




Abstract
Colorectal tumors caused by failure of the DNA mismatch repair system commonly show microsatellite instability. Our goals were to compare the performance of two panels of markers (a panel previously recommended by the National Cancer Institute [NCI] and a pentaplex of mononucleotide repeats) and to devise the simplest diagnostic strategy for identification of patients with colorectal cancer characterized by defects in mismatch repair.
We recruited 1058 patients who were newly diagnosed with colorectal cancer. DNA from fresh-frozen and paraffin-embedded tumors was tested for microsatellite instability, using the NCI-recommended panel of microsatellite markers and the pentaplex panel of mononucleotide repeats, respectively, as templates for polymerase chain reactions (PCRs). Microsatellite instability in fresh-frozen tumors was also assessed using the pentaplex panel of mononucleotides in a crossover analysis. The expression of mismatch repair proteins (MLH1, MSH2, MSH6, and PMS2) in the tumors was determined immunohistochemically. The sensitivity and specificity with which the marker panels identified tumors with deficiencies in the expression of mismatch repair proteins were calculated. All statistical tests were two-sided.
The sensitivity and positive predictive value of the NCI panel were 76.5% (95% confidence interval [CI] = 61% to 92%) and 65.0% (95% CI = 49% to 81%), respectively; corresponding values for the mononucleotide pentaplex panel were 95.8% (95% CI = 89% to 103%) and 88.5% (95% CI = 79% to 98%), respectively. A panel consisting of the mononucleotide repeat markers BAT26 and NR24 alone had the same predictive value as the pentaplex panel of mononucleotide repeats.
The pentaplex panel of mononucleotide repeats performs better than the NCI panel for the detection of mismatch repair–deficient tumors. Simultaneous assessment of the instability of BAT26 and NR24 is as effective as use of the pentaplex panel for diagnosing mismatch repair deficiency.
There is a need to identify colorectal cancers that are due to defects in DNA mismatch repair because the presence of these defects helps to define two important types of colorectal cancers. One of these is an inherited condition called Lynch syndrome. However, there are also sporadic colorectal cancers with similar defects, and patients with this form of colorectal cancer have a unique prognosis and respond differently to particular drug therapies.
Tumor samples from a multicenter cohort of colorectal cancer patients were analyzed for concordance between microsatellite instability, as detected by different tests, and mismatch repair protein expression.
The authors have improved the accuracy of a testing method that identifies those patients whose colorectal cancer is caused by defects in mismatch repair. The test is based on sequencing stretches of the patient's DNA called microsatellite sequences to identify characteristic changes (mutations).
The improved test could result in more patients being assigned to proper treatment based on their disease profile.
The ultimate effect of a test for the presence of defects in mismatch repair in sporadic colorectal cancers on patient outcomes remains to be determined.
Colorectal cancer is the second-leading cause of cancer-related death in the Western World ( 1 ). The majority of colorectal cancers develop through the suppressor pathway and are characterized by chromosomal instability and frequent cytogenetic alterations and allelic losses ( 2 ). In the less common mutator pathway ( 3 – 5 ) of colorectal cancer development, tumors display DNA insertions and deletions at microsatellite sequences, and they are said to have microsatellite instability. Microsatellites consist of stretches of repeating units of 1–5 base pairs (bp) that are distributed throughout the genome. In mononucleotide repeats, a single base is repeated, and in dinucleotide repeats, the repeating unit is two bases. The microsatellite instability phenotype results from a failure of the DNA mismatch repair system, an enzymatic machinery that recognizes and repairs DNA damaged by erroneous insertions and deletions and misincorporation of bases.
Germline mutations of mismatch repair genes, most often of MLH1 or MSH2 and much less commonly of MSH6 and PMS2, are the underlying cause of tumors with a microsatellite-unstable phenotype from families with hereditary nonpolyposis colorectal cancer (HNPCC, also known as Lynch syndrome) ( 6 ), the most common form of hereditary colorectal cancer. These mutations, however, are absent in the approximately half of HNPCC families whose tumors do not show microsatellite instability ( 7 ). Between 8% and 15% of sporadic colorectal cancers also exhibit microsatellite instability, but in these cancers, instability is due mostly to MLH1 silencing through promoter methylation ( 8 ). Patients with sporadic colorectal cancers with microsatellite instability appear to have a better prognosis than patients with stable tumors ( 9 ), but they do not respond to 5-fluorouracil ( 10 ).
The utility of microsatellite instability analysis as the first step in identifying patients who should be tested for mismatch repair gene mutations was affirmed in an International Workshop on HNPCC sponsored by the National Cancer Institute (NCI). This conference developed the Bethesda guidelines, a set of clinical criteria that would prompt the performance of microsatellite instability testing ( 11 ). The conference recommended the use of a panel of five microsatellite sequences to assess instability. This panel consisted of two mononucleotide (BAT25 and BAT26) and three dinucleotide (D2S123, D5S346, and D17S250) repeat sequences. It was stipulated that testing should compare DNA from tumor and normal tissue. If two or more of the five microsatellite sequences were mutated, the tumor would be classified as high microsatellite instability (MSI-H); if only one was mutated, it would be low microsatellite instability (MSI-L); and if none were mutated, it would be microsatellite stable (MSS) ( 12 ).
In a follow-up workshop, the Bethesda guidelines were revised to take into account some common pathologic features and to include patients who could be older but still susceptible to have mismatch repair gene mutations ( 13 ). We subsequently validated the usefulness of the revised guidelines as the initial screening tool for the identification of mismatch repair gene mutations ( 14 ). In the same workshop, it was agreed to continue using the NCI set of markers despite controversy concerning the diagnostic utility of the MSI-L phenotype and the validity of a categorization of a tumor as one showing microsatellite instability based exclusively on the presence of mutations in dinucleotide repeats. However, it was also suggested that a pentaplex of five quasimonomorphic (i.e., the same number of repeats is found in an overwhelming proportion of humans) mononucleotide repeats could be more sensitive for detection of MSI-H tumors and that its use would obviate the need to perform separate tests on DNA from both the tumor and normal tissue of a patient ( 15 ).
The main objectives of this study were to ascertain the performance of the NCI panel for the detection of colorectal cancer with failure of the mismatch repair system, to compare its performance with the pentaplex panel of mononucleotide repeats, and to devise the simplest strategy, based on microsatellite instability analysis, for the detection of mismatch repair–deficient tumors. We also addressed the controversial issues regarding the use of the NCI panel, such as the inclusion of dinucleotide repeats.
Methods
Study Population and Data Collection
The 1058 patients included in this study were part of a larger series of 1978 patients with newly diagnosed colorectal cancer who were recruited prospectively between November 1, 2000, and October 31, 2001, from 20 hospitals in Spain. Nonconsenting patients; patients with unavailable tumor DNA; and patients with familial adenomatous polyposis, a personal history of inflammatory bowel disease, or an incomplete family history were excluded. Participating centers collected demographic, clinical, and pathology data from probands as well as their detailed family history of cancer. Depending on the institution, the centers collected either freshly frozen normal and tumor tissue (from biopsies during colonoscopy or from surgical specimens) or paraffin-embedded tumor tissue from tumors processed by the corresponding pathology departments. Each type of analysis (microsatellite instability, immunohistochemistry, and mutational testing) was performed in a single institution. The Institutional Ethics Committee of each participating hospital approved the study, and written informed consent was obtained from all patients.
Microsatellite Instability Analysis
For microsatellite instability analysis, the NCI panel, composed of BAT26, BAT25, D5S346, D2S123, and D17S250 markers ( 12 ), was tested in all tumors (N = 531) from hospitals that collected fresh-frozen samples of tumor and noninvolved mucosa. The sequences of the primers used for amplification have been published elsewhere ( 16 ). The pentaplex panel of mononucleotide repeats was used to test all tumors (N = 527) from hospitals that collected paraffin-embedded tumor tissue. This panel, suggested by Suraweera et al. ( 15 ), is composed of five mononucleotide markers: BAT26, BAT25, NR21, NR22, and NR24. The primers used for amplification of microsatellite sequences in this panel were those used previously ( 15 ). All forward primers (Applied Biosystems, Foster City, CA) had a fluorescent tag (6FAM, HEX, NED) at the 5′ end to allow microsatellite detection by the ABI Prism 3100 Avant Genetic Analyzer (Applied Biosystems).
Polymerase chain reaction (PCR) conditions for all markers were the following: 10 mM Tris–HCl, pH 8.3; 50 mM KCl; 2 mM MgCl 2 ; 0.03 U/μL Taq polymerase; 0.5 μM of each primer; 0.2 mM dNTPs; and 2 μL of DNA in a final volume of 11 μL. After initial denaturation at 94 °C for 2 minutes, PCR steps were as follows: 94 °C for 30 seconds, T °C of annealing for 30 seconds, and 72 °C for 30 seconds, for 36 cycles. Final extension was at 72 °C for 10 minutes. Annealing temperatures were as follows: 55 °C for D5S346 and mononucleotide repeat markers, 50 °C for D2S123, and 48 °C for D17S250. PCRs were performed on a 2700 GeneAmp PCR system (Perkin Elmer). After successful amplifications, PCR products (≤2 μL) were mixed with 18 μL of Hi-Di formamide (Applied Biosystems) and 0.3 μL of size standards (ROX; Applied Biosystems). Samples were denatured by incubation at 95 °C for 3 minutes and placed on ice until analysis on an ABI Prism 3100 Avant Genetic Analyzer using GeneScan Analysis software (Applied Biosystems).
When the analysis was performed using the NCI panel of microsatellites, the presence of peaks in the fluorescence profile of the amplified microsatellite DNA that were absent in a corresponding profile derived from normal mucosa of the same patient was interpreted as microsatellite instability. For analysis of instability using the pentaplex panel of mononucleotide markers, microsatellites were considered unstable if PCR fragments showed deletions of at least 3 bp at a given locus.
To further define the distinct performance of the panels, we also analyzed both sets of markers in the same tumors. We included in this analysis all fresh-frozen and paraffin-embedded tumors that were MSI-L, MSI-H, and all MSS tumors that had an altered mismatch repair protein expression as determined by immunohistochemistry (see below). Finally, we also included 100 randomly chosen fresh-frozen tumors (that had been analyzed with the NCI panel of markers initially) that were MSS and had normal mismatch repair protein expression.
MLH1, MSH2, MSH6, and PMS2 Protein Expression
Immunohistochemical analysis of MLH1, MSH2, and MSH6 expression was performed in all tumors. PMS2 expression was evaluated in a majority (719/1058) of tumors. Sections (4 μm) were de-waxed and rehydrated using xylene and alcohol. Before immunostaining, antigen retrieval was performed by immersing sections in citrate buffer (10 mmol/L, pH 6.0) and boiling in a pressure cooker for 5 minutes. Sections were then incubated for 20 minutes at room temperature with mouse monoclonal antibodies against MLH1 (clone G168-15, dilution 1 : 30; BD PharMingen, San Diego, CA), MSH2 (clone FE11, dilution 1 : 30; Oncogene Research Products, Boston, MA), MSH6 (clone 44 MSH6-GTBP, dilution 1 : 100; BD Biosciences, Erembodegem, Belgium), or PMS2 (clone A16-4, dilution 1 : 100; BD PharMingen). The Ultra-Vision streptavidin–biotin peroxidase detection kit (Dako, Carpinteria, CA) was used as the secondary detection system according to manufacturer's instructions. Loss of MLH1, MSH2, MSH6, or PMS2 expression was recorded when there was a complete absence of nuclear staining in neoplastic cells. Nuclear staining in normal epithelial cells, lymphocytes, and stromal cells in each slide served as an internal positive control. When there was no nuclear staining of internal control in three separate experiments, the results were considered inconclusive.
MSH2/MLH1 Germline Mutation Analysis
All patients found to have tumors with microsatellite instability and/or lack of MSH2 or MLH1 protein expression underwent germline genetic testing for MSH2 and MLH1 by both sequencing and multiple ligation probe amplification analysis. This analysis was performed as part of a different project using this series of patients ( 14 ), but these data were also used in this study to further support our findings in some controversial cases. Multiple ligation probe amplification was performed using the SALSA MLPA kit P003 for MLH1/MSH2 exon deletion assay (MRC-Holland, Amsterdam, The Netherlands), which allows the detection of rearrangements in MLH1 and MSH2. Ligation products were amplified by PCR using a fluorescently labeled primer and analyzed in an ABI 3100 sequencer using GeneScan and Genotyper Analysis software (Applied Biosystems). The peak height of each fragment was compared with the corresponding measurements of a control sample, and deletions were suspected when peak height was 60% or less than that of healthy control subjects Multiple ligation probe amplification results were confirmed by reverse transcription–PCR encompassing contiguous exons of the suspected deletion.
Statistical Analysis
Associations between clinicopathologic features and degree of microsatellite instability were analyzed using either the chi-square test or the Fisher's exact test. Sex, tumor location, and mucin production variables were analyzed using the Fisher's exact test. Family history, Duke's stage, and differentiation were analyzed using the chi-square test. Differences in continuous variables were analyzed by the Student's t test. All reported P values correspond to two-sided tests. Differences were considered statistically significant if the P value was less than .05. All calculations were performed using the 12.0 SPSS software package (SPSS Inc, Chicago, IL).
Results
Colorectal Cancer Classification Based on Use of the National Cancer Institute and Pentaplex of Mononucleotide Repeat Panels to Detect Microsatellite Instability
Of the 1058 tumors analyzed in this study, 531 were tested with the NCI panel and 527 were tested with the pentaplex panel of mononucleotide repeats. There were no statistically significant differences between the two groups of tumors regarding clinical features, pathology, or family history of cancer in the patients from which they were derived ( Table 1 ). Approximately 90% of the tumors tested with either panel were MSS. The remaining were all MSI-H in the pentaplex panel of mononucleotide repeat–tested group, whereas in the NCI panel–tested group, 7.5% of the tumors tested were MSI-H and 5.5% were MSI-L. Among tumors assigned to the MSI-H group by testing with the NCI panel, 9.5% fulfilled the Amsterdam criteria of HNPCC. Among tumors assigned to the MSI-H group by testing with the pentaplex panel of mononucleotides, 12.0% fulfilled these criteria.
Clinical, pathologic, and family history features *
| Patient features | NCI MSS | PMRP MSS | P | NCI MSI-H | PMRP MSI-H | P |
|---|---|---|---|---|---|---|
| No. of patients | 462 | 475 | 40 | 52 | ||
| Mean age † | 70.5 (69.5 to 71.5) | 69.6 (68.6 to 70.6) | .218 ‡ | 69.5 (64.9 to 74.1) | 67.8 (63.9 to 71.6) | .561 ‡ |
| Sex | .306 § | .828 § | ||||
| Male | 61.1% | 64.5% | 33.3% | 37.3% | ||
| Female | 38.9% | 35.5% | 66.7% | 62.7% | ||
| Location | .817 § | .660 § | ||||
| Right | 23.8% | 24.5% | 63.4% | 68.6% | ||
| Left | 76.2% | 75.5% | 36.6% | 31.4% | ||
| Differentiation | .002 ‡ | .371 ‡ | ||||
| Well | 19.2% | 28.3% | 11.4% | 22.9% | ||
| Moderate | 74.6% | 63.5% | 68.8% | 56.3% | ||
| Poor | 6.2% | 28.3% | 20.0% | 20.8% | ||
| Duke's stage | .481 ‡ | .202 ‡ | ||||
| A1 + B1 | 14.2% | 14.8% | 10.8% | 2.0% | ||
| B2 + B3 + C1 + C2 | 65.1% | 61.4% | 73.0% | 82.4% | ||
| C3 + D | 20.7% | 23.7% | 16.2% | 15.7% | ||
| Mucin production ‖ | 10.2% | 10.0% | .913 § | 24.3% | 24.0% | 1.000 § |
| Family history | ||||||
| HNPCC | 0.9% | 1.3% | 9.5% | 12.0% | ||
| CRC | 16.4% | 11.9% | 7.1% | 10.0% | ||
| No cancer ¶ | 52.5% | 48.6% | .067 ‡ | 59.5% | 42.0% | .090 ‡ |
| Patient features | NCI MSS | PMRP MSS | P | NCI MSI-H | PMRP MSI-H | P |
|---|---|---|---|---|---|---|
| No. of patients | 462 | 475 | 40 | 52 | ||
| Mean age † | 70.5 (69.5 to 71.5) | 69.6 (68.6 to 70.6) | .218 ‡ | 69.5 (64.9 to 74.1) | 67.8 (63.9 to 71.6) | .561 ‡ |
| Sex | .306 § | .828 § | ||||
| Male | 61.1% | 64.5% | 33.3% | 37.3% | ||
| Female | 38.9% | 35.5% | 66.7% | 62.7% | ||
| Location | .817 § | .660 § | ||||
| Right | 23.8% | 24.5% | 63.4% | 68.6% | ||
| Left | 76.2% | 75.5% | 36.6% | 31.4% | ||
| Differentiation | .002 ‡ | .371 ‡ | ||||
| Well | 19.2% | 28.3% | 11.4% | 22.9% | ||
| Moderate | 74.6% | 63.5% | 68.8% | 56.3% | ||
| Poor | 6.2% | 28.3% | 20.0% | 20.8% | ||
| Duke's stage | .481 ‡ | .202 ‡ | ||||
| A1 + B1 | 14.2% | 14.8% | 10.8% | 2.0% | ||
| B2 + B3 + C1 + C2 | 65.1% | 61.4% | 73.0% | 82.4% | ||
| C3 + D | 20.7% | 23.7% | 16.2% | 15.7% | ||
| Mucin production ‖ | 10.2% | 10.0% | .913 § | 24.3% | 24.0% | 1.000 § |
| Family history | ||||||
| HNPCC | 0.9% | 1.3% | 9.5% | 12.0% | ||
| CRC | 16.4% | 11.9% | 7.1% | 10.0% | ||
| No cancer ¶ | 52.5% | 48.6% | .067 ‡ | 59.5% | 42.0% | .090 ‡ |
NCI MSS = stable tumors tested with the National Cancer Institute (NCI) panel; PMRP MSS = stable tumors tested with the pentaplex mononucleotide repeat panel; NCI MSI-H = unstable tumors with high microsatellite instability tested with the NCI panel; PMRP MSI-H = unstable tumors with high microsatellite instability tested with the pentaplex mononucleotide repeat panel; HNPCC = hereditary nonpolyposis colorectal cancer; CRC = colorectal cancer.
Age of patient's diagnosis was expressed as mean with the 95% confidence interval.
P values correspond to two-sided chi-square test.
P values correspond to two-sided Fisher's exact test.
Presence of more than 50% of extracellular mucin in a tumor.
No family history of any cancer.
Clinical, pathologic, and family history features *
| Patient features | NCI MSS | PMRP MSS | P | NCI MSI-H | PMRP MSI-H | P |
|---|---|---|---|---|---|---|
| No. of patients | 462 | 475 | 40 | 52 | ||
| Mean age † | 70.5 (69.5 to 71.5) | 69.6 (68.6 to 70.6) | .218 ‡ | 69.5 (64.9 to 74.1) | 67.8 (63.9 to 71.6) | .561 ‡ |
| Sex | .306 § | .828 § | ||||
| Male | 61.1% | 64.5% | 33.3% | 37.3% | ||
| Female | 38.9% | 35.5% | 66.7% | 62.7% | ||
| Location | .817 § | .660 § | ||||
| Right | 23.8% | 24.5% | 63.4% | 68.6% | ||
| Left | 76.2% | 75.5% | 36.6% | 31.4% | ||
| Differentiation | .002 ‡ | .371 ‡ | ||||
| Well | 19.2% | 28.3% | 11.4% | 22.9% | ||
| Moderate | 74.6% | 63.5% | 68.8% | 56.3% | ||
| Poor | 6.2% | 28.3% | 20.0% | 20.8% | ||
| Duke's stage | .481 ‡ | .202 ‡ | ||||
| A1 + B1 | 14.2% | 14.8% | 10.8% | 2.0% | ||
| B2 + B3 + C1 + C2 | 65.1% | 61.4% | 73.0% | 82.4% | ||
| C3 + D | 20.7% | 23.7% | 16.2% | 15.7% | ||
| Mucin production ‖ | 10.2% | 10.0% | .913 § | 24.3% | 24.0% | 1.000 § |
| Family history | ||||||
| HNPCC | 0.9% | 1.3% | 9.5% | 12.0% | ||
| CRC | 16.4% | 11.9% | 7.1% | 10.0% | ||
| No cancer ¶ | 52.5% | 48.6% | .067 ‡ | 59.5% | 42.0% | .090 ‡ |
| Patient features | NCI MSS | PMRP MSS | P | NCI MSI-H | PMRP MSI-H | P |
|---|---|---|---|---|---|---|
| No. of patients | 462 | 475 | 40 | 52 | ||
| Mean age † | 70.5 (69.5 to 71.5) | 69.6 (68.6 to 70.6) | .218 ‡ | 69.5 (64.9 to 74.1) | 67.8 (63.9 to 71.6) | .561 ‡ |
| Sex | .306 § | .828 § | ||||
| Male | 61.1% | 64.5% | 33.3% | 37.3% | ||
| Female | 38.9% | 35.5% | 66.7% | 62.7% | ||
| Location | .817 § | .660 § | ||||
| Right | 23.8% | 24.5% | 63.4% | 68.6% | ||
| Left | 76.2% | 75.5% | 36.6% | 31.4% | ||
| Differentiation | .002 ‡ | .371 ‡ | ||||
| Well | 19.2% | 28.3% | 11.4% | 22.9% | ||
| Moderate | 74.6% | 63.5% | 68.8% | 56.3% | ||
| Poor | 6.2% | 28.3% | 20.0% | 20.8% | ||
| Duke's stage | .481 ‡ | .202 ‡ | ||||
| A1 + B1 | 14.2% | 14.8% | 10.8% | 2.0% | ||
| B2 + B3 + C1 + C2 | 65.1% | 61.4% | 73.0% | 82.4% | ||
| C3 + D | 20.7% | 23.7% | 16.2% | 15.7% | ||
| Mucin production ‖ | 10.2% | 10.0% | .913 § | 24.3% | 24.0% | 1.000 § |
| Family history | ||||||
| HNPCC | 0.9% | 1.3% | 9.5% | 12.0% | ||
| CRC | 16.4% | 11.9% | 7.1% | 10.0% | ||
| No cancer ¶ | 52.5% | 48.6% | .067 ‡ | 59.5% | 42.0% | .090 ‡ |
NCI MSS = stable tumors tested with the National Cancer Institute (NCI) panel; PMRP MSS = stable tumors tested with the pentaplex mononucleotide repeat panel; NCI MSI-H = unstable tumors with high microsatellite instability tested with the NCI panel; PMRP MSI-H = unstable tumors with high microsatellite instability tested with the pentaplex mononucleotide repeat panel; HNPCC = hereditary nonpolyposis colorectal cancer; CRC = colorectal cancer.
Age of patient's diagnosis was expressed as mean with the 95% confidence interval.
P values correspond to two-sided chi-square test.
P values correspond to two-sided Fisher's exact test.
Presence of more than 50% of extracellular mucin in a tumor.
No family history of any cancer.
For each category of microsatellite instability (MSI-H, MSI-L, or MSS), we calculated the percentages of tumors that lacked a given mismatch repair protein (MLH1, MSH2, or MSH6; Table 2 ). Among the MSI-H tumors analyzed with the NCI panel, 55% exhibited loss of MLH1, 10% exhibited loss of MSH2, and 2.5% exhibited loss of MSH6. Among the MSI-H tumors analyzed with the pentaplex panel of mononucleotide repeats, 62.3% had loss of MLH1, 23.1% had loss of MSH2, and 15.4% had loss of MSH6. Of the total 10 tumors that showed loss of MSH6 expression, nine were negative for MSH2 expression as well. None of the 719 tumors examined for PMS2 showed loss of its expression (data not shown). Among all tumors lacking MSH2 or MLH1 expression, there were four tumors with a MLH1 pathologic point mutation, six tumors with a MSH2 pathologic point mutation, and one tumor with a MSH2 exon deletion in both series.
Lack of MMR protein expression according to microsatellite instability in tumors analyzed by the NCI panel and the PMRP *
| MMR protein status | ||||||
|---|---|---|---|---|---|---|
| Tumor category | No. of tumors (%) | Loss of MLH1 | Loss of MSH2 | Loss of MSH6 | ||
| Tested with NCI panel | ||||||
| All | 531 | (26) 4.9% | (8) 1.5% | (1) 0.19% | ||
| MSI-H | (40) 7.5% | (22) 55% | (4) 10% | (1) 2.5% | ||
| MSI-L | (29) 5.5% | 0% | (1) 3.4% | 0% | ||
| MSS | (462) 87% | (4) 0.9% | (3) 0.6% | 0% | ||
| Tested with PMRP | ||||||
| All | 527 | (33) 6.3% | (14) 2.7% | (9) 1.7% | ||
| MSI-H | (52) 9.9% | (33) 62.3% | (12) 23.1% | (8) 15.4% | ||
| MSS | (475) 90.1% | 0% | (2) 0.42% | (1) 0.21% | ||
| MMR protein status | ||||||
|---|---|---|---|---|---|---|
| Tumor category | No. of tumors (%) | Loss of MLH1 | Loss of MSH2 | Loss of MSH6 | ||
| Tested with NCI panel | ||||||
| All | 531 | (26) 4.9% | (8) 1.5% | (1) 0.19% | ||
| MSI-H | (40) 7.5% | (22) 55% | (4) 10% | (1) 2.5% | ||
| MSI-L | (29) 5.5% | 0% | (1) 3.4% | 0% | ||
| MSS | (462) 87% | (4) 0.9% | (3) 0.6% | 0% | ||
| Tested with PMRP | ||||||
| All | 527 | (33) 6.3% | (14) 2.7% | (9) 1.7% | ||
| MSI-H | (52) 9.9% | (33) 62.3% | (12) 23.1% | (8) 15.4% | ||
| MSS | (475) 90.1% | 0% | (2) 0.42% | (1) 0.21% | ||
MMR = mismatch repair; NCI = National Cancer Institute; PMRP = pentaplex mononucleotide repeat panel; MLH1 = loss of MLH1 protein expression; MSH2 = loss of MSH2 protein expression; MSH6 = loss of MSH6 protein expression; MSI-H = high microsatellite instability; MSI-L = low microsatellite instability; MSS = microsatellite stable.
Lack of MMR protein expression according to microsatellite instability in tumors analyzed by the NCI panel and the PMRP *
| MMR protein status | ||||||
|---|---|---|---|---|---|---|
| Tumor category | No. of tumors (%) | Loss of MLH1 | Loss of MSH2 | Loss of MSH6 | ||
| Tested with NCI panel | ||||||
| All | 531 | (26) 4.9% | (8) 1.5% | (1) 0.19% | ||
| MSI-H | (40) 7.5% | (22) 55% | (4) 10% | (1) 2.5% | ||
| MSI-L | (29) 5.5% | 0% | (1) 3.4% | 0% | ||
| MSS | (462) 87% | (4) 0.9% | (3) 0.6% | 0% | ||
| Tested with PMRP | ||||||
| All | 527 | (33) 6.3% | (14) 2.7% | (9) 1.7% | ||
| MSI-H | (52) 9.9% | (33) 62.3% | (12) 23.1% | (8) 15.4% | ||
| MSS | (475) 90.1% | 0% | (2) 0.42% | (1) 0.21% | ||
| MMR protein status | ||||||
|---|---|---|---|---|---|---|
| Tumor category | No. of tumors (%) | Loss of MLH1 | Loss of MSH2 | Loss of MSH6 | ||
| Tested with NCI panel | ||||||
| All | 531 | (26) 4.9% | (8) 1.5% | (1) 0.19% | ||
| MSI-H | (40) 7.5% | (22) 55% | (4) 10% | (1) 2.5% | ||
| MSI-L | (29) 5.5% | 0% | (1) 3.4% | 0% | ||
| MSS | (462) 87% | (4) 0.9% | (3) 0.6% | 0% | ||
| Tested with PMRP | ||||||
| All | 527 | (33) 6.3% | (14) 2.7% | (9) 1.7% | ||
| MSI-H | (52) 9.9% | (33) 62.3% | (12) 23.1% | (8) 15.4% | ||
| MSS | (475) 90.1% | 0% | (2) 0.42% | (1) 0.21% | ||
MMR = mismatch repair; NCI = National Cancer Institute; PMRP = pentaplex mononucleotide repeat panel; MLH1 = loss of MLH1 protein expression; MSH2 = loss of MSH2 protein expression; MSH6 = loss of MSH6 protein expression; MSI-H = high microsatellite instability; MSI-L = low microsatellite instability; MSS = microsatellite stable.
Specificity and Sensitivity of Instability in Single Markers for Detection of Mismatch Repair Deficiency
The specificity with which individual markers detected mismatch repair deficiency (defined as loss of expression of any mismatch repair protein) was very high, ranging from 96.0% (D17S250) to more than 99% (NR22 and NR24). However, the sensitivity of the same markers was lower, ranging from 76.5% (NR21) to 91.3% (NR22) among mononucleotide markers, and below 57% for dinucleotide markers ( Fig. 1 ).
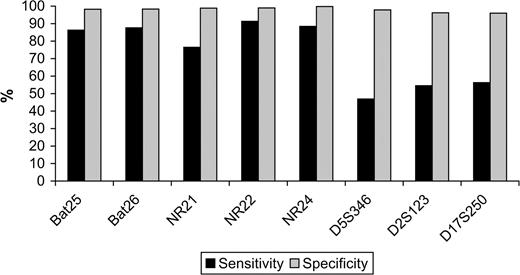
Sensitivity and specificity of each microsatellite in identifying mismatch repair protein deficiency. Sensitivity is the proportion of tumors with instability in a specific marker among the tumors with mismatch repair protein deficiency. Specificity is the proportion of tumors without instability in a specific marker among the tumors without mismatch repair protein deficiency. Mononucleotide markers: BAT25, BAT26, NR21, NR22, and NR24. Dinucleotide markers: D5S346, D2S123, and D17S250. The National Cancer Institute panel consists of the dinucleotide markers plus BAT25 and BAT26.
Diagnostic Utility of Low-Level Microsatellite Instability
The follow-up NCI conference that revised the Bethesda guidelines ( 13 ) also recommended testing all MSI-L tumors with an additional set of microsatellites to accurately characterize them. Of the 531 tumors analyzed with the NCI panel, 29 (5.5%) were MSI-L, all but one due to an unstable dinucleotide repeat. The MSI-L tumors exhibiting an unstable dinucleotide repeat were tested with an additional set of mononucleotide markers (NR21, NR22, and NR24). None of these additional mononucleotide markers was unstable (data not shown), and all showed normal expression of MLH1, MSH2, MSH6, and PMS2 proteins (data not shown). The tumor designated MSI-L due to the presence of an unstable mononucleotide repeat (BAT26) did not express MSH2 and harbored a mutation in that gene (an adenosine insertion at position 1703 in codon 568 that caused the substitution of serine for phenylalanine and the introduction of a termination signal at codon 571).
Efficacy of the National Cancer Institute and Pentaplex of Mononucleotides Repeats Panels in Detecting Absence of Mismatch Repair Protein Expression
We evaluated the sensitivity, specificity, and positive and negative predictive values (PPV and NPV) of both panels to detect absence of expression of at least one mismatch repair protein ( Table 3 ). Specificity and NPV of the pentaplex panel of mononucleotide repeats were 98.7% (95% confidence interval [CI] = 98% to 100%) and 99.6% (95% CI = 99% to 100%), respectively. Corresponding values for specificity and NPV of the NCI panel were 97.2% (95% CI = 96% to 99%) and 98.4% (95% CI = 97% to 100%), respectively. The sensitivity and PPV of the pentaplex panel of mononucleotide repeats were 95.8% (95% CI = 89% to 103%) and 88.5% (95% CI = 79% to 98%), respectively, and corresponding values for the NCI panel were 76.5% (95% = 61% to 92%) and 65.0% (95% CI = 49% to 81%), respectively. Thus, specificity and NPV were high for both panels, but the sensitivity and PPV of the pentaplex panel of mononucleotide repeats were much higher than those of the NCI panel ( Table 3 ).
Predictive values of the microsatellite panels in detecting tumors with absence of mismatch repair protein expression *
| Microsatellite markers | Sensitivity † | Specificity ‡ | PPV § | NPV ‖ |
|---|---|---|---|---|
| NCI panel ¶ | 76.5 (61 to 92) | 97.2 (96 to 99) | 65.0 (49 to 81) | 98.4 (97 to 100) |
| PMRP # | 95.8 (89 to 103) | 98.7 (98 to 100) | 88.5 (79 to 98) | 99.6 (99 to 100) |
| BAT26 + NR24 | 95.8 (89 to 103) | 98.7 (98 to 100) | 88.5 (79 to 98) | 99.6 (99 to 100) |
| Microsatellite markers | Sensitivity † | Specificity ‡ | PPV § | NPV ‖ |
|---|---|---|---|---|
| NCI panel ¶ | 76.5 (61 to 92) | 97.2 (96 to 99) | 65.0 (49 to 81) | 98.4 (97 to 100) |
| PMRP # | 95.8 (89 to 103) | 98.7 (98 to 100) | 88.5 (79 to 98) | 99.6 (99 to 100) |
| BAT26 + NR24 | 95.8 (89 to 103) | 98.7 (98 to 100) | 88.5 (79 to 98) | 99.6 (99 to 100) |
Results are expressed as percentages, with 95% confidence intervals in parentheses. PPV = positive predictive value; NPV = negative predictive value; NCI = National Cancer Institute; PMRP = pentaplex mononucleotide repeat panel.
Sensitivity = (high microsatellite instability [MSI-H] tumors with lack of mismatch repair protein expression/total number of tumors with lack of mismatch repair protein expression) × 100.
Specificity = (non–MSI-H tumors with normal expression of mismatch repair proteins/total tumors with normal mismatch repair protein expression) × 100.
PPV = (MSI-H tumors with lack of mismatch repair protein expression/all MSI-H tumors) × 100.
NPV = (non–MSI-H tumors with normal mismatch repair protein/all non-MSI-H tumors) × 100.
Composed of markers D5S346, D2S123, D17S250, BAT25, and BAT26.
Composed of BAT26, BAT25, NR21, NR22, and NR24.
Predictive values of the microsatellite panels in detecting tumors with absence of mismatch repair protein expression *
| Microsatellite markers | Sensitivity † | Specificity ‡ | PPV § | NPV ‖ |
|---|---|---|---|---|
| NCI panel ¶ | 76.5 (61 to 92) | 97.2 (96 to 99) | 65.0 (49 to 81) | 98.4 (97 to 100) |
| PMRP # | 95.8 (89 to 103) | 98.7 (98 to 100) | 88.5 (79 to 98) | 99.6 (99 to 100) |
| BAT26 + NR24 | 95.8 (89 to 103) | 98.7 (98 to 100) | 88.5 (79 to 98) | 99.6 (99 to 100) |
| Microsatellite markers | Sensitivity † | Specificity ‡ | PPV § | NPV ‖ |
|---|---|---|---|---|
| NCI panel ¶ | 76.5 (61 to 92) | 97.2 (96 to 99) | 65.0 (49 to 81) | 98.4 (97 to 100) |
| PMRP # | 95.8 (89 to 103) | 98.7 (98 to 100) | 88.5 (79 to 98) | 99.6 (99 to 100) |
| BAT26 + NR24 | 95.8 (89 to 103) | 98.7 (98 to 100) | 88.5 (79 to 98) | 99.6 (99 to 100) |
Results are expressed as percentages, with 95% confidence intervals in parentheses. PPV = positive predictive value; NPV = negative predictive value; NCI = National Cancer Institute; PMRP = pentaplex mononucleotide repeat panel.
Sensitivity = (high microsatellite instability [MSI-H] tumors with lack of mismatch repair protein expression/total number of tumors with lack of mismatch repair protein expression) × 100.
Specificity = (non–MSI-H tumors with normal expression of mismatch repair proteins/total tumors with normal mismatch repair protein expression) × 100.
PPV = (MSI-H tumors with lack of mismatch repair protein expression/all MSI-H tumors) × 100.
NPV = (non–MSI-H tumors with normal mismatch repair protein/all non-MSI-H tumors) × 100.
Composed of markers D5S346, D2S123, D17S250, BAT25, and BAT26.
Composed of BAT26, BAT25, NR21, NR22, and NR24.
Crossover Analysis Evaluation
To further define the distinct performance of the panels, we analyzed both sets of markers in the same tumors. We included in this analysis all fresh-frozen and paraffin-embedded tumors that were MSI-L and MSI-H and all MSS tumors that had altered mismatch repair protein expression. Finally, we also included 100 randomly chosen fresh-frozen tumors (therefore initially analyzed with the NCI panel of markers) that were MSS and had normal mismatch repair protein expression. In this last group, all tumors were found to be stable for all markers of the pentaplex panel. Of the seven MSS tumors, as determined by the NCI panel, that lacked mismatch repair protein expression, one had all mononucleotide repeat markers mutated, did not express MSH2, and harbored a mutation (435 T to G transversion causing a substitution of methionine for isoleucine, codon 145) in the MSH2 gene. The rest were stable for all mononucleotide repeat markers. The majority of MSI-L tumors studied with the NCI panel that normally expressed mismatch repair proteins were stable when tested with the pentaplex panel of mononucleotide repeats.
The NCI follow-up workshop recommended the testing of a secondary panel of markers with mononucleotide repeats when a tumor is classified as MSI-H based only on mutated dinucleotide repeats, to exclude MSI-L ( 13 ). Among tumors tested with the NCI panel, two MSI-H tumors (classified as such due to at least two mutated dinucleotide repeats) with normal mismatch repair protein expression were found to have normal mononucleotide repeat markers. Furthermore, five tumors that were MSI-H (with only one mononucleotide-mutated and one or more dinucleotide-mutated repeats) and normally expressed mismatch repair proteins were also found to have normal NR markers.
Evaluation of the Results Obtained With the Pentaplex Panel of Mononucleotide Repeats
It was suggested ( 17 ) that, due to the existence of polymorphisms, for a tumor to be classified as MSI-H with the pentaplex panel of mononucleotide repeats panel it should be required that three of the five markers be unstable. In our series, of 52 tumors classified as MSI-H using the panel of mononucleotide markers, there were three that had instability in only two markers, and in the two that were available for study, mismatch repair protein expression was lost. Therefore, two or more unstable markers could be sufficient to designate a tumor as MSI-H using the pentaplex panel of mononucleotide repeats. Fourteen tumors showed instability in a single marker (NR21 in all 14). Because it has been reported that there are substantial percentages of NR21 polymorphisms in most populations in the world ( 17 ), we compared the amplification products obtained when testing for instability of the NR21 marker in the tumor with amplification products obtained when the template was DNA from normal tissue. In all cases in which the NR21 marker had been presumed to be unstable, polymorphisms were present.
Evaluation of a Simplified Approach Using Markers From the Pentaplex Panel of Mononucleotide Repeats to Detect Loss of Mismatch Repair Protein Expression
To establish the easiest yet most reliable approach for the detection of tumors with loss of mismatch repair protein expression, we examined if a combination of a reduced number of markers was as effective as the entire pentaplex panel of mononucleotide repeats. We identified all MSI-H tumors that had one or two stable markers and observed that the combination of stable BAT25 or BAT26 with stable NR21 or NR24 was not observed in any tumor with loss of mismatch repair protein expression. Thus, assaying two markers, one of which was BAT25 or BAT26 and one of which was NR21 or NR24, would be sufficient to detect all tumors with loss of mismatch repair protein expression. Considering that NR24 is consistently monomorphic in most world populations and that in our series there was only one tumor with mismatch repair deficiency that was exclusively unstable for BAT26, we decided to evaluate the performance of a panel consisting of just BAT26 and NR24. As shown in Table 3 , this approach yielded values for sensitivity and specificity that were identical to those obtained when testing with the entire panel of pentaplex mononucleotide markers, and therefore, it was as reliable for the detection of mismatch repair–deficient tumors. An added advantage of testing with a panel consisting of BAT26 and NR24 is that the PCR products resulting from amplification of both these markers can be separated in a single sequencing run ( Fig. 2 , bottom panel).
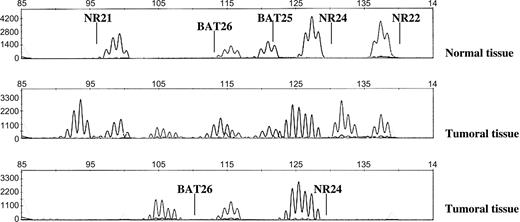
Microsatellite instability analysis of a tumor with instability in five markers. (NR21, BAT26, BAT25, NR24, and NR22).Polymerase chain reaction (PCR) products from mononucleotide pentaplex analysis of DNA from a patient's normal tissue ( upper panel ) and the DNA from a tumor in the same patient that was unstable for all mononucleotide markers ( middle panel ) were separated using an ABI Prism 3100 Avant Genetic Analyzer (Applied Biosystems). Lower panel : separation of PCR products obtained after amplification of only BAT26 and NR24 using the same tumor DNA as above.
Discussion
The observation of a new class of mutations in colorectal cancer with insertions and deletions at microsatellite sequences ( 3 – 5 ) led to the discovery of the MSI-H phenotype ( 12 ), which is due to an alteration in the mismatch repair system ( 18 – 21 ). A workshop convened by the NCI proposed a panel of markers for uniform analysis of microsatellite instability to detect defects in the mismatch repair system ( 12 ). At a follow-up meeting ( 13 ) convened by the NCI, it was recommended to continue using the same set of markers, but some potential limitations of this approach were recognized. Furthermore, it was suggested that testing for microsatellite instability with a pentaplex panel of mononucleotide repeats be examined to determine if this panel could be more sensitive for the detection of MSI-H tumors ( 15 ). The goal of this study was to ascertain and compare the performance of both panels and to devise a simple yet highly reliable strategy for the detection of mismatch repair deficiency.
One of the potential limitations of this study was the fact that samples were assigned to be tested with the NCI panel or the pentaplex panel of mononucleotide repeats depending on the type of tissue (fresh-frozen or paraffin-embedded, respectively) that each hospital obtained. The overall quality of DNA, and thus that of the microsatellite instability analysis, could have been less optimal using paraffin-embedded samples and have therefore led to underestimation of the sensitivity and specificity of the pentaplex panel of mononucleotide repeats panel. However, contrary to expectations based on this possibility, sensitivity and specificity in detecting mismatch repair deficiency were better in the paraffin-embedded samples. Furthermore, the crossover analysis of all tumors found to be unstable for at least one marker, and all tumors that showed discrepancy between mismatch repair protein expression and microsatellite instability, showed the shortcomings of the NCI panel in comparison to the performance of the pentaplex panel of mononucleotide repeats.
One noticeable difference between the two panels was the detection of a substantial number of tumors with an MSI-L phenotype in the NCI panel and the absence of this phenotype as assessed by the pentaplex panel of mononucleotide repeats. One might presume that this difference could be accounted for by the inability of the pentaplex panel to distinguish between MSI-H and MSI-L tumors and that the latter were misclassified as MSI-H rather than MSI-L. However, this explanation is not supported by our findings, which show a strong association of the MSI-H phenotype with lack of mismatch repair protein expression in the pentaplex mononucleotide panel–tested group.
MSI-L tumors are believed to have clinical and pathologic features similar to MSS tumors, but some studies have suggested that the tumors in these groups may have distinct molecular alterations ( 22 – 24 ), leading to distinct prognoses ( 25 , 26 ). Furthermore, some authors have identified MSH6 germline mutations which they suggest are responsible for a substantial group of MSI-L tumors ( 27 ). In our series, all MSI-L tumors (except one that was unstable for BAT26) were unstable for only dinucleotide markers, and they had normal expression of MLH1, MSH2, MSH6, and PMS2. Testing them with an additional set of microsatellites did not change their status. Therefore, detection of this class of tumors was not useful for identification of tumors with mutator pathway alterations, and further testing of these tumors with additional markers would not be useful, contrary to what was suggested by the NCI conference ( 13 ).
Adding to the lack of usefulness of dinucleotide markers, all tumors that were found to be MSI-H due only to mutated dinucleotide repeats were positive for mismatch repair protein expression. When we examined an additional set of mononucleotide repeat markers in these tumors, as was suggested in the NCI conference ( 13 ), all of them were stable. The same was true for the three tumors assigned an MSI-H phenotype based on instability in one mononucleotide and in one dinucleotide repeat. Cases in which tumors were designated as MSI-H based on mutated dinucleotide repeats and one or no mutated mononucleotide repeats were rare, but they were consistently found to be positive for expression of mismatch repair proteins. Therefore, we believe that the use of dinucleotide repeats as markers to detect mismatch repair–deficient tumors should be discouraged.
The performance of a pentaplex panel of mononucleotide repeats that would not require the comparison with normal DNA and presumably facilitate testing and make it more affordable was suggested by Suraweera et al. ( 15 ). Its usefulness has been recently confirmed in a series of sporadic endometrial adenocarcinomas ( 28 ). To our knowledge, the present analysis is the first large, population-based prospective study on colorectal cancer that attempts to evaluate the pentaplex panel of mononucleotide repeats and compare it with the NCI panel. We have been able to confirm not only the accuracy with which the pentaplex panel detects deficiency in expression of mismatch repair proteins but also that it is superior to the NCI panel in this regard.
There is a limitation of using pentaplex panel without comparison with normal DNA as was done in this study that is due to the frequency of polymorphisms in various populations. It has been reported that polymorphisms in BAT25 and BAT26 are not uncommon in African ethnic groups and African Americans ( 29 , 30 ). More recently, Buhard et al. ( 17 ) evaluated the incidence of polymorphisms in different populations around the world including polymorphisms of four of the five mononucleotide markers originally suggested by Suraweera et al. ( 15 ) as well as those of NR27. They found that BAT25 and BAT26 were polymorphic to a statistically significant degree only in African populations (up to 15% of individuals affected). NR24 was found to be monomorphic in almost all individuals of the worldwide series, and there were substantial percentages of polymorphisms in NR21 in Africa and Oceania ( 17 ). Taking these results into consideration, the authors suggested that a tumor be designated MSI-H if at least three of the five markers of the pentaplex panel were unstable. In our series, which was composed mostly of Caucasians, the only marker that showed a substantial percentage of polymorphisms was NR21. The absence of polymorphisms in BAT25 and BAT26 could be explained by the negligible number of patients of African origin in our series.
In our study, the combination of BAT25 or BAT26 with NR21 or NR24 performed as well as the entire pentaplex panel in identifying tumors with defects in mismatch repair. Given that NR21 is unreliable due to a substantial number of polymorphisms and that there was but a single case that was unstable only for BAT26, the most suitable combination of markers for general screening would be BAT26 and NR24. Use of these markers is a simple strategy that would require only a single PCR and one analysis, with no need to test normal DNA. In our study, PCR fragment sizes were separated enough so that results were unambiguous ( Fig. 2 ). Based on our results, instability in either marker alone is a highly reliable indicator of mismatch repair deficiency, and if both markers were assayed for instability, the odds that a single case would be missed would be extremely low. This strategy can presumably be extended to most populations in the world, except Africans. Even in that population, the very high sensitivity and specificity of NR24 for mismatch repair deficiency and its monomorphic nature could make this approach highly effective. In any case, a study that targets Africans and tests the effectiveness of the strategy based on these two markers should be supported.
In summary, while the pentaplex panel of mononucleotide repeats is superior to the NCI panel for the detection of tumors with an altered mismatch repair system, a screen based on assessment of only BAT26 and NR24 could be equally effective.
Appendix
Writing Committee and Working Group: Xavier Llor, Antoni Castells, and Montserrat Andreu. Xavier Llor, Antoni Castells, and Montserrat Andreu conceived the idea for the study; Xavier Llor, Rosa M. Xicola, Elisenda Pons, Antoni Castells, Montserrat Andreu, Virgínia Piñol, and Artemio Payá designed the protocol; Rosa M. Xicola, Virgínia Piñol, Sergi Castellví-Bel, Cristina Alenda, Rodrigo Jover, and Xavier Bessa supervised the execution of the study; Virgínia Piñol was responsible for database design and management.
All participants listed below were fully involved in the study.
Hospital 12 de Octubre, Madrid: Juan Diego Morillas (local coordinator), Raquel Muñoz, Marisa Manzano, Francisco Colina, Jose Díaz, Carolina Ibarrola, Guadalupe López, Alberto Ibáñez; Hospital Clínic, Barcelona: Antoni Castells (local coordinator), Virgínia Piñol, Sergi Castellví-Bel, Francisco Rodríguez-Moranta, Francesc Balaguer, Antonio Soriano, Rosa Cuadrado, Maria Pellisé, Rosa Miquel, J. Ignasi Elizalde, Josep M. Piqué; Hospital Clínico Universitario, Zaragoza: Ángel Lanas (local coordinator), Javier Alcedo, Javier Ortego; Hospital Cristal-Piñor, Complexo Hospitalario de Ourense, Ourense: Joaquin Cubiella (local coordinator), Ma Soledad Díez, Mercedes Salgado, Eloy Sánchez, Mariano Vega; Hospital del Mar, Barcelona: Montserrat Andreu (local coordinator), Xavier Bessa, Agustín Panadés, Asumpta Munné, Felipe Bory, Miguel Nieto, Agustín Seoane; Hospital Donosti, San Sebastián: Luis Bujanda (local coordinator), Juan Ignacio Arenas, Isabel Montalvo, Julio Torrado, Ángel Cosme; Hospital General Universitario de Alicante, Alicante: Artemio Payá (local coordinator), Rodrigo Jover, Juan Carlos Penalva, Cristina Alenda; Hospital General de Granollers, Granollers: Joaquim Rigau (local coordinator), Ángel Serrano, Anna Giménez; Hospital General de Vic, Vic: Joan Saló (local coordinator), Eduard Batiste-Alentorn, Josefina Autonell, Ramon Barniol; Hospital General Universitario de Guadalajara, Guadalajara: Ana María García (local coordinator), Fernando Carballo, Antonio Bienvenido, Eduardo Sanz, Fernando González, Jaime Sánchez; Hospital General Universitario de Valencia, Valencia: Enrique Medina (local coordinator), Jaime Cuquerella, Pilar Canelles, Miguel Martorell, José Ángel García, Francisco Quiles, Elisa Orti; Hospital do Meixoeiro, Vigo: Juan Clofent (local coordinator), Jaime Seoane, Antoni Tardío, Eugenia Sanchez; Hospital San Eloy, Baracaldo: Luis Bujanda (local coordinator), Carmen Muñoz, María del Mar Ramírez, Araceli Sánchez; Hospital Universitari Germans Trias i Pujol, Badalona: Xavier Llor (local coordinator), Rosa M. Xicola, Marta Piñol, Mercè Rosinach, Anna Roca, Elisenda Pons, José M. Hernández, Miquel A. Gassull; Hospital Universitari Mútua de Terrassa, Terrassa: Fernando Fernández-Bañares (local coordinator), Josep M. Viver, Antonio Salas, Jorge Espinós, Montserrat Forné, Maria Esteve; Hospital Universitari Arnau de Vilanova, Lleida: Josep M. Reñé (local coordinator), Carmen Piñol, Juan Buenestado, Joan Viñas; Hospital Universitario de Canarias, Santa Cruz de Tenerife: Enrique Quintero (local coordinator), David Nicolás, Adolfo Parra, Antonio Martín; Hospital Universitario La Fe, Valencia: Lidia Argüello (local coordinator), Vicente Pons, Virginia Pertejo, Teresa Sala; Hospital Universitario Reina Sofía, Córdoba: Antonio Naranjo (local coordinator), María del Valle García, Patricia López, Fernando López, Rosa Ortega, Javier Briceño, Javier Padillo; Fundació Hospital Son Llatzer, Palma de Mallorca: Àngels Vilella (local coordinator), Carlos Dolz, Hernan Andreu.
This work was supported by grants from the Fondo de Investigación Sanitaria (FIS 01/0104) and from the Instituto de Salud Carlos III (RC03/02 and RC03/10). R. M. Xicola is a recipient of a Formacío D'investigadors grant of the Departament d'Universitats, Recerca i Societat de la Informació from Generalitat de Catalunya and the European Social Fund. X. Llor is a recipient of a Ramon y Cajal grant from Ministerio de Ciencia y Tecnología of the Spanish government. V. Piñol received a research grant from the Institut d'Investigacions Biomèdiques August Pi i Sunyer. The funding agencies had no role in the design of this study, data collecting, analysis and interpretation of the results, or the writing of the manuscript.
References
Greenlee RT, Murray T, Bolden S, Wingo PA. Cancer statistics, 2000.
Vogelstein B, Fearon ER, Kern SE, Hamilton SR, Preisinger AC, Nakamura Y, et al. Allelotype of colorectal carcinomas.
Peltomäki P, Aaltonen LA, Sistonen P, Pylkkänen L, Mecklin J-P, Järvinen HJ, et al. Genetic mapping of a locus predisposing to human colorectal cancer.
Ionov Y, Peinado MA, Malkhosyan S, Shibata D, Perucho M. Ubiquitous somatic mutations in simple repeated sequences reveal a new mechanism for colonic carcinogenesis.
Thibodeau SN, Bren G, Schaid D. Microsatellite instability in cancer of the proximal colon.
Peltomaki P. Deficient DNA mismatch repair: a common etiologic factor for colon cancer.
Llor X, Pons E, Xicola RM, Castells A, Alenda C, Pinol V, et al. Differential features of colorectal cancers fulfilling Amsterdam criteria without involvement of the mutator pathway.
Gryfe R, Kim H, Hsieh ETK, Aronson MD, Holowaty EJ, Bull SB, et al. Tumor microsatellite instability and clinical outcome in young patients with colorectal cancer.
Jover R, Zapater P, Castells A, Llor X, Andreu M, Cubiella J, et al. Mismatch repair status in the prediction of benefit from adjuvant fluorouracil chemotherapy in colorectal cancer.
Rodriguez-Bigas MA, Boland CR, Hamilton SR, Henson DE, Jass JR, Khan PM, et al. A National Cancer Institute Workshop on Hereditary Nonpolyposis Colorectal Cancer Syndrome:meeting highlights and Bethesda guidelines.
Boland CR, Thibodeau SN, Hamilton SR, Sidransky D, Eshleman JR, Burt RW, et al. A National Cancer Institute Workshop on microsatellite instability for cancer detection and familial predisposition: development of international criteria for the determination of microsatellite instability in colorectal cancer.
Umar A, Boland CR, Terdiman JP, Syngal S, de la Chapelle A, Ruschoff J, et al. Revised Bethesda Guidelines for hereditary nonpolyposis colorectal cancer (Lynch syndrome) and microsatellite instability.
Pinol V, Castells A, Andreu M, Castellvi-Bel S, Alenda C, Llor X, et al. Accuracy of revised Bethesda guidelines, microsatellite instability, and immunohistochemistry for the identification of patients with hereditary nonpolyposis colorectal cancer.
Suraweera N, Duval A, Reperant M, Vaury C, Furlan D, Leroy K, et al. Evaluation of tumor microsatellite instability using five quasimonomorphic mononucleotide repeats and pentaplex PCR.
Dietmaier W, Wallinger S, Bocker T, Kullmann F, Fishel R, Ruschoff J. Diagnostic microsatellite instability: definition and correlation with mismatch repair protein expression.
Buhard O, Cattaneo F, Wong YF, Yim SF, Friedman E, Flejou JF, et al. Multipopulation analysis of polymorphisms in five mononucleotide repeats used to determine the microsatellite instability status of human tumors.
Leach FS, Nicolaides NC, Papadopoulos N, Liu B, Jen J, Parsons R, et al. Mutations of a mut S homolog in hereditary nonpolyposis colorectal cancer.
Fishel R, Lescoe MK, Rao MRS, Copeland NG, Jenkins M, Garber J, et al. The human mutator gene homolog MSH2 and its association with hereditary nonpolyposis colon cancer.
Papadopoulos N, Nicolaides NC, Wei F-S, Ruben SM, Carter KC, Rosen CA, et al. Mutations of a mutL homolog in hereditary colon cancer.
Bronner CE, Baker SM, Morrison PT, Warren G, Smith LG, Lescoe MK, et al. Mutation in the DNA mismatch repair gene homologue hMLH1 is associated with hereditary non-polyposis colon cancer.
Jass JR, Biden KG, Cummings MC, Simms LA, Walsh M, Schoch E, et al. Characterisation of a subtype of colorectal cancer combining features of the suppressor and mild mutator pathways.
Biden KG, Simms LA, Cummings M, Buttenshaw R, Schoch E, Searle J, et al. Expression of Bcl-2 protein is decreased in colorectal adenocarcinomas with microsatellite instability.
Michael-Robinson JM, Biemer-Huttmann A, Purdie DM, Walsh MD, Simms LA, Biden KG, et al. Tumour infiltrating lymphocytes and apoptosis are independent features in colorectal cancer stratified according to microsatellite instability status.
Kohonen-Corish MR, Daniel JJ, Chan C, Lin BP, Kwun SY, Dent OF, et al. Low microsatellite instability is associated with poor prognosis in stage C colon cancer.
Wright CM, Dent OF, Newland RC, Barker M, Chapuis PH, Bokey EL, et al. Low level microsatellite instability may be associated with reduced cancer specific survival in sporadic stage C colorectal carcinoma.
Kolodner RD, Tytell JD, Schmeits JL, Kane MF, Gupta RD, Weger J, et al. Germ-line msh6 mutations in colorectal cancer.
Wong YF, Cheung TH, Lo KW, Yim SF, Chan LK, Buhard O, et al. Detection of microsatellite instability in endometrial cancer: advantages of a panel of five mononucleotide repeats over the National Cancer Institute panel of markers.
Samowitz WS, Slattery ML, Potter JD, Leppert MF. BAT-26 and BAT-40 instability in colorectal adenomas and carcinomas and germline polymorphisms.


{kind=link}
{kind=link}