





Abstract
Erdheim–Chester disease (ECD) is a rare nonLangerhans cell histiocytosis. Although approximately 50% of cases eventually involve the central nervous system (CNS), the CNS has seldom been reported as the initial biopsy site. The diagnosis of CNS ECD can be challenging due to morphologic overlap with reactive histiocytic proliferation, Langerhans cell histiocytosis (LCH), and extranodal Rosai–Dorfman disease (RDD). We present 3 cases from our files that illustrate the protean manifestations of ECD. Case 1 was a 47-year-old man with ataxia, dysarthria, and intermittent ophthalmoplegia whose cerebellar biopsy had shown only profuse, nonspecific Rosenthal fiber-rich piloid gliosis; ECD was diagnosed only at autopsy. The gliosis and marked variations in histiocyte morphology in different anatomical sites added to the diagnostic challenge. Case 2 was a 67-year-old female with chronic progressive symptoms and a pontine lesion that had been considered to be CLIPPERS by neuroimaging. Identification of a BRAFV600E mutation allowed an ECD diagnosis and treatment with the specific BRAFV600E inhibitor vemurafenib, which resulted in a marked sustained clinical response. Case 3 was diagnosed as ECD after positive bone biopsy with typical foamy histiocytes. Six years later, there was massive dural involvement that showed RDD-like, BRAF-mutation-negative histiocytosis. These cases highlight the clinical and histologic overlap that can occur among these disorders.
INTRODUCTION
Erdheim–Chester disease (ECD) is a rare nonLangerhans cell histiocytosis characterized by xanthomatous or xanthogranulomatous infiltrates in tissues (1). The number of cases has increased recently due to more recognition of this entity, improved diagnostic criteria, and reliable molecular studies. In the 2016 World Health Organization Classification, ECD has been reclassified as a distinct entity and separated from disseminated juvenile xanthogranuloma (2). The clinical course of ECD is mainly dependent on the disease extent and distribution, ranging from asymptomatic bone involvement to systemic life-threatening disease with poor prognosis, particularly in the cases with central nervous system (CNS) or cardiovascular involvement (3).
The diagnosis of ECD is typically based on the characteristic radiologic findings and pathologic features in the appropriate clinical context. However, recognition of CNS ECD can be challenging due to variable clinical courses, often limited tissue biopsies and mimicries with other reactive and neoplastic histiocytic lesions; this is particularly problematic in localized CNS ECD cases without stereotypic radiographic changes elsewhere. Here, we review the major clinicopathologic characteristics of CNS ECD with emphasis on the diagnostic workups and differential diagnosis through several illustrative case presentations.
Case 1. ECD Diagnosed at Autopsy After Inconclusive Cerebellar Biopsy With Profuse Piloid Gliosis
The decedent was a 47-year-old Hispanic man with a history of a left cerebellar lesion since 2006. Symptoms consisted of ataxia, dysarthria, and intermittent ophthalmoplegia. The patient had no other chronic medical problems but had been hospitalized in the spring of 2009 for pneumonia. In May 2009, he developed decreasing mental status and was taken to the emergency department in the southern part of Colorado where computerized tomography imaging revealed obstructive hydrocephalus. Magnetic resonance imaging (MRI) showed a combination of bilateral cerebellar lesions epicentered on the dentate nuclei with enhancement (Fig. 1A), upper cervical cord lesions (Fig. 1B), and, in retrospect, enhancement in the pituitary stalk (Fig. 1C). An external ventricular drain was placed and the patient was transferred to the University of Colorado Hospital Neuro ICU soon thereafter. He was suspected to have neurosarcoidosis and respiratory failure.
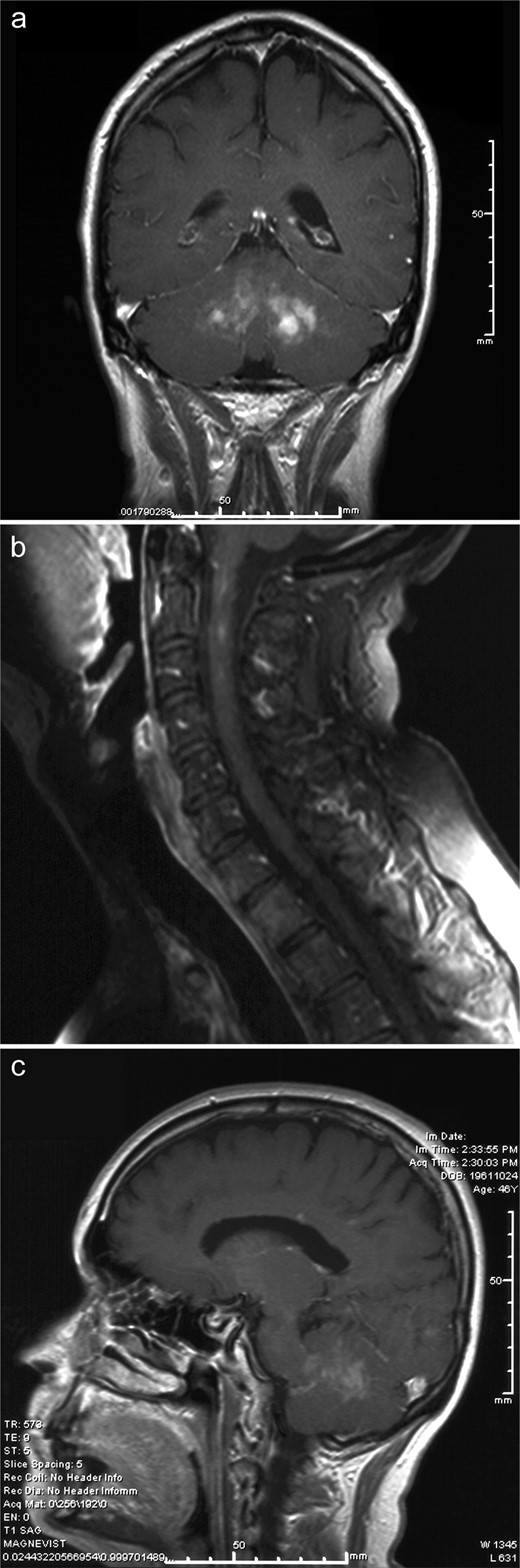
Case 1. (A) T1-weighted coronal magnetic resonance imaging (MRI) with gadolinium illustrates the bilateral enhancing cerebellar lesions with epicenters near the dentate nuclei. (B) T1-weighted sagittal MRI with gadolinium of the spine shows patchy, somewhat linear enhancement in the upper cervical cord, without cord expansion. (C) T1-weighted sagittal MRI with gadolinium reveals increased signal in the infundibular stalk and posterior pituitary gland, which can be seen in patients with ECD even if diabetes insipidus is not clinically evident. Posterior fossa lesions in this patient are also seen well on this sagittal image.
Biopsy of the cerebellar mass revealed no foamy macrophages or granuloma formation. The most conspicuous feature was profuse Rosenthal fiber-rich piloid gliosis (Fig. 2A), with modest collections of cytologically bland, CD68-positive histiocytes without granuloma formation (Fig. 2B). The findings were not diagnostic for a specific disease process. The patient’s respiratory status remained poor and he was placed on comfort care measures before dying 10 days later. Permission was given for a full autopsy.
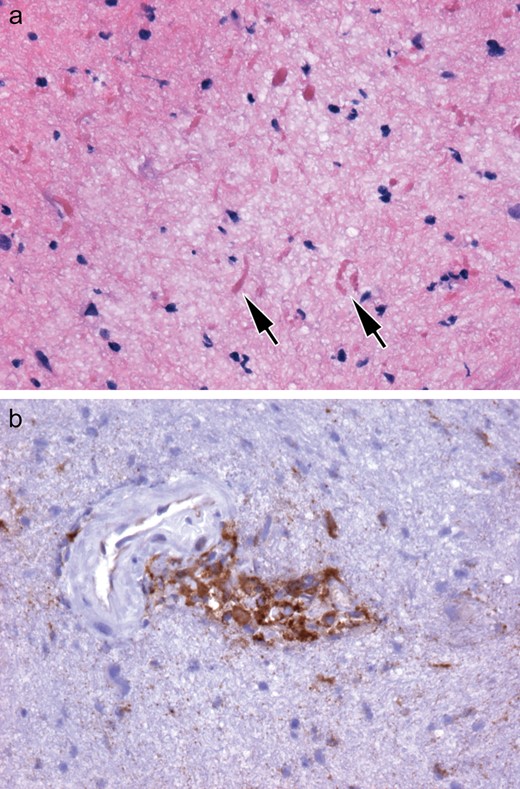
Case 1. (A, B) The nondiagnostic biopsy showed prominent piloid gliosis with profuse numbers of Rosenthal fibers (arrows, A) not accompanied by neoplastic piloid tumor cells. Small collections of cytologically bland, CD68-immunopositive histiocytes are less conspicuous (B).
At autopsy, there was bilateral pneumonia and atrophy of the testes. Although numerous systemic organs microscopically contained small, amorphously grouped, nongranulomatous collections of histiocytes, these small collections had been overlooked by the general autopsy pathologist. Indeed, only after neuropathological examination of the brain yielded a postmortem diagnosis of ECD did rereview confirm the findings of ECD in the lung, testis, parathyroid capsule, bone, adventitia of coronary arteries, and very focally in the kidney, the latter with a rare Touton giant cell (Fig. 3A–F). As is typical of ECD, the histiocytes showed cytologically bland nuclei, inconspicuous nucleoli, and quite varying degrees of foamy (Fig. 3C) versus eosinophilic cytoplasm (Fig. 3D). Admixed lymphocytes and plasma cells were also variable in number, but in some sites, they were more pronounced than the histiocytes (Fig. 3E).
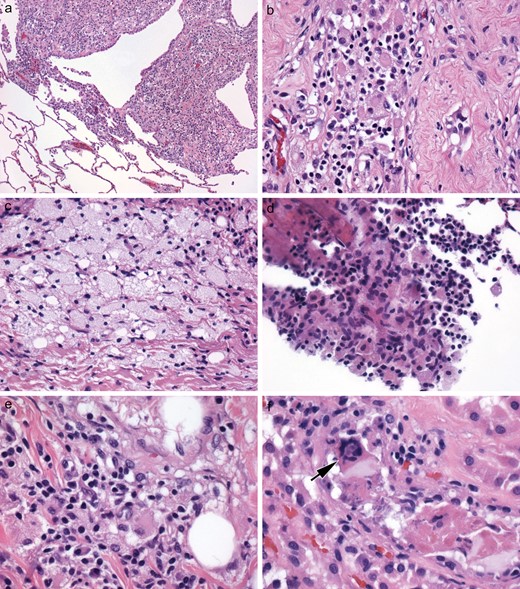
Case 1. (A–F) The diagnosis of ECD was made only at autopsy, based on the much more striking brain findings than in the biopsy. Interestingly, histiocytic collections in systemic organs had been overlooked by the general pathologist but on retrospective review, non-CNS organs were clearly affected by ECD, yielding small multifocal nodular histiocytic collections in the lung (A), histiocytes with eosinophilic cytoplasm in the testis (B), and foamy histiocytes in the parathyroid (C). Bone marrow was focally involved, even if not clinically apparent (D); and in a few locations, the numbers of other types of mononuclear inflammatory cells exceeded that of the histiocytes, as in the kidney (E). Touton giant cells (arrow), a characteristic finding in ECD when present, were quite rare; this one was identified in the kidney, however (F).
The most striking macroscopic finding was in the brain. The cerebellum manifested bright yellow lesions measuring 1.0 cm in greatest diameter, involving the dentate nuclei of the cerebellum bilaterally (Fig. 4). Although Rosenthal fibers were still prominent, microscopic examination of the brain and spinal cord at autopsy demonstrated massive lipogranulomatous disease. Maximally affected areas included the central portion of the cerebellar white matter near the dentate nucleus bilaterally (Fig. 5A–D), hypothalamus (Fig. 5E), spinal cord (Fig. 5F), and portions of the brainstem. These areas contained cohesive collections of lipid-laden histiocytes both within parenchyma and around blood vessels that were highlighted by periodic acid Schiff stain (Fig. 5A) and CD68 immunohistochemistry (IHC) (Fig. 5D). They were composed of epithelioid histiocytes with small, round, eccentrically placed nuclei and abundant eosinophilic to focally foamy cytoplasm (Fig. 5B, E). In a few areas, diffuse microglial activation was even more prominent than the perivascular CD68 IHC-positive histiocytic collections (Fig. 5F). There was no evidence in the brain of an infectious process or tight small granulomas. No necrosis, multinucleated giant cells or Touton giant cells were found in the CNS, unlike the rare example of the latter in systemic organs (Fig. 5F), although the latter is not essential for the diagnosis of ECD. No cholesterol clefts were identified anywhere in the brain or systemic organs, excluding diagnostic consideration of cerebrotendinous xanthomatosis. IHC for BRAF VE1, performed in retrospect, was positive.
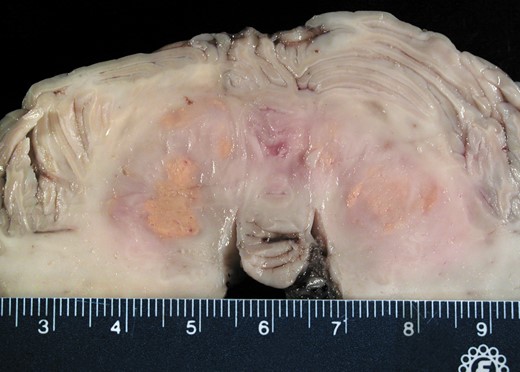
Case 1. The most striking gross finding at autopsy was multifocal bright yellow histiocytic deposits in bilateral cerebellar hemispheres, encompassing the dentate nuclei. Compare with premortem neuroimaging in Figure 1A, C.
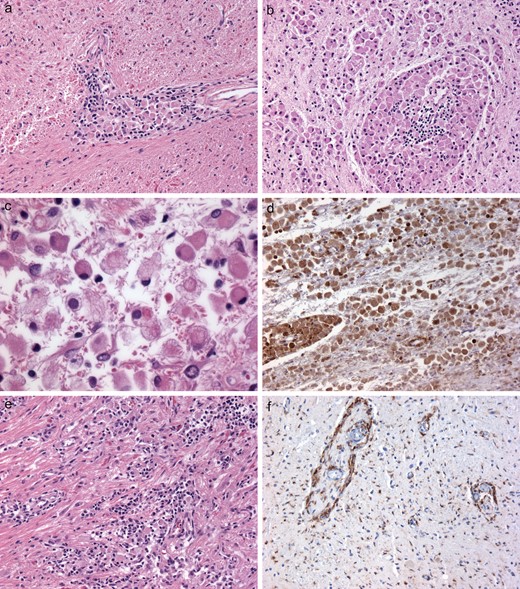
Case 1. (A–F) Cerebellum at autopsy demonstrated numerous Rosenthal fibers in surrounding gliotic brain tissue but there were more prominent perivascular histiocytic collections (A, B), as well as diffuse parenchymal histiocytic infiltrates (B). A range of morphological features was seen within a single aggregate of the ECD histiocytes, with some showing mild foamy cytoplasmic features and others showing completely eosinophilic cytoplasm (C). The rounded cytoplasmic profiles distinguished these from stellate, reactive gemistocytic astrocytes, as did CD68 immunoreactivity (D). Histiocytic collections were identified in the hypothalamus, another frequently involved anatomical CNS site in ECD (E). Although lesions had been identified on premortem imaging in the spinal cord (see Fig. 1B), even at autopsy many areas of involvement were subtle, as seen with anti-CD68 immunohistochemistry (F). As in the premortem biopsy, such areas were clearly abnormal but not fully and specifically diagnostic for ECD without corroborating the lesion with findings in other areas in the same brain (A–E).
Case 2. ECD Diagnosed at Biopsy
A 67-year-old female was found to have an enhancing pontine lesion of unclear etiology, which had been stable for 3 years. She initially experienced diplopia, which was improved with prism glasses; later she developed worsening ataxia, balance deficits, and intermittent dysarthria. On workup, lumbar puncture pressure and cerebrospinal fluid analysis were normal. MRI studies detected a heterogeneous multifocal lesion centered in the pons (Fig. 6A). This case occurred soon after the seminal report on chronic lymphocytic inflammation with pontine perivascular enhancement responsive to steroids (CLIPPERS), and after we had encountered a case of CLIPPERS at our institution seen by the same clinical service (4). The current patient’s condition was, therefore, initially clinically felt to be CLIPPERS. No enhancing lesions were noted elsewhere in the brain.
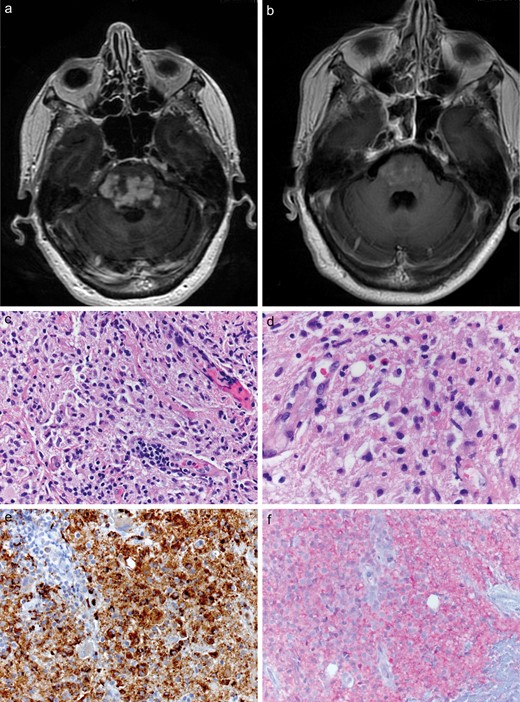
Case 2. (A, B) Axial MRI, T-weighted, with contrast scan(s) detected a heterogeneous expansile lesion in the pons of the patient (A). After vemurafenib treatment there has been a sustained, near-total regression of the pontine lesion, as seen on this MRI scan taken 1 month after start of therapy following diagnosis of ECD made on biopsy (B). (C–F) Biopsy of the pontine lesion revealed a prominent histiocytic infiltration with scattered lymphocytes and plasma cells (C). Histiocytes had no overt cytologic atypia and abundant pale, nonfoamy cytoplasm (D), CD68 immunoreactivity (E), and diffuse cytoplasmic positivity to anti-BRAF VE1 antibody (F).
The patient underwent biopsies of the pontine lesion. Histologically, there was a prominent histiocytic infiltrate in a background of reactive lymphocytes and plasma cells (Fig. 6C). The large histiocytes showed round to ovoid nuclei with fine chromatin, occasional small nucleoli, and abundant pale cytoplasm (Fig. 6D). No microorganisms, hemophagocytosis, or emperipolesis was seen. Histiocytes revealed no significant cytologic atypia, increased mitotic rate, or areas of granuloma formation or necrosis. Histiocytes were immunopositive for CD68 (Fig. 6E), but negative for S100 and CD1a. Notably, the lesional cells had a diffuse cytoplasmic staining for BRAF VE1 (Fig. 6F), an antibody reactive to the mutant protein, BRAFV600E. Molecular assays with polymerase chain reaction and subsequent sequencing detected BRAFV600E mutation in this case. Although BRAF mutation is not essential for diagnosis (see below), when identified in conjunction with the correct clinicopathological features as in the case, the diagnosis of ECD is solidified.
The patient was further evaluated with positron emission tomography–computed tomography and was found to have no evidence of systemic disease, including perirenal soft tissues or suspicious bone involvement. She was treated with vemurafenib and had a marked response 1 month later with dramatic decrease of the lesion in the pons (Fig. 6B, placed immediately adjacent to the initial pretreatment scan). She has remained disease free on the drug for 3 years.
Case 3. Patient With Clinical Features of ECD Including Bone Involvement but Histological Features in Dura of RDD
A 46-year-old female experienced the onset of headaches associated with facial tingling on the left cheek in 2009. Headaches were progressive, with gradual loss of sensation of taste on the left side of her tongue and mouth. MRI scan of the brain showed an enhancing mass in the left middle cranial fossa, left cerebellopontine angle, and hyperostosis of the petrous apex. Further and systemic studies then revealed axillary and right inguinal lymph nodes, and soft tissue nodules in the low anterior pelvis, concerning for metastases.
Excisional biopsies of the left trigeminal nerve and mass in 2009 demonstrated massive expansion and distortion of the ganglion by a mixed inflammatory infiltrate in which reactive lymphocytes and plasma cells were just as numerous as foamy histiocytes. Multinucleated or Touton giant cells were not found, nor were there tight granulomas. There was no molecular evidence for a monoclonal T or B cell neoplasm (i.e. no neurolymphomatosis) and the diagnosis was descriptive only. A bone biopsy was performed soon thereafter in 2009 and this demonstrated small collections of foamy histiocytes. A bone scan was performed in 2010 (Fig. 7A); ECD was suspected given the histiocytic features in the bone biopsy combined with positive bone scans, particularly the increased bilateral uptake in the distal femur, proximal tibia, and humeral head (Fig. 7A).
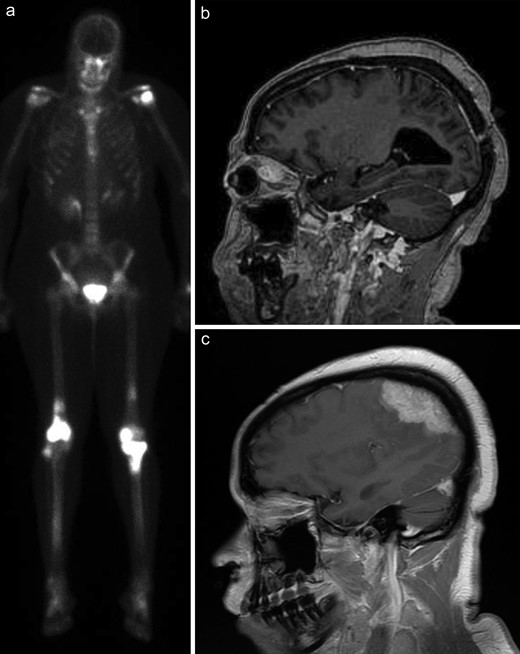
Case 3. (A) One year after presentation, the patient underwent nuclear medicine bone scans that showed bilateral increased uptake in the distal femur, proximal tibia, and humeral head. (B) Five years later, she developed eye fullness and visual changes that prompted an MRI scan (sagittal, T-weighted, with contrast), which demonstrated a left intraconal mass intimately associated with the ocular musculature. (C) Six years later, new neurological symptoms prompted another MRI scan that showed an enhancing convexity dural mass.
The patient initially had a clinical response to Gleevec, but then showed persistent and progressive disease. She then developed left eye fullness, vision changes and diplopia, and imaging in 2014 demonstrated a left intraconal mass, 13 × 15 × 15 cm. That displaced the lateral rectus muscle and optic nerve sheath complex, without definite signal abnormalities or enhancement in the optic nerve (Fig. 7B). This mass was not approached surgically.
In 2015, she developed more neurological symptoms, resulting in imaging studies performed at an outside hospital that showed a new, massive dural lesion over the convexity (Fig. 7C). An excision of this dural mass was performed at the outside local hospital 7 years after the initial presentation. The resection specimen was referred to our institution for consultative diagnosis.
Previous bone and trigeminal biopsies were retrieved for comparison and showed a range of morphological features (Fig. 8A–D). The bone biopsy contained very small nests of foamy histiocytes with nuclei showing indistinct nucleoli (Fig. 8A), whereas the left trigeminal nerve and ganglion biopsy showed fewer histiocytes, albeit also with relatively indistinct nucleoli and no emperipolesis; lymphocytes and plasma cells were more conspicuous (Fig. 8B). Morphologically, the new meningeal lesion manifested placode-like to bulbous expansion of dura and contained sheets of histiocytes with slightly different cytological features than previous biopsies. Notably, there were sheets of abnormal histiocytes that had enlarged nuclei with fine chromatin and frequent large nucleoli. The cytoplasm was abundant and often dense pale or pink staining with frequent viable intracytoplasmic lymphocytes and plasma cells, so called “emperipolesis” (Fig. 8C). A subset of histiocytes also showed foamy cytoplasm. The histiocytes were diffusely immunopositive for CD68 and S100 (Fig. 8D) on both trigeminal and dural biopsies but were negative for CD1a and BRAF VE1. Molecular studies on the dural mass detected no significant gene mutations, including BRAF, NRAS, MAP2K1, and PIK3CA. Overall, the CNS disease had overlapping clinicopathologic features of RDD and ECD, although the presence of cells with large nucleoli was thought to be archetypal for the former.
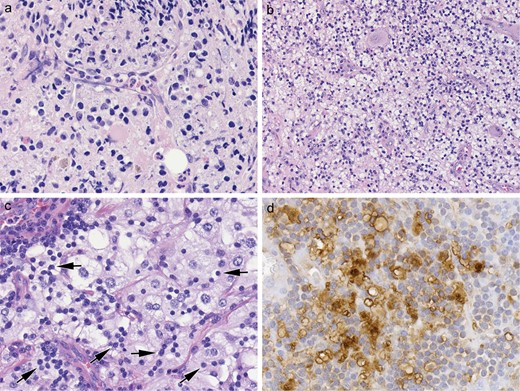
Case 3. (A–C) Over the course of her disease, 3 anatomical sites have had biopsy/excisional material: bone (A), left trigeminal nerve and ganglion (B), and dura (C). Side-by-side comparison of the histiocytic features from these 3 sites shows an ECD to RDD spectrum. The bone (A) and the left trigeminal nerve and ganglion (B) contained foamy histiocytes but had no apparent emperipolesis, consistent with ECD; histiocytes were similar in the trigeminal biopsy but accompanying nonneoplastic lymphocytes and plasma cells were more conspicuous (B). In contrast, the dural mass showed histiocytes with frequent emperipolesis (C, arrows) and histiocytes manifested more open chromatin pattern, larger nuclei and more distinct nucleoli (C). The histiocytes were diffusely positive for S100 both on the trigeminal biopsy (D) and the dural mass, with negative immunostaining for CD1a and BRAF VE1 on the latter.
The patient is still alive with slowly enlarging brain lesions in multiple sites on recent MRI imaging. Recent clinical laboratory studies showed normal serum interleukin-6 and IgG4 levels.
DISCUSSION
Based on the presence of BRAFV600E and other related mutations, ECD, along with LCH, is recognized today to be a true neoplastic, rather than inflammatory disorder (5). Haroche et al have long reported the overlapping clinical features seen in some patients. This is an interesting observation, given the fact that ECD and LCH, but not RDD, share similar genetic underpinnings. Case 3 in this series underscores the clinical and even histologic overlap between ECD and RDD that can occur. The most frequent overlap, however, is between ECD and LCH, with some cases initially showing LCH and eventuating in ECD (6). LCH is predominantly a disorder of childhood whereas most ECD patients are middle-aged to elderly, ranging from 40 to 80 years with a mean age of 55 years. ECD shows a strong male predilection, with a male to female ratio of 3:1 (3).
Like other histiocytoses, ECD can virtually affect every organ system. The resultant variable clinical presentations depend upon the extent and distribution of the disease. Skeletal involvement is nearly always present and eventually develops in approximately 96% of ECD cases with characteristic bilateral osteosclerotic lesions of long bones in the lower extremities. Generally, the clinical features are chronic, and sometimes of sufficiently long duration that ECD is not in the differential considerations. The major extraskeletal manifestations include neurological symptoms, diabetes insipidus, cardiovascular involvement with circumferential thickening of the aorta (“coated aorta”), and retroperitoneal fibrosis (“hairy kidney”). It is when patients present with prominent neurological symptoms that overshadow or predate the systemic signs and symptoms that diagnostic confusion often arises.
CNS involvement is present in up to 50% of ECD cases and less than one third of ECD cases initially present with neurological symptoms (7–9). The major CNS manifestations are cerebellar and pyramidal syndromes, with central diabetes insipidus as the most common CNS manifestation, reported in approximately 25% of patients. Other symptoms include seizures, headaches, neuropsychiatric manifestations, cognitive impairment, sensory disturbances, and cranial nerve paralysis. The CNS symptoms may last for several years before the diagnosis, as noted in our Cases 1 and 2. In the French series, over a decade of unexplained diabetes insipidus was present in some patients before other symptoms developed that led to ECD diagnosis (10). CNS involvement is an independent, adverse prognostic factor of ECD (3). Due to the high prevalence of CNS involvement, all new ECD patients should be systematically examined on the brain with imaging studies, even if they are asymptomatic.
CNS ECD may occur throughout the neuraxis, with both intraaxial and extraaxial infiltrations. Pachymeninges-based lesions can be expansile and gadolinium-enhancing along the cerebral hemispheres or in the cerebellar tentorium, which can mimic meningioma, granulomatous disease, meningeal RDD, and LCH (11, 12). Intraaxially, ECD most commonly involves the cerebellar dentate nuclei and the pons (12), causing cerebellar symptoms (i.e. ataxia and dysarthria) and brainstem symptoms, which may resemble metastatic tumor, demyelinating disease, and/or inflammatory processes. Unilateral or bilateral infiltration of the orbits occurs in approximately 25% of patients (13) and typically shows bilateral intraconal masses (Fig. 7B), a hypointense signal on both T1- and T2-weighted images, and intense enhancement on gadolinium-enhanced T1-weighted images. Patients can clinically present with exophthalmos, retro-orbital pain, oculomotor palsies, or blindness, resembling Graves’ disease, lymphoma, IgG4-related sclerosing disease, and granulomatous disease.
Radiographic Studies
Bone involvement is present in almost all ECD patients and represents the most common clinical feature. Plain radiograph shows bilateral, symmetric cortical osteosclerosis of the diaphyseal and/or metaphyseal regions of the distal ends of lower extremities, which can be highlighted by the sensitive bone scintigraphy using technetium Tc 99 m with symmetric uptakes in the long bones (Fig. 7A).
In the brain, the hypothalamic-pituitary axis is most frequently involved, and the high signal intensity of the posterior pituitary lobe is normally seen on T1-weighted MR images (Fig. 1C) (9). Meningeal involvement occurs in 23% of patients, presenting as single or multiple dural masses or diffuse pachymeningeal thickening. The intraaxial involvement shows one or multiple mass lesions in the supratentorial or infratentorial area (Fig. 1A). These meningeal or intraaxial lesions typically reveal an iso- or hypointense signal on T2-weighted images, and intense homogeneous enhancement on gadolinium-enhanced T1-weighted images. ECD in the dentate nucleus demonstrates bilateral symmetric high signal intensity areas on T2-weighted MR images and corresponding low signal intensity on T1-weighted images.
Clinical Features
Clinical features have been reviewed by Parisi et al (14), with symptoms referable to this posterior fossa/cerebellar predilection, including central diabetes insipidus, cerebellar syndromes, orbital lesions, and extraaxial masses involving the dura. When only the brainstem is involved, the differential diagnosis may be broader, as illustrated by Case 2.
Histomorphologic Features
The diagnosis of ECD is made based upon the appropriate clinical and radiologic context, characteristic morphologic features, and necessary immunohistochemical and molecular studies (1). Lesional tissue typically contains foamy or lipid-laden macrophages with mixed fibrosis and frequent plasma cells in the background. The histiocytes are often organized into polymorphic xanthogranulomatous nests; and they have round nuclei and abundant foamy cytoplasm with no apparent cytologic atypia or increase in mitosis or apoptosis. Touton giant cells are often, but not invariably, present (e.g. Fig. 3F). As illustrated by our Case 1, the variability of foamy versus pale eosinophilic cytoplasm and presence of Touton giant cells is best appreciated on cases that come to autopsy. Difficulty with diagnosis on small biopsy or suboptimally targeted biopsies is also underscored by Case 1.
By immunohistochemical staining, ECD histiocytes are positive for CD68, CD163, and factor XIIIa, whereas immunostains for CD1a, S100, and Langerin (CD207) are negative. Immunohistochemical staining on paraffin sections with the BRAFV600E mutant-specific antibody (VE1) can be used to assist in the diagnosis of ECD, although it must be emphasized that absence of mutation does not exclude ECD.
Molecular Studies
BRAFV600E mutation is detected in about 55%–60% of ECD cases (15–20). BRAF is a serine/threonine protein kinase, with a crucial role in the regulation of cell proliferation and survival, as it contributes to the RAS/RAF-MEK-ERK protein kinase pathway. The mutation causes the amino acid substitution of glutamic acid for valine at position 600 of the BRAF protein (V600E). Recently, it has been shown that ECD patients treated with vemurafenib, a selective BRAFV600E inhibitor, had dramatic clinical and radiographic improvement (21, 22), as observed in our Case 2. In ECD patients with wild type BRAF, 10.9% and 3.7% of patients have mutations in PIK3CA and NRAS, respectively. PIK3CA and NRAS are both implicated in MAPK activation pathway which further highlights the importance of MAPK signaling to ECD pathogenesis (23, 24).
Differential Diagnosis
CNS ECD must be distinguished from reactive and neoplastic processes with a histiocytic component, particularly extranodal RDD and LCH (Table). Like CNS ECD, CNS RDD usually contains histiocytic infiltration, prominent fibrosis, and abundant plasma cells in the tissue (Fig. 6C, D). However, the histiocytes in RDD are usually larger and have a large nucleus with a prominent nucleolus and mild cytologic atypia (Fig. 8C). The cytoplasm is typically homogeneous pale or pink staining with characteristic emperipolesis, the presence of viable intracytoplasmic lymphocytes, plasma cells, and/or neutrophils, although it may not be as obvious as the nodal disease. Immunostaining for S100 is positive in RDD but not ECD.
Differential Diagnosis of Erdheim–Chester Disease with Langerhans Cell Histiocytosis and Rosai–Dorfman Disease
| Erdheim–Chester Disease | Langerhans Cell Histiocytosis | Rosai–Dorfman Disease | |
|---|---|---|---|
| Locations in CNS | Cerebellum, pons, dura, brain parenchyma | Cerebellum, pons, dura, brain parenchyma | Dura |
| Morphology | |||
| Nucleus | Round | Epithelioid, frequent grooves | Often enlarged with mild atypia |
| Nucleolus | Small | Variable | Variable. May be prominent |
| Cytoplasm | Abundant, foamy, or lipid-laden | Moderate, pink | Abundant, dense pale or pink, frequent emperipolesis |
| Mitosis/necrosis | None | May be present | None |
| Other features | Touton cells | Occasional osteoclast-like giant cells | |
| Background | Fibrosis, plasma cells | Abundant eosinophils | Fibrosis and plasma cells |
| Immunophenotype | |||
| CD1a | − | + | − |
| CD68 | + | + | + |
| CD207 (langerin) | − | + | − |
| Factor XIIIa | + | − | − |
| S100 | − | + | − |
| VE1 | +* | +* | − |
| BRAF V600E | 55%–60% | ∼64% | − |
| Other Mutations |
| MAP2K1 12%–27.5% (mutually exclusive to BRAF V600E) |
|
| Erdheim–Chester Disease | Langerhans Cell Histiocytosis | Rosai–Dorfman Disease | |
|---|---|---|---|
| Locations in CNS | Cerebellum, pons, dura, brain parenchyma | Cerebellum, pons, dura, brain parenchyma | Dura |
| Morphology | |||
| Nucleus | Round | Epithelioid, frequent grooves | Often enlarged with mild atypia |
| Nucleolus | Small | Variable | Variable. May be prominent |
| Cytoplasm | Abundant, foamy, or lipid-laden | Moderate, pink | Abundant, dense pale or pink, frequent emperipolesis |
| Mitosis/necrosis | None | May be present | None |
| Other features | Touton cells | Occasional osteoclast-like giant cells | |
| Background | Fibrosis, plasma cells | Abundant eosinophils | Fibrosis and plasma cells |
| Immunophenotype | |||
| CD1a | − | + | − |
| CD68 | + | + | + |
| CD207 (langerin) | − | + | − |
| Factor XIIIa | + | − | − |
| S100 | − | + | − |
| VE1 | +* | +* | − |
| BRAF V600E | 55%–60% | ∼64% | − |
| Other Mutations |
| MAP2K1 12%–27.5% (mutually exclusive to BRAF V600E) |
|
Positive in the cases with BRAF V600E mutation.
Differential Diagnosis of Erdheim–Chester Disease with Langerhans Cell Histiocytosis and Rosai–Dorfman Disease
| Erdheim–Chester Disease | Langerhans Cell Histiocytosis | Rosai–Dorfman Disease | |
|---|---|---|---|
| Locations in CNS | Cerebellum, pons, dura, brain parenchyma | Cerebellum, pons, dura, brain parenchyma | Dura |
| Morphology | |||
| Nucleus | Round | Epithelioid, frequent grooves | Often enlarged with mild atypia |
| Nucleolus | Small | Variable | Variable. May be prominent |
| Cytoplasm | Abundant, foamy, or lipid-laden | Moderate, pink | Abundant, dense pale or pink, frequent emperipolesis |
| Mitosis/necrosis | None | May be present | None |
| Other features | Touton cells | Occasional osteoclast-like giant cells | |
| Background | Fibrosis, plasma cells | Abundant eosinophils | Fibrosis and plasma cells |
| Immunophenotype | |||
| CD1a | − | + | − |
| CD68 | + | + | + |
| CD207 (langerin) | − | + | − |
| Factor XIIIa | + | − | − |
| S100 | − | + | − |
| VE1 | +* | +* | − |
| BRAF V600E | 55%–60% | ∼64% | − |
| Other Mutations |
| MAP2K1 12%–27.5% (mutually exclusive to BRAF V600E) |
|
| Erdheim–Chester Disease | Langerhans Cell Histiocytosis | Rosai–Dorfman Disease | |
|---|---|---|---|
| Locations in CNS | Cerebellum, pons, dura, brain parenchyma | Cerebellum, pons, dura, brain parenchyma | Dura |
| Morphology | |||
| Nucleus | Round | Epithelioid, frequent grooves | Often enlarged with mild atypia |
| Nucleolus | Small | Variable | Variable. May be prominent |
| Cytoplasm | Abundant, foamy, or lipid-laden | Moderate, pink | Abundant, dense pale or pink, frequent emperipolesis |
| Mitosis/necrosis | None | May be present | None |
| Other features | Touton cells | Occasional osteoclast-like giant cells | |
| Background | Fibrosis, plasma cells | Abundant eosinophils | Fibrosis and plasma cells |
| Immunophenotype | |||
| CD1a | − | + | − |
| CD68 | + | + | + |
| CD207 (langerin) | − | + | − |
| Factor XIIIa | + | − | − |
| S100 | − | + | − |
| VE1 | +* | +* | − |
| BRAF V600E | 55%–60% | ∼64% | − |
| Other Mutations |
| MAP2K1 12%–27.5% (mutually exclusive to BRAF V600E) |
|
Positive in the cases with BRAF V600E mutation.
CNS LCH has similar distribution and radiographic appearances to CNS ECD, although ECD is often expansile in meninges, and spinal cord is often spared in LCH. Both lesions may share the features of diabetes insipidus. Morphologically, LCH cells are epithelioid with obvious cytologic atypia and frequent nuclear grooves. Abundant background eosinophils, increased mitoses and apoptotic bodies, and areas of necrosis are commonly seen in LCH. LCH cells express CD1a, S100 and Langerin. Despite these differences mentioned above, both ECD and LCH commonly have a BRAFV600E mutation; and 12%–19% of ECD cases have concurrent LCH, suggesting that they are clonally related in some cases (6, 25).
IgG4-related sclerosing disease may involve the CNS and mostly manifests as hypertrophic pachymeningitis and hypophysitis with infrequent direct parenchymal brain involvement (26). Histologically, IgG4-related disease contains prominent fibrosis and lymphoplasmacytic infiltration, which is like ECD, whereas features of foamy histiocytes and xanthogranuloma are generally absent in IgG4-related disease. However, immunostaining for IgG4 may be considered in the initial workup if no apparent etiology is uncovered.
Prominent histiocytic and/or granulomatous reaction is frequently seen in the brain following infection and necrosis due to infarction or hemorrhage, which may resemble ECD; therefore, essential clinical examinations, careful morphologic evaluation, and necessary ancillary studies are required for a correct diagnosis.
Finally, sellar region histiocytic/xanthomatous lesions secondary to ruptured Rathke cleft cyst, leakage of adamantinomatous craniopharyngioma cyst contents, or uncertain causes must be distinguished from histiocytic disorders that affect the sellar region/pituitary stalk (27).
Management and Therapy
Many different therapeutic approaches for ECD have been explored so far; these include steroids, cytotoxic drugs, and autologous hematopoietic stem cell transplant, but the clinical efficacy is limited. Interferon-α (IFN-α) became the first-line drug in the management of symptomatic ECD, and the therapeutic modality with the largest amount of supporting evidence in ECD is IFN-α and pegylated IFN-α (28). Treatment with the recombinant interleukin 1-receptor antagonist anakinra has been attempted and found effective in some patients (29). The discovery of BRAFV600E mutation in ECD has led to the very promising treatment with vemurafenib, an inhibitor of the BRAFV600E mutation, with dramatic and unprecedented clinical and radiographic responses (21, 30). Although anecdotal, the response seen in Case 2 in this series underscores that unlike targeted therapies for some conditions, the response appears to be better sustained in BRAF-mutated ECD patients treated with targeted drug (21, 22, 31–33).
Conclusion
In summary, for a brain lesion with prominent histiocytic proliferation, an initial workup is necessary to rule out reactive processes based on the clinical presentations and morphologic features. For persistent and progressive lesions, special considerations include ECD, LCH, and RDD, and further immunohistochemical stains can be utilized to separate these entities. Additional radiologic examinations and molecular studies may be required to confirm the diagnosis of CNS ECD and open the possibility of targeted therapy treatments. Take home messages include the fact that the neoplastic histiocytes may be quite variable in number and cytoplasmic content, and that the hypothalamic/pituitary and posterior fossa are epicenters of involvement in the CNS, although certainly disease can extend beyond these sites. Diabetes insipidus may antedate other symptoms by years and although ECD presenting with neurological, not systemic organ clinical manifestations, is the most challenging to diagnose, initial CNS presentation is certainly not rare and must be recognized. Overlap between ECD and especially LCH has been better appreciated by clinician experts in histiocytic disorders than by pathologists, but occasional morphological overlap also occurs.
ACKNOWLEDGMENTS
The authors thank Ms. Lisa Litzenberger for photographic expertise and Mrs. Diane Hutchinson for manuscript preparation.
REFERENCES
Author notes
The authors declare no specific funding.
The authors have no duality or conflicts of interest to declare.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}