





Abstract
Paracoccidioides brasiliensis is the etiologic agent of paracoccidioidomycosis, a disease confined to Latin America and of marked importance in the endemic areas due to its frequency and severity. This species is considered to be clonal according to mycological criteria and has been shown to vary in virulence. To characterize natural genetic variation and reproductive mode in this fungus, we analyzed P. brasiliensis phylogenetically in search of cryptic species and possible recombination using concordance and nondiscordance of gene genealogies with respect to phylogenies of eight regions in five nuclear loci. Our data indicate that this fungus consists of at least three distinct, previously unrecognized species: S1 (species 1 with 38 isolates), PS2 (phylogenetic species 2 with six isolates), and PS3 (phylogenetic species 3 with 21 isolates). Genealogies of four of the regions studied strongly supported the PS2 clade, composed of five Brazilian and one Venezuelan isolate. The second clade, PS3, composed solely of 21 Colombian isolates, was strongly supported by the α-tubulin genealogy. The remaining 38 individuals formed S1. Two of the three lineages of P. brasiliensis, S1 and PS2, are sympatric across their range, suggesting barriers to gene flow other than geographic isolation. Our study provides the first evidence for possible sexual reproduction in P. brasiliensis S1, but does not rule it out in the other two species.
Introduction
Paracoccidioides brasiliensis is the agent responsible for paracoccidioidomycosis (PCM), one of the most important human systemic mycosis in Latin America (Restrepo 2003). It is endemic to an area extending from Mexico to Argentina, and it is estimated that at least 10 million people are infected by the fungus (Brummer, Castaneda, and Restrepo 1993). The annual incidence rate in Brazil is 10–30 per million inhabitants, and the mean mortality rate is 1.4 per million per year (Restrepo 2003). Paracoccidioides brasiliensis was described by Almeida in 1930 (Franco et al. 1994) and has been considered to be a single species since that time. As is the case with many other human pathogenic fungi, no sexual stage (teleomorph) has been reported. This fungus has shown extensive genetic variability when analyzed by molecular tools such as random amplified polymorphic DNA, restriction fragment length polymorphism (Soares et al. 1995; Nino-Vega et al. 2000), and electrophoretic karyotyping (Montoya et al. 1997).
Defining species boundaries is especially difficult in fungi. The parameters most often utilized are those involving morphological characters and, less commonly, the ability to produce fertile offspring (Futuyma 1998). Unfortunately, in fungi, those morphological characters that differentiate species are not easy to find fungal species may become genetically isolated before the development of such features (Taylor, Jacobson, and Fisher 1999). It has been estimated that the use of phenotype as the sole parameter defining fungal species underestimates their number by a factor of at least two when compared to species recognized by phylogenetics (Koufopanou et al. 2001). Additionally, not all fungi can be cultured or mated in vitro. It is known that at least one-fifth of described fungi have no known sexual stage (Geiser, Pitt, and Taylor 1998) and, consequently, performing successful crosses is not possible. Yet, inability to demonstrate the sexual stage does not mean that it does not exist. In several cases, sexual reproduction has been demonstrated from crosses of fungi thought to be asexual (McDonough and Lewis 1967; Kwon-Chung 1972; Hull, Raisner, and Johnson 2000; B. B. Magee and P. T. Magee 2000) and recombination has been detected from population studies using nucleic acid markers (Burt et al. 1996; Geiser, Pitt, and Taylor 1998). While it is possible that some species lack a sexual stage and have evolved other strategies to overcome the resulting deficit of genetic recombination (Lynch et al. 1993; Butcher 1995), it is difficult to conclusively demonstrate such an absence (Leslie and Klein 1996; Sanders 1999; Pawlowska and Taylor 2004). For these reasons, the phylogenetic analysis of molecular markers has been adopted as a technique for species recognition and determination of reproductive mode (Tibayrenc 1996).
In this study, we have applied a phylogenetic approach to study cryptic speciation and recombination in the fungus P. brasiliensis, using sequences of eight regions of five nuclear loci. Our data indicate that P. brasiliensis consists of at least three distinct and previously unrecognized species, in a manner similar to what has been reported for other onygenalean human pathogenic fungi such as Histoplasma capsulatum (Kasuga et al. 2003) and Coccidioides immitis (Koufopanou, Burt, and Taylor 1997). Our study also provides the first evidence showing that at least one of these species possibly reproduces sexually. These results are particularly interesting because two of the three lineages of P. brasiliensis are sympatric across their range, suggesting barriers to gene flow other than geographic isolation.
Materials and Methods
Paracoccidioides brasiliensis Isolates and Growth Conditions
Sixty-five P. brasiliensis isolates representing six recognized endemic areas of PCM were studied (Supplementary Material 1 online). The cultures were grown as reported previously (Diez et al. 1999). Total DNA was extracted from the yeast culture with protocols using glass beads (Van Burik et al. 1998) or maceration of frozen cells (Morais et al. 2000).
Polymerase Chain Reaction and Sequencing Conditions
Eight regions from five nuclear coding genes were selected: (1) promoter-exon 1 and (2) exons 2–4 from chitin synthase (CHS2) (Nino-Vega et al. 1998); (3) exon 2 and (4) exon 3 from β-glucan synthase (FKS) (Pereira et al. 2000); (5) exons 2–4 from α-tubulin (Kasuga, White, and Taylor 2002); (6) exons 2–3 from adenyl ribosylation factor (Kasuga, White, and Taylor 2002); and (7) promoter-exon 1 and (8) exon 2 from PbGP43 (Cisalpino et al. 1996; Morais et al. 2000). We used OLIGO 4.0 (National Biosystems, Plymouth, Minn.) to design the oligonucleotide primers (Operon Technologies Inc., Alameda, Calif). The primers and their respective annealing temperatures are presented in the Supplementary Material 2 online. The eight regions were polymerase chain reaction amplified from genomic DNA. The nucleotide sequences were determined with the cyclic reaction termination method using the BigDye Terminator Cycle Sequencing Kits (Applied Biosystems, Foster City, Calif). Sequence data were collected from both strands and examined with Sequence Navigator v. 1.0.1 (Applied Biosystems).
The sequences were aligned manually, and the coding sequences were assigned by visual inspection of each gene region. DNASp v. 4.0.6 (Rozas et al. 2003) was used to estimate fixation index (Fst), nucleotidic polymorphism (π), average pairwise divergence of isolates, and the average pairwise divergence between groups (Dxy) (Nei 1987).
Phylogenetic Reconstruction
Maximum-parsimony and statistical-parsimony genealogies were constructed manually and confirmed using PAUP* (Swofford 2002) and TCS v. 1.13 (Clement, Posada, and Crandall 2000), respectively. No weighting was introduced in the single-locus analysis and all character state transformations were treated as unordered. For the combined data, the weight assigned to each nucleotide of a particular region was inversely proportional to the total number of phylogenetically informative sites at that region. In this way, the signal of highly polymorphic regions did not obscure the signal from the less polymorphic ones; incongruence between regions therefore resulted in a loss of resolution on the resulting phylogeny. All gaps were treated as missing data and then coded for their presence or absence.
Bayesian analyses were performed using MrBayes v. 3.0b4 (Ronquist and Huelsenbeck 2003) for each region and for the combined data. The best-fitted evolutionary model was estimated using MrModeltest v. 1.01 (Nylander 2002). Each run consisted of three incrementally heated Markov chains with heating values set to default (0.2). Markov chains were initialized from a random tree and were run for 1,000,000s of generations. All analyses were performed twice to check for entrapment at local maxima. To assess confidence of phylogenetic analyses, maximum-parsimony bootstrap (MPBB) (Hillis and Bull 1993) values were calculated using PAUP* and Bayesian posterior probabilities (BPPs) (Rannala and Yang 1996) were estimated using MrBayes (Ronquist and Huelsenbeck 2003).
Detection of Recombination
In the absence of recombination, all gene trees should be compatible and alleles at different gene regions should be associated. The observed tree lengths for the combined data set were compared with the minimum possible length (the sum of tree lengths for all individual regions) in order to identify possible homoplasies within trees and incompatibilities among them (Burt et al. 1996; Koufopanou, Burt, and Taylor 1997). Gene trees were visually inspected to find incongruent pairs of trees. The partition homogeneity test inconguence length difference test (PHT/ILD) (Farris et al. 1995) was performed with the combined alignment (heuristic searches with 100,000 replications) in order to detect any conflict between the regions using P < 0.001 as the threshold to reject the null hypothesis of compatibility, following Cunningham (1997).
In addition, the split decomposition method (Bandelt and Dress 1992) was used to visualize incompatibility generated by recombination (SplitsTree v. 4b06) (Huson 1998) from pairwise genetic distances estimated in PAUP* using the Kimura three-parameter model (Kimura 1981).
Heterogeneity rates can mimic the effects of recombination (Hagelberg 2003), so the informative sites test (IST) was used to compare the proportion of observed informative sites to the proportion found in 1,000 simulated data sets obtained by observation of evolutionary parameters and clonality constraint (Worobey 2001). IST was implemented using the program PIST (http://evolve.zoo.ox.ac.uk/software/PIST/PIST.html) and maximum likelihood estimates of tree topologies and model parameters obtained previously with PAUP* and Modeltest v. 3.06 (Posada and Crandall 1998), respectively.
Association among alleles, as expected under clonality, was investigated using the index of association (IA) (Avise and Wollenberg 1997). Alleles at each region were coded by picking the polymorphic nucleotide with the most balanced distribution of alleles in order to make a multilocus data set for the IA. The regions that came from the same gene were treated as a single locus. The IA was calculated (Maynard Smith et al. 1993) with Multilocus v. 1.3b (Agapow and Burt 2001), either for the full data set or once the data set was reduced to unique haplotypes. Significance was assessed by comparing the original observed data set with those in which alleles had been resampled without replacement to mimic the effects of recombination.
Results
Polymorphism Data
The sequences obtained during this study have been deposited in the GenBank database under the following accession numbers: CHS2 promoter-exon 1 (DQ004114–DQ004178), CHS2 exons 2–4 (DQ004179–DQ004243), FKS exon 2 (DQ003919–DQ003983), FKS exon 3(DQ003854–DQ003918), α-tubulin (DQ003789–DQ003853), adenyl ribosylation factor (DQ004049–DQ004113), PbGP43 promoter-exon 1 (DQ003984–DQ004048), and PbGP43 exon 2 (DQ003724–DQ003788).
Combined data for the eight gene regions tested totaled 3,492 sites, of which 85 were variable (2.43%); of these, 57 were informative and 28 were uninformative (including the only insertion/deletion [indel] found), while 32 represented nonsynonymous substitutions. Only one site had more than two alleles (Supplementary Material 2 online). The most polymorphic gene region was PbGP43 exon 2 (π = 0.00853) and the least polymorphic was FKS exon 2 (π = 0.00014). The only indel found was located in the PbGP43 promoter region of isolate A1, which consisted of 7 bp.
Phylogeny of Multilocus Haplotypes and Identification of Phylogenetic Species
The genealogy of each of the eight gene regions was best described by a single, unrooted most-parsimonious tree (fig. 1). Relationships among the clades were topologically identical in the maximum-parsimony and Bayesian consensus trees; therefore, the maximum-parsimony tree is shown with BPPs given for those well-supported branches (fig. 1). Each tree had a minimal length equal to the number of polymorphic sites in that gene region, showing no evidence of homoplasy.
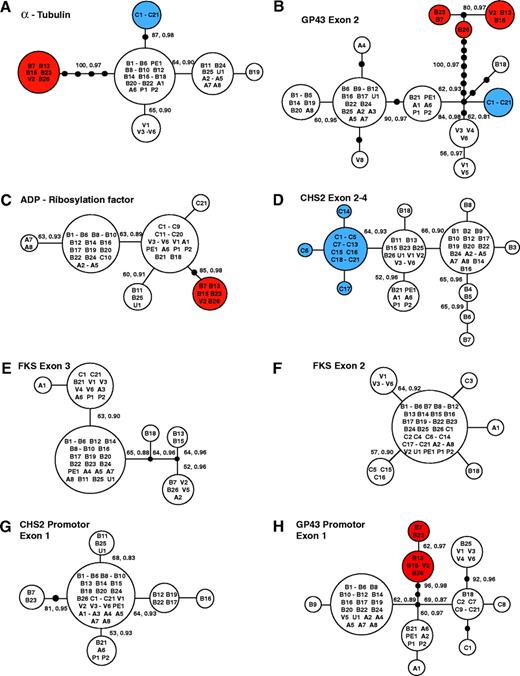
(A–H) Genealogies from fragments of the eight gene regions studied in the 65 isolates of Paracoccidioides brasiliensis. All the parsimony trees were constructed manually and the topologies were confirmed with TCS. All the trees illustrated present a minimum possible length (i.e., no homoplasy) indicating no intralocus recombination. The values along the branches represent their support: the first is the weighted high bootstrap and the second is the posterior probabilities. The dots represent missing haplotypes. Refer to figure 2 for color codes.
Three distinct groups were observed (fig. 2): S1 (species 1), encompasing 38 isolates; PS2 (phylogenetic species 2), having six isolates; and PS3 (phylogenetic species 3), which included 21 isolates. The genealogies of four of the eight regions studied strongly supported the PS2 clade, composed of five Brazilian (from the states of São Paulo and Minas Gerais) and one Venezuelan (from Caracas) isolate. This clade was not present in the genealogies of the other four gene regions (fig. 1). The second clade, PS3, was composed solely of 21 Colombian isolates. This clade was found in three of the topologies and was strongly supported by one genealogy (α-tubulin, fig. 1) with high MPBB and BPP values. The clades in those gene trees that were not congruent with this partition were supported by bootstrap values below 70% or posterior probability values below 0.95, which meant that clades contradicting this group in other genes were not well supported.
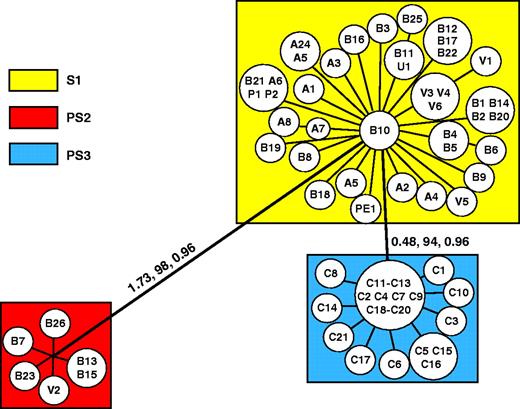
Unrooted tree showing the partitions found in Paracoccidioides brasiliensis, based on combined data obtained with weighted maximum parsimony. The values above the branches represent their individual support: the first is the tree length, the second is the weighted high bootstrap, and the third is the posterior probabilities. The branches with support values below 70% for high bootstrap or 0.95 for posterior probabilities or those that do not satisfy the genealogical concordance or the genealogical nondiscordance criteria are not shown. When the eight gene regions are considered together, the structure of the genealogies collapses into two branches, subdividing the population into three phylogenetic species. The branch lengths are drawn in scale only in the branches that connect the phylogenetic species.
Two other groups exhibited a high degree of support in at least one genealogy and in the total phylogeny: the B11, U1, and B25 clade and the V1 and V3–V6 clade. However, we do not consider either clade to be an independent lineage because their partitions were contradicted by a highly supported clade in the PbGP43 promoter genealogy (fig. 1H).
When the combined data set was evaluated, a tree with 115 steps was found, which had a consistency index (CI) = 0.746 and an homoplasy index (HI) = 0.254. The highest −lnL value obtained for the topology when the Bayesian analyses were performed was −6564.47. The partitions observed in the individual gene genealogies were supported by high weighted bootstrap and posterior probabilities values (fig. 2). Eighteen fixed differences were found between PS2 and S1 and four between PS3 and S1.
The mean divergences of sequences between the three major groups are represented in table 1. The pairwise distances within species were equal to 2.13 × 10−3 (σ = 2.2 × 10−4) for S1, 1.32 × 10−3 (σ = 3.5 × 10−4) for PS2, and 2.22 × 10−3 (σ = 6.0 × 10−5) for PS3. Distances between individuals from different species ranged from 3.3 to 7.7 × 10−3 and the mean, 6.24 × 10−3, was threefold higher than the intraspecific distances (table 1). Similarly, the Fst values, all of them above 0.6, were also consistent with low gene flow and high genetic structure among S1, PS2, and PS3 (table 1).
Fst and Dxy Values for the Pairwise Comparisons of the Three Paracoccidioides brasiliensis Phylogenetic Species
Clade 1 | Clade 2 | Fst | Dxy (×10−3) | Standard Deviation of Dxy (×10−4) |
|---|---|---|---|---|
| S1 | PS2 | 0.76843 | 7.77 | 7.4 |
| S1 | PS3 | 0.63472 | 3.32 | 1.9 |
| PS2 | PS3 | 0.88551 | 7.71 | 9.7 |
Clade 1 | Clade 2 | Fst | Dxy (×10−3) | Standard Deviation of Dxy (×10−4) |
|---|---|---|---|---|
| S1 | PS2 | 0.76843 | 7.77 | 7.4 |
| S1 | PS3 | 0.63472 | 3.32 | 1.9 |
| PS2 | PS3 | 0.88551 | 7.71 | 9.7 |
Fst and Dxy Values for the Pairwise Comparisons of the Three Paracoccidioides brasiliensis Phylogenetic Species
Clade 1 | Clade 2 | Fst | Dxy (×10−3) | Standard Deviation of Dxy (×10−4) |
|---|---|---|---|---|
| S1 | PS2 | 0.76843 | 7.77 | 7.4 |
| S1 | PS3 | 0.63472 | 3.32 | 1.9 |
| PS2 | PS3 | 0.88551 | 7.71 | 9.7 |
Clade 1 | Clade 2 | Fst | Dxy (×10−3) | Standard Deviation of Dxy (×10−4) |
|---|---|---|---|---|
| S1 | PS2 | 0.76843 | 7.77 | 7.4 |
| S1 | PS3 | 0.63472 | 3.32 | 1.9 |
| PS2 | PS3 | 0.88551 | 7.71 | 9.7 |
Evaluation of Panmixis Within the P. brasiliensis Partitions
Although both phylogenies and Fst values support three genetically isolated groups, we challenged this hypothesis by evaluating whether the polymorphism distribution was due to independent evolutionary histories or to random sorting of genetic variation. The probability of there being three or more groups of isolates that, by chance, do not share polymorphisms was calculated by randomly shuffling the gene sequences among isolates, leaving the linkage of sites within each gene region intact (i.e., the regions were randomized as blocks). This procedure was performed within and across the species, which, respectively, correspond to the partitioned and nonpartitioned data sets.
Within the 10,000 randomizations done, no partitions were found that would create groups sharing no polymorphisms. Among the 65 isolates, the shortest trees obtained with the nonpartitioned, randomized data set were significantly longer than the tree obtained with the randomized data set respecting the partitions (the three species) (260 vs. 175 steps, P < 0.0001) (fig. 3).
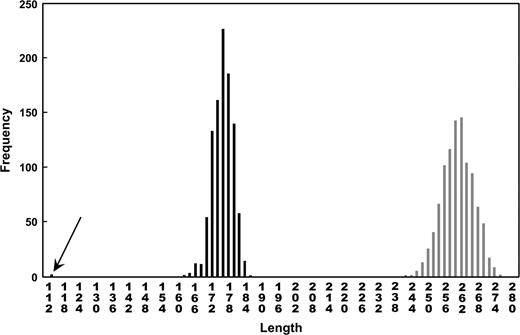
Frequency distributions of the tree lengths of maximum-parsimony trees fitted to randomized data sets (1,000 randomizations in each case). White bars correspond to the tree length distribution when the genotypes are shuffled among the isolates in the entire population. Black bars correspond to the tree length distribution when the genotypes are shuffled among isolates within each of the three groups, respecting the partition. The arrow shows the tree length from the original observed data set.
Presence of Recombination in P. brasiliensis
The 28 possible pairwise comparisons of the eight gene genealogies yielded only six fully compatible pairs (21.43%), all of them corresponding to pairs of regions located in different genes (Supplementary Material 3 online). This result suggested a lack of association among alleles consistent with recombination, even for those that were in the same gene (e.g., CHS2, FKS, and PbGP43). To test for incompatibility among gene genealogies, we compared the sum of the lengths of the most-parsimonious trees of each region to the sum of the length of trees obtained from shuffled data using the PHT/ILD test (Farris et al. 1995). We could reject the null hypothesis of compatibility (P = 0.0001) for all isolates and all gene regions, again consistent with a lack of association among alleles compatible with recombination.
Recombination produces networks of sequences rather than bifurcating evolutionary trees (Fitch 1997), which should be revealed in an analysis of the combined data. The split decomposition analysis (fig. 4) provided evidence for networked relationships and gave graphical support for the recombination hypothesis within S1 (Bandelt and Dress 1992; Holmes, Worobey, and Rambaut 1999).
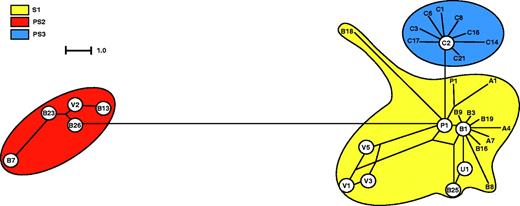
Split decomposition analysis of the Paracoccidioides brasiliensis combined data set. The observation that multiple pathways form an interconnected network linking isolates, rather than a single bifurcating tree, is consistent with recombination. All branch lengths are drawn to scale. C2 represents C2, C4, C7, C9, C10, C11, C12, C13, C18, C19, C20. B1 represents B1, B2, B4 B5, B6, B10, B14, B20, B24, A3, A5. B22 represents B12, B17, B22. B13 represents B15, B13. V3 represents V3, V4, V6. U1 represents B11, U1. P1 represents P1, P2, B21, A6. C5 represents C5, C15, C16.
The amount of polymorphism was sufficient for the IST only in S1; the other species had low levels of polymorphism and very few informative sites (one in the PS3 and three in the PS2). The IST results for S1 had a value of 0.965517 (P < 0.001), showing a significant conflict between informative sites compared to that expected for a clonal species; therefore, the IST result is consistent with recombination in S1.
The recombination detected by the analysis of gene genealogies may not be exclusively intraspecific as several individuals appear to be the result of recombination between species. For example, accessions A2 and V5, which group with S1 in most genes, contain sequences unique to PS2 in the FKS exon 3 (fig. 1E).
We also used the IA to examine recombination in S1 for all genotypes and for the reduced-by-haplotypes data set (Maynard Smith et al. 1993). The null hypothesis that assumes a lack of association could be rejected for all genotypes (IA = 1.312, P < 0.001) and for only unique genotypes and the resulting fewer informative gene regions (IA = 1.292, P < 0.001).
Finally, we evaluated the tree length obtained from randomized data when the regions were randomized within S1, PS2, and PS3, but not among them, to compare the lengths of both observed and randomized trees. The tree length obtained from randomized data was significantly longer than the original tree (175 vs. 115 steps, P < 0.001), indicating that panmixis was not the case within all species.
Discussion
Cryptic Species in P. brasiliensis
Dettman et al. (2003) (Dettman, Jacobson, and Taylor 2003) proposed a simple approach, consisting of fulfilling either of two criteria, to identify independent evolutionary lineages and phylogenetic species from multilocus genealogies. The first criterion, genealogical concordance, stipulates that the clade must be present in the majority of the single-locus genealogies. The second criterion, genealogical nondiscordance, recognizes a clade as an independent evolutionary lineage if it is well supported by at least one single-locus genealogy, as judged by both high bootstrap and posterior probabilities values above 70% and 0.95%, respectively, and if it is not contradicted by any other single-locus genealogy determined by the same methods. Boundaries of fungal species recognized by this approach have been shown to be in good agreement with those identified by mating tests (Dettman et al. 2003).
Avise and Wollenberg (1997) noted that an appropriately constructed phylogenetic species concept is not contrary to the biological species concept and that both concepts, which reflect genetic isolation and reproductive isolation, respectively, should be reconcilable.
Four of the eight studied gene regions here and the total phylogeny analysis strongly support (MPBB > 70%, BPP > 0.95) the existence of an independent evolutionary lineage of P. brasiliensis PS2 comprising five Brazilian (São Paulo and Minas Gerais states) and a Venezuelan isolate. Two regions showed topologies that were incongruent due to the genotypes of B7 and B23. In neither case was the topology supported by high bootstrap values at or above 70% or posterior probabilities at or above 0.95. Therefore, these conflicts are not significant in accordance with the genealogical nondiscordance criterion. In addition, the average pairwise divergence between S1 and PS2 was more than threefold larger than the average pairwise divergence of isolates within the groups. The differentiation of these P. brasiliensis species appears to be of the same magnitude as that found in the C. immitis phylogenetic species (C. immitis sensu stricto and Coccidioides posadasii), although the Paracoccidioides species are in sympatry, while the Coccidioides species overlap geographically only in Baja California, Mexico (Fisher et al. 2002).
A second partition, PS3, was found in three gene regions (figs. 1A, 1B, and 1D) and in the total phylogeny (fig. 2). The support of the partition in the α-tubulin genealogy was strong, and in the five genealogies in which this clade was absent, the support for any incompatible branches was weak. PS3, therefore, is considered to be an independent evolutionary lineage by the criterion of nondiscordance. Although PS3 is not as distinct as PS2, that is, the Dxy value between S1 and PS3 is only 1.5 times larger than the π within the S1, the difference between these two values is significant (P < 0.01). Considering that PS3 is geographically restricted to Colombia, this speciation process may be attributed to dispersal or some other event leading to the genetic isolation of PS3 from S1, which is distributed in Brazil, Argentina, Paraguay, Peru, and Venezuela.
S1 is also considered an independent species, although PS2 and PS3 make it paraphyletic. Species are known to pass through stages of polyphyly and paraphyly on the way to eventual reciprocal monophyly (Avise and Ball 1990) and, thus, it is feasible to find true biological species that are paraphyletic (Rosenberg 2003) and lack the synapomorphies required to define them as a phylogenetic species.
The clade comprising V1, V3, V4, and V6 was not considered to group isolates of an independent phylogenetic species for three reasons: (1) these isolates do not form a well-supported monophyletic group, (2) when they are part of a highly supported group, the latter includes other isolates, such as V5 and B25, and (3) the PbGP43 genealogy presents a significant contradiction.
A special case is the apparent isolation of B18, reflected in four of the genealogies showing that it is different from all the isolates studied. Until more isolates are found that form a monophyletic group with B18, it should not be considered as a separate species.
It is important to remember that the criteria of genealogical concordance and nondiscordance that are used to detect phylogenetic species are strongly dependent on the polymorphism of the sequenced gene region, as are high bootstrap and posterior probabilities measures of significance (Crandall 1994). It also is important to remember that the number of analyzed regions and the degree of polymorphism may bias the genealogical concordance criterion. In light of these admonitions, it is possible that more genetically isolated P. brasiliensis groups may exist.
Variation in virulence and gene expression of P. brasiliensis have previously been found between P. brasiliensis isolates belonging to our S1 and PS2 species (Hebeler-Barbosa, Montenegro, and Bagagli 2003; Carvalho et al. 2005), but their phenotypic differences were not associated with cryptic species. Nevertheless, the two species share host species, humans and armadillos, and both are capable of inducing disease (Carvalho et al. 2005).
Recombination in P. brasiliensis
Several lines of evidence indicate that P. brasiliensis recombines in nature. The first evidence is our observation that phylogenetic trees obtained from the combined data were much longer than the minimum possible (83 vs. 115 steps using all sites) and had significant homoplasy (CI = 0.746, HI = 0.254) despite the fact that individual gene genealogies showed no homoplasy (CI = 1.000, HI = 0.000). The very low divergence and the low proportion of multiple alleles observed for each of the nucleotide sites indicate that homoplasy due to multiple hits is improbable (Crandall 1994) and cannot explain the incompatibly between the topologies. The PHT test told the same story, that is, there was significant conflict among the gene genealogies (P < 0.05) (Cunningham 1997).
The second evidence for recombination comes from split decomposition analysis (fig. 3), which revealed the existence of networked relationships within S1, a result that is not consistent with a totally clonal species. The third type of evidence was found using informative-sites index, which rejected the null hypothesis of clonality (P < 0.001), as has been shown for fungi known to have a sexual stage, for example, Fusarium graminearum (teleomorph = Gibberella zeae) (Ward et al. 2002). The fourth evidence for recombination is the fact that several individuals appear to be the result of recombination between species. The extent of this interspecific recombination is unknown.
Contrary to these results, the IA test rejected the null hypothesis of recombination for S1. A key difference between the IA and IST is that only one polymorphic nucleotide position is used to represent each region in IA, which greatly reduces the number of unique genotypes and may leave too few data for a meaningful test. It is also possible that S1 is not panmictic and that there are genetically differentiated groups within the species. Genotyping of more individuals from S1 may settle the matter. More genotyped individuals also are needed for PS2 and PS3 to begin to address the question of the reproductive mode in these species.
Conclusions
We report here the existence of at least three different phylogenetic species in P. brasiliensis, a taxon formerly considered to be a single clonal species. The results follow the trend of detecting cryptic speciation using concordance and nondiscordance of gene genealogies with respect to phylogenies of nuclear gene sequences, as has been shown in Neurospora (Dettman, Jacobson, and Taylor 2003) and other fungi (Kasuga et al. 2003). In each of the three species within P. brasilensis, either the genealogical nondiscordance rule (Dettman, Jacobson, and Taylor 2003) or the genealogical concordance criterion has been fulfilled, revealing that there are significant barriers to gene flow among these species. Among the three species, some gene flow is occurring as concluded from the hybrid genotypes evidence. Two independent speciation events must have happened in the past to account for the present distribution of the species of P. brasiliensis.
Some of the closest relatives of P. brasiliensis, Blastomyces dermatitidis (Ajellomyces dermatitidis) (McDonough and Lewis 1967), and H. capsulatum (Ajellomyces capsulatus) (Kwon-Chung 1972) have been shown to possess sexual stages; nevertheless, recombination events had never been demonstrated for P. brasiliensis, a microorganism considered to be clonal (Morais et al. 2000). The present findings reveal that recombination is part of P. brasiliensis life cycle.
In general, the comparison of genealogies of single-copy nuclear genes has proven to be a powerful tool to detect recombination and in this case also to measure the extent of its importance in the life history of the P. brasiliensis species. This approach has been used in organisms like viruses and bacteria (Worobey 2001), but in eukaryotes very few cases of recombination have been evaluated from multilocus sequences (Geiser, Pitt, and Taylor 1998; Ward et al. 2002).
Now that cryptic species have been identified, studies are needed concerning the incidence and phenotype of the diseases caused by each of the three species aimed at determining if virulence differs among them. In addition, the type of reproductive behavior of two of the P. brasiliensis species remains veiled. With data from more individuals, we hope to acquire better knowledge about the natural history, epidemiology, and life cycle of P. brasiliensis.
These authors contributed equally to this work.
Laura Katz, Associate Editor
We thank R. Negroni, E. Castañeda, B. Wanke, R. D. Naiff, M. Silva-Vergara, M. S. Ferreira, P. E. Santos, and E. Heins-Vaccari for providing the isolates of P. brasiliensis; T. Kasuga for allowing us to design primers from unpublished sequences; and I. Torres, T. Szaro, A. Caicedo, and T. Pawloska for technical assistance. This work was supported by Fogarty International Research Collaboration Award grant R03TW01308, Comite de Investigaciones de la Universidad de Antioquia grant 2044, and Corporación para Investigaciones Biologicas.
References
Agapow, P., and A. Burt.
Avise, J. C., and R. M. Ball.
Avise, J. C., and K. Wollenberg.
Bandelt, H. J., and A. W. Dress.
Brummer, E., E. Castaneda, and A. Restrepo.
Burt, A., D. A. Carter, G. L. Koenig, T. J. White, and J. W. Taylor.
Carvalho, K. C., L. Ganiko, W. L. Batista, F. V. Moraisa, E. R. Marques, G. H. Goldman, M. F. Franco, and R. Puccia.
Cisalpino, P. S., R. Puccia, L. M. Yamauchi, M. I. Cano, J. F. da Silveira, and L. R. Travassos.
Clement, M., D. Posada, and K. A. Crandall.
Crandall, K. A.
Cunningham, C. W.
Dettman, J. R., D. J. Jacobson, and J. W. Taylor.
Dettman, J. R., D. J. Jacobson, E. Turner, A. Pringle, and J. W. Taylor.
Diez, S., E. A. Garcia, P. A. Pino, S. Botero, G. G. Corredor, L. A. Peralta, J. H. Castano, A. Restrepo, and J. G. McEwen.
Farris, J. S., M. Kallersjo, A. G. Kluge, and C. Bult.
Fisher, M. C., G. L. Koenig, T. J. White, and J. W. Taylor.
Franco, M., C. S. Lacaz, A. Restrepo, and G. Del Negro.
Geiser, D. M., J. I. Pitt, and J. W. Taylor.
Hagelberg, E.
Hebeler-Barbosa, F., M. R. Montenegro, and E. Bagagli.
Hillis, D. M., and J. J. Bull.
Holmes, E. C., M. Worobey, and A. Rambaut.
Hull, C. M., R. M. Raisner, and A. D. Johnson.
Huson, D. H.
Kasuga, T., T. J. White, G. Koenig, J et al. (19 co-authors).
Kasuga, T., T. J. White, and J. W. Taylor.
Kimura, M.
Koufopanou, V., A. Burt, T. Szaro, and J. W. Taylor.
Koufopanou, V., A. Burt, and J. W. Taylor.
Leslie, J. F., and K. K. Klein.
Lynch, M., R. Burger, D. Butcher, and W. Gabriel.
Magee, B. B., and P. T. Magee.
Maynard Smith, J., N. H. Smith, M. O'Rourke, and B. G. Spratt.
McDonough, E. S., and A. L. Lewis.
Montoya, A. E., M. N. Moreno, A. Restrepo, and J. G. McEwen.
Morais, F. V., T. F. Barros, M. K. Fukada, P. S. Cisalpino, and R. Puccia.
Nino-Vega, G. A., E. T. Buurman, G. W. Gooday, G. San-Blas, and N. A. Gow.
Nino-Vega, G. A., A. M. Calcagno, G. San-Blas, F. San-Blas, G. W. Gooday, and N. A. Gow.
Nylander, J. A. A.
Pawlowska, T. E., and J. W. Taylor.
Pereira, M., M. S. Felipe, M. M. Brigido, C. M. Soares, and M. O. Azevedo.
Posada, D., and K. A. Crandall.
Rannala, B., and Z. Yang.
Restrepo, A.
Ronquist, F., and J. P. Huelsenbeck.
Rosenberg, N. A.
Rozas, J., J. C. Sanchez-DelBarrio, X. Messeguer, and R. Rozas.
Soares, C. M., E. E. Madlun, S. P. da Silva, M. Pereira, and M. S. Felipe.
Swofford, D. L.
Taylor, J., D. Jacobson, and M. Fisher.
Tibayrenc, M.
Van Burik, J. A., R. W. Schreckhise, T. C. White, R. A. Bowden, and D. Myerson.
Ward, T. J., J. P. Bielawski, H. C. Kistler, E. Sullivan, and K. O'Donnell.
Author notes
*Corporación para Investigaciones Biológicas, Medellín, Colombia; †Universidad de los Andes, Bogotá, Colombia; ‡Facultad de Medicina, Universidad de Antioquia, Medellín, Colombia; §Departamento de Microbiologia, Imunologia e Parasitologia, Universidade Federal de São Paulo, São Paulo, SP, Brazil; |Mycology Laboratory, Venezuelan Institute for Scientific Research (IVIC), Caracas, Venezuela; ¶Departamento de Microbiologia e Imunologia, Instituto de Biociência, Universidade Estadual Paulista, Botucatu, SP, Brazil; #Laboratorio de Botánica y Sistemática, Universidad de los Andes, Bogotá, Colombia; and **Department of Plant and Microbial Biology, University of California, Berkeley

{kind=link}
{kind=link}
{kind=link}
{kind=link}