur at drug concentrations that are substantially lower than those required for a cytotoxic effect ( 54 ) . In addition, it has also been shown that genes demethylated by DNA methyltransferase inhibitors can be remethylated within a few days ( 65 , 66 ) . As a consequence, treatment schedules have been modified to include multiple courses of treatment to sustain demethylation and reduced drug concentrations to decrease the severity of side effects ( 63 , 67 ) . Initial reports suggest that these regimens appear to have been well tolerated, with possibly increased response rates ( 53 ) . However, the clinical feasibility of these schedules has still to be fully evaluated, and their clinical significance for improving survival has not been established in large randomized clinical trials yet. Additional limitations of single-agent aza-nucleoside therapies include low drug stability ( 68 ) , poor oral bioavailability, rapid elimination by patients ( 69 ) , and, therefore, frequent hospital stays.
Present treatment schedules, including multiple courses of low-dose treatment, do not lead to the continuous demethylation of a patient's DNA ( 19 , 51 ) and so may not achieve stable epigenetic reprogramming in tumor cells. An alternative approach is therefore to use the period of demethylation (before remethylation can occur) as a window of epigenetic sensitization for combination therapies. More specifically, demethylation may sensitize tumors to existing cytostatic therapies or make cells permissive for epigenetic reprogramming with other epigenetic drugs, such as histone deacetylase inhibitors.
For example, resistance of human tumor xenografts to treatment with cisplatin, carboplatin, temozolomide, and epirubicin was decreased by adding nontoxic doses of decitabine. Importantly, the timing of drug administration appears to be associated with therapeutic response ( 47 ) ( Fig. 4 ). To be effective, decitabine had to be given 6–12 days before the cytotoxic drug; if decitabine was given at the same time or after the cytotoxic drug was administered, sensitization was lost. This observation provides strong support for the notion that decitabine sensitizes tumors by epigenetic reactivation of proapoptotic genes that potentiate the effects of cytotoxic drugs ( 47 , 70 ) . In this respect, it is important that previous combination treatment designs involved concurrent drug administration and did not incorporate the kinetics of demethylation and dependence of chemosensitization into the schedule ( 71 – 73 ) . In future clinical combination trials with aza-nucleosides, the administration of the second drug should, therefore, be delayed for several days to achieve the highest possible demethylation of tumor DNA.
Because DNA methylation and histone acetylation can act synergistically to silence tumor suppressor genes in cancer cell lines, DNA methyltransferase inhibitors have also been combined with histone deacetylase inhibitors to enhance reversal of epigenetic silencing ( 74 ) . The balance between the activity of histone deacetylases and the activity of histone acetyltransferases determines the acetylation level and acetylation pattern of histones. In addition, these enzymes regulate the expression of various genes by acetylating or deacetylating transcription factors such as GATA-1, TFIIE, and p53 ( 75 ) . Expression of histone acetyltransferases and histone deacetylases or the patterns of gene-specific histone acetylation have not been extensively studied in cancer cells, although certain mutations in histone acetyltransferases or aberrant recruitment of histone deacetylases have been associated with cancer-prone syndromes and tumors ( 76 ) . Histone deacetylase inhibitors, such as hydroxamic acid derivatives, inhibit proliferation of tumor cell lines in vitro and induce apoptosis, although with variable efficacy that is dependant on the cell type examined ( 77 , 78 ) . In addition, DNA methyltransferase and histone deacetylase inhibitors exert genome-wide effects and are not specific to particular genes. However, the combination of a histone deacetylase inhibitor and a DNA methyltransferase inhibitor becomes an attractive treatment option for epigenetic cancer therapies if the maximal epigenetic effects of DNA methyltransferase inhibitors are limited by toxic effects.
Directions for Further Developments
For a new epigenetic therapy to improve the treatment of cancer patients, it should have sufficient tumor cell versus normal cell selectivity so that a useful therapeutic index (i.e., the dose producing biological effects in tumor compared with the maximum tolerated dose) can be obtained. Examples of epigenetic therapies with a comparatively high therapeutic index include some of the histone deacetylase inhibitors currently undergoing clinical evaluation ( 77 ) . Also, low-dose aza-nucleoside treatments appear to have a sufficiently high therapeutic index to warrant further consideration and optimization. However, myelosuppression may limit the clinical use of aza-nucleotides at their maximum biologically effective dose. Nonnucleoside inhibitors may be less toxic because they are not incorporated into DNA. As described above, nonnucleoside inhibitors, such as EGCG, RG108, and procaine, should now be evaluated in preclinical and clinical studies, and additional nonnucleoside inhibitors should be developed.
As our understanding of the regulation of chromatin remodeling and epigenetic mechanisms for the regulation of transcription increases, new molecular targets will be identified and new inhibitors may be discovered. The histone methyltransferases are such new molecular targets. Another target may be the aberrant chromatin structure formed by tumor-specific modifications of the DNA and histones in tumors. This aberrant structure, rather than levels or activities of specific enzymes, may prove to be of great utility in differentiating between tumor and normal cells and thus may represent an exciting target for new therapeutic approaches.
As epigenetic therapies are being evaluated in clinical trials, new surrogate endpoints need to be defined and validated in early-phase clinical trials. In particular, the use of pharmacodynamic endpoints defined by the optimal biologic effect rather than by the maximum tolerated dose will help to identify the best doses and schedules for later trials. Although surrogate endpoints (such as demethylation) in surrogate tissues (such as blood cells) will be useful in guiding such studies and confirming that a drug is effective on its expected target, these endpoints provide only limited information about effects in the tumor. Because repeated sampling of the tumor will be restricted to only certain malignancies, such as leukemias, a major challenge will be identifying appropriate noninvasive means of monitoring the drug's effect on the tumor. Molecular imaging approaches and the assessment of tumor biomarkers released into body fluids may be able to determine drug effects in a clinically feasible manner and should be investigated further.
Once the recommended dose and treatment schedule for an epigenetic therapy has been established, the appropriate design for a randomized clinical trial within the context of existing therapies must be selected. Studies in experimental models have demonstrated that demethylating agents are not effective against all tumor types. For example, although colon tumor xenografts with methylated MLH1 can be sensitized to the chemotherapeutic cytotoxic effects of demethylating agents, colon xenografts with mutated MLH1 cannot be sensitized to these effects ( 47 ) . Consequently, appropriate biomarkers will need to be found to identify patient populations that may benefit from certain epigenetic therapies. Most studies that have investigated methylation of individual genes and patterns of methylation as a biomarker have been small and retrospective. Large prospective studies should be done to evaluate the potential of such biomarkers further.
DNA methylation patterns appear to be increasingly important in the management of cancer patients. These patterns are being examined as a means of early diagnosis of cancer ( 79 ), and the detection of methylated DNA in body fluids such as plasma of cancer patients may provide a noninvasive means of diagnosis or of monitoring response to treatment, as mentioned above. For example, acquired methylation of MLH1 at relapse in ovarian cancer patients has been associated with poor survival after conventional carboplatin–taxane chemotherapy ( 80 ) . The polymerase chain reaction amplification of bisulfite-modified DNA is a relatively simple and rapid method to characterize methylation patterns in tumors and requires small amounts of tumor tissue. Aberrant methylation patterns have been used to identify patients for epigenetic therapies ( 81 – 83 ) , and this approach needs to be examined further in randomized trials.
Editor's note: Dr. Brown is a member of the Topotargets Clinical Advisory Board and work in his laboratory is sponsored, in part, by Topotargets (Abingdon).
References
Goll MG, Bestor TH. Eukaryotic cytosine methyltransferases. Annu Rev Biochem
Hendrich B, Tweedie S. The methyl-CpG binding domain and the evolving role of DNA methylation in animals.
Li E. Chromatin modification and epigenetic reprogramming in mammalian development.
Di Croce L, Raker VA, Corsaro M, Fazi F, Fanelli M, Faretta M, et al. Methyltransferase recruitment and DNA hypermethylation of target promoters by an oncogenic transcription factor.
Ehrlich M. DNA methylation in cancer: too much, but also too little.
Jones PA, Baylin SB. The fundamental role of epigenetic events in cancer.
Herman JG, Umar A, Polyak K, Graff JR, Ahuja N, Issa JP, et al. Incidence and functional consequences of hMLH1 promoter hypermethylation in colorectal carcinoma.
Strathdee G. Epigenetic versus genetic alterations in the inactivation of E-cadherin.
Egger G, Liang G, Aparicio A, Jones PA. Epigenetics in human disease and prospects for epigenetic therapy.
Sorm F, Piskala A, Cihak A, Vesely J. 5-Azacytidine, a new, highly effective cancerostatic.
Jones PA, Taylor SM. Cellular differentiation, cytidine analogs and DNA methylation.
Santi DV, Norment A, Garrett CE. Covalent bond formation between a DNA-cytosine methyltransferase and DNA containing 5-azacytosine.
Momparler RL, Momparler LF, Samson J. Comparison of the antileukemic activity of 5-AZA-2′-deoxycytidine, 1-beta-D-arabinofuranosylcytosine and 5-azacytidine against L1210 leukemia.
Santini V, Kantarjian HM, Issa JP. Changes in DNA methylation in neoplasia: pathophysiology and therapeutic implications.
Leone G, Voso MT, Teofili L, Lubbert M. Inhibitors of DNA methylation in the treatment of hematological malignancies and MDS.
Issa JP, Gharibyan V, Cortes J, Jelinek J, Morris G, Verstovsek S, et al. Phase II study of low-dose decitabine in patients with chronic myelogenous leukemia resistant to imatinib mesylate.
Juttermann R, Li E, Jaenisch R. Toxicity of 5-aza-2′-deoxycytidine to mammalian cells is mediated primarily by covalent trapping of DNA methyltransferase rather than DNA demethylation.
Zhou L, Cheng X, Connolly BA, Dickman MJ, Hurd PJ, Hornby DP. Zebularine: a novel DNA methylation inhibitor that forms a covalent complex with DNA methyltransferases.
Cheng JC, Matsen CB, Gonzales FA, Ye W, Greer S, Marquez VE, et al. Inhibition of DNA methylation and reactivation of silenced genes by zebularine.
Holleran JL, Parise RA, Joseph E, Eiseman JL, Covey JM, Glaze ER, et al. Plasma pharmacokinetics, oral bioavailability, and interspecies scaling of the DNA methyltransferase inhibitor, zebularine.
Cheng JC, Yoo CB, Weisenberger DJ, Chuang J, Wozniak C, Liang G, et al. Preferential response of cancer cells to zebularine.
Moyers SB, Kumar NB. Green tea polyphenols and cancer chemoprevention: multiple mechanisms and endpoints for phase II trials.
Fang MZ, Wang Y, Ai N, Hou Z, Sun Y, Lu H, et al. Tea polyphenol (-)-epigallocatechin-3-gallate inhibits DNA methyltransferase and reactivates methylation-silenced genes in cancer cell lines.
Nakagawa H, Hasumi K, Woo JT, Nagai K, Wachi M. Generation of hydrogen peroxide primarily contributes to the induction of Fe(II)-dependent apoptosis in Jurkat cells by (-)-epigallocatechin gallate.
Fiala ES, Staretz ME, Pandya GA, El-Bayoumy K, Hamilton SR. Inhibition of DNA cytosine methyltransferase by chemopreventive selenium compounds, determined by an improved assay for DNA cytosine methyltransferase and DNA cytosine methylation.
Siedlecki P, Boy RG, Comagic S, Schirrmacher R, Wiessler M, Zielenkiewicz P, et al. Establishment and functional validation of a structural homology model for human DNA methyltransferase 1.
Brueckner B, Garcia Boy R, Siedlecki P, Musch T, Kliem HC, Zielenkiewicz P, et al. Epigenetic reactivation of tumor suppressor genes by a novel small-molecule inhibitor of human DNA methyltransferases.
Lin X, Asgari K, Putzi MJ, Gage WR, Yu X, Cornblatt BS, et al. Reversal of GSTP1 CpG island hypermethylation and reactivation of pi-class glutathione S-transferase (GSTP1) expression in human prostate cancer cells by treatment with procainamide.
Villar-Garea A, Fraga MF, Espada J, Esteller M. Procaine is a DNA-demethylating agent with growth-inhibitory effects in human cancer cells.
Nieto M, Samper E, Fraga MF, Gonzalez de Buitrago G, Esteller M, Serrano M. The absence of p53 is critical for the induction of apoptosis by 5-aza-2′-deoxycytidine.
Pina IC, Gautschi JT, Wang GY, Sanders ML, Schmitz FJ, France D, et al. Psammaplins from the sponge Pseudoceratina purpurea: inhibition of both histone deacetylase and DNA methyltransferase.
Flynn J, Fang JY, Mikovits JA, Reich NO. A potent cell-active allosteric inhibitor of murine DNA cytosine C5 methyltransferase.
Robert MF, Morin S, Beaulieu N, Gauthier F, Chute IC, Barsalou A, et al. DNMT1 is required to maintain CpG methylation and aberrant gene silencing in human cancer cells.
Ting AH, Jair KW, Suzuki H, Yen RW, Baylin SB, Schuebel KE. CpG island hypermethylation is maintained in human colorectal cancer cells after RNAi-mediated depletion of DNMT1.
Frommer M, McDonald LE, Millar DS, Collis CM, Watt F, Grigg GW, et al. A genomic sequencing protocol that yields a positive display of 5-methylcytosine residues in individual DNA strands.
Herman JG, Graff JR, Myohanen S, Nelkin BD, Baylin SB. Methylation-specific PCR: a novel PCR assay for methylation status of CpG islands.
Xiong Z, Laird PW. COBRA: a sensitive and quantitative DNA methylation assay.
Ramsahoye BH. Measurement of genome wide DNA methylation by reversed-phase high-performance liquid chromatography.
Fraga MF, Uriol E, Diego LB, Berdasco M, Esteller M, Canal MJ, et al. High-performance capillary electrophoretic method for the quantification of 5-methyl 2′-deoxycytidine in genomic DNA: application to plant, animal and human cancer tissues.
Stach D, Schmitz OJ, Stilgenbauer S, Benner A, Dohner H, Wiessler M, et al. Capillary electrophoretic analysis of genomic DNA methylation levels.
Adorjan P, Distler J, Lipscher E, Model F, Muller J, Pelet C, et al. Tumour class prediction and discovery by microarray-based DNA methylation analysis.
Shi H, Maier S, Nimmrich I, Yan PS, Caldwell CW, Olek A, et al. Oligonucleotide-based microarray for DNA methylation analysis: principles and applications.
Mund C, Beier V, Bewerunge P, Dahms M, Lyko F, Hoheisel JD. Array-based analysis of genomic DNA methylation patterns of the tumour suppressor gene p16INK4A promoter in colon carcinoma cell lines.
Plumb JA, Strathdee G, Sludden J, Kaye SB, Brown R. Reversal of drug resistance in human tumor xenografts by 2′-deoxy-5-azacytidine-induced demethylation of the hMLH1 gene promoter.
Samlowski WE, Leachman SA, Wade M, Cassidy P, Porter-Gill P, Busby L, et al. Evaluation of a 7-day continuous intravenous infusion of decitabine: inhibition of promoter-specific and global genomic dna methylation.
De Smet C, De Backer O, Faraoni I, Lurquin C, Brasseur F, Boon T. The activation of human gene MAGE-1 in tumor cells is correlated with genome-wide demethylation.
De Smet C, Loriot A, Boon T. Promoter-dependent mechanism leading to selective hypomethylation within the 5′ region of gene MAGE-A1 in tumor cells.
Mund C, Hackanson B, Stresemann C, Lubbert M, Lyko F. Characterization of DNA demethylation effects induced by 5-aza-2′-deoxycytidine in patients with myelodysplastic syndrome.
Daskalakis M, Nguyen TT, Nguyen C, Guldberg P, Kohler G, Wijermans P, et al. Demethylation of a hypermethylated P15/INK4B gene in patients with myelodysplastic syndrome by 5-Aza-2′-deoxycytidine (decitabine) treatment.
Issa JP, Garcia-Manero G, Giles FJ, Mannari R, Thomas D, Faderl S, et al. Phase 1 study of low-dose prolonged exposure schedules of the hypomethylating agent 5-aza-2′-deoxycytidine (decitabine) in hematopoietic malignancies.
Bender CM, Pao MM, Jones PA. Inhibition of DNA methylation by 5-aza-2′-deoxycytidine suppresses the growth of human tumor cell lines.
DeSimone J, Koshy M, Dorn L, Lavelle D, Bressler L, Molokie R, et al. Maintenance of elevated fetal hemoglobin levels by decitabine during dose interval treatment of sickle cell anemia.
Gius D, Cui H, Bradbury CM, Cook J, Smart DK, Zhao S, et al. Distinct effects on gene expression of chemical and genetic manipulation of the cancer epigenome revealed by a multimodality approach.
Laird PW, Jackson-Grusby L, Fazeli A, Dickinson SL, Jung WE, Li E, et al. Suppression of intestinal neoplasia by DNA hypomethylation.
Trinh BN, Long TI, Nickel AE, Shibata D, Laird PW. DNA methyltransferase deficiency modifies cancer susceptibility in mice lacking DNA mismatch repair.
Gaudet F, Hodgson JG, Eden A, Jackson-Grusby L, Dausman J, Gray JW, et al. Induction of tumors in mice by genomic hypomethylation.
Momparler RL, Bouffard DY, Momparler LF, Dionne J, Belanger K, Ayoub J. Pilot phase I-II study on 5-aza-2′-deoxycytidine (Decitabine) in patients with metastatic lung cancer.
Lubbert M. DNA methylation inhibitors in the treatment of leukemias, myelodysplastic syndromes and hemoglobinopathies: clinical results and possible mechanisms of action.
Aparicio A, Weber JS. Review of the clinical experience with 5-azacytidine and 5-aza-2′- deoxycytidine in solid tumors.
Silverman LR, Demakos EP, Peterson BL, Kornblith AB, Holland JC, Odchimar-Reissig R, et al. Randomized controlled trial of azacitidine in patients with the myelodysplastic syndrome: a study of the cancer and leukemia group B.
Thibault A, Figg WD, Bergan RC, Lush RM, Myers CE, Tompkins A, et al. A phase II study of 5-aza-2'deoxycytidine (decitabine) in hormone independent metastatic (D2) prostate cancer.
Bender CM, Gonzalgo ML, Gonzales FA, Nguyen CT, Robertson KD, Jones PA. Roles of cell division and gene transcription in the methylation of CpG islands.
Velicescu M, Weisenberger DJ, Gonzales FA, Tsai YC, Nguyen CT, Jones PA. Cell division is required for de novo methylation of CpG islands in bladder cancer cells.
Wijermans P, Lübbert M, Verhoef G, Bosly A, Ravoet C, Andre M, et al. Low-dose 5-aza-2′-deoxycytidine, a DNA hypomethylating agent, for the treatment of high-risk myelodysplastic syndrome: a multicenter phase II study in elderly patients.
Lin KT, Momparler RL, Rivard GE. High-performance liquid chromatographic analysis of chemical stability of 5-aza-2′-deoxycytidine.
van Groeningen CJ, Leyva A, O'Brien AM, Gall HE, Pinedo HM. Phase I and pharmacokinetic study of 5-aza-2′-deoxycytidine (NSC 127716) in cancer patients.
Soengas MS, Capodieci P, Polsky D, Mora J, Esteller M, Opitz-Araya X, et al. Inactivation of the apoptosis effector Apaf-1 in malignant melanoma.
Frost P, Abbruzzese JL, Hunt B, Lee D, Ellis M. Synergistic cytotoxicity using 2′-deoxy-5-azacytidine and cisplatin or 4-hydroperoxycyclophosphamide with human tumor cells.
Lenzi R, Raber MN, Gravel D, Frost P, Abbruzzese JL. Phase I and II trials of a laboratory-derived synergistic combination of cisplatin and 2′-deoxy-5-azacytidine.
Schwartsmann G, Schunemann H, Gorini CN, Filho AF, Garbino C, Sabini G, et al. A phase I trial of cisplatin plus decitabine, a new DNA-hypomethylating agent, in patients with advanced solid tumors and a follow-up early phase II evaluation in patients with inoperable non-small cell lung cancer.
Cameron EE, Bachman KE, Myohanen S, Herman JG, Baylin SB. Synergy of demethylation and histone deacetylase inhibition in the re- expression of genes silenced in cancer.
Bode AM, Dong Z. Post-translational modification of p53 in tumorigenesis.
Plumb JA, Finn PW, Williams RJ, Bandara MJ, Romero MR, Watkins CJ, et al. Pharmacodynamic response and inhibition of growth of human tumor xenografts by the novel histone deacetylase inhibitor PXD101.
Marks PA, Jiang X. Histone deacetylase inhibitors in programmed cell death and cancer therapy.
Esteller M. Relevance of DNA methylation in the management of cancer.
Gifford G, Paul J, Vasey PA, Kaye SB, Brown R. The acquisition of hMLH1 methylation in plasma DNA after chemotherapy predicts poor survival for ovarian cancer patients.
Wei SH, Chen CM, Strathdee G, Harnsomburana J, Shyu CR, Rahmatpanah F, et al. Methylation microarray analysis of late-stage ovarian carcinomas distinguishes progression-free survival in patients and identifies candidate epigenetic markers.
Lyko F, Stach D, Brenner A, Stilgenbauer S, Dohner H, Wirtz M, et al. Quantitative analysis of DNA methylation in chronic lymphocytic leukemia patients.


{kind=link}
{kind=link}
{kind=link}
{kind=link}