





Abstract
Eukaryotic 5′ mRNA cap structures participate to the post-transcriptional control of gene expression before being released by the two main mRNA decay pathways. In the 3′-5′ pathway, the exosome generates free cap dinucleotides (m7GpppN) or capped oligoribonucleotides that are hydrolyzed by the Scavenger Decapping Enzyme (DcpS) forming m7GMP. In the 5′-3′ pathway, the decapping enzyme Dcp2 generates m7GDP. We investigated the fate of m7GDP and m7GpppN produced by RNA decay in extracts and cells. This defined a pathway involving DcpS, NTPs and the nucleoside diphosphate kinase for m7GDP elimination. Interestingly, we identified and characterized in vitro and in vivo a new scavenger decapping enzyme involved in m7GpppN degradation. We show that activities mediating cap elimination identified in yeast are essentially conserved in human. Their alteration may contribute to pathologies, possibly through the interference of cap (di)nucleotide with cellular function.
INTRODUCTION
In eukaryotes, transcripts produced by RNA polymerase II and by some viruses are cotranscriptionally modified by the incorporation of a 5′ cap structure and, in most cases, by addition of a 3′ polyA tail (1,2). The cap structure contains minimally a guanosine methylated on position 7 attached through a 5′-5′ triphosphate linkage to the first residue of the transcript (m7G(5′)ppp(5′)N with N being the first residue of the transcribed RNA). The cap structure can be further modified by addition of other methyl groups on various positions, depending on the species (3–6). The mRNA cap is a distinctive mark that allows cells to recognize bonafide mRNA 5′ end and in some instances to discriminate against transcript emanating from infectious genetic elements. This structure plays an essential role in the post-transcriptional control of gene expression. In the nucleus, it interacts with the cap binding complex and participates to mRNA splicing, 3′ end maturation and nuclear export (7). In the cytoplasm, the cap interacts with the eIF4F complex playing a critical role in translation initiation (7). Finally, the mRNA cap protects transcripts from degradation by blocking access to exonucleases (8–10). mRNA turnover is a tightly regulated process that allows cells to rapidly adapt to environmental changes as well as to eliminate defective transcripts. Bulk mRNA decay usually initiates with the removal of the 3′ polyA tail (11). This rate-limiting step is most often followed by cap removal (a process named decapping) that constitutes the first step of the 5′-3′ mRNA decay pathway. Decapping is mostly performed by Dcp2 after stimulation by accessory factors. This Nudix protein family member hydrolyzes mRNA caps to generate m7GDP and 5′ monophosphorylated RNA (12–14). Additional enzymes have been shown to catalyze cap removal in vitro, including several members of the Nudix protein family such as Nudt16 (15,16) as well as Rai1 and Dxo1 (17,18). However, evidence that such proteins participate to cap removal in cells has so far only been reported for Nudt16, a protein present in metazoan that also produces m7GDP (15). 5′ monophosphorylated RNAs produced by Dcp2 or Nudt16 are exonucleolytically digested by Xrn1 in the 5′ to 3′ direction. Deadenylated mRNAs that are not subject to decapping enters the 3′-5′ mRNA decay pathway where the mRNA body is exonucleolytically degraded by the exosome. When the exosome reaches the mRNA 5′ end, it releases free cap dinucleotides (m7GpppN) and/or capped oligoribonucleotides (19). Aberrant mRNAs are degraded by a variety of pathways that are activated as a consequence of molecular defects or distinctive features present in the transcript (premature termination codon, abasic site, structure impairing translation, absence of in frame stop codon, aberrantly terminated cryptic transcripts etc.) (20–22). While initiated by dedicated preliminary steps, these pathways invariably lead to activation of decapping or 3′-5′ exonucleolytic digestion of the RNA and thus generate m7GDP or m7GpppN. In vitro assays revealed that m7GpppN cap structures resulting from exosome-mediated mRNA degradation were further hydrolyzed in mammalian cell extracts by an activity bored by DcpS (19,23). DcpS is a member of the histidine triad (HIT) superfamily whose members contain conserved histidines and hydrolyze nucleotide derivatives (24,25). Mammalian DcpS, as well as its yeast homolog Dcs1, form homodimers that cleave the m7GpppN dinucleotides, or short capped oligoribonucleotides, between the γ and β phosphates of the cap releasing m7GMP (19,23,26). Several lines of evidence indicate that DcpS also interacts with 5′-3′ mRNA decay. Hence, DcpS was reported to facilitate 5′-3′ mRNA degradation in yeast and 5′-3′ miRNA degradation in Caenorhabditis elegans (27,28), possibly through the stimulation of Xrn1 (28,29). Moreover, m7GDP resulting from decapping by Dcp2 is converted into m7GMP and downstream by-products when incubated in yeast extracts in a process dependent upon Dcs1 (30). The exact role of DcpS in this reaction is however controversial because m7GDP was reported to be either a substrate or a high affinity inhibitor of this enzyme using recombinant proteins from yeast, human or C. elegans (23,30–33). Consistent with a role in the degradation of cap structures released by mRNA decay, Saccharomyces cerevisiae Dcs1 is principally cytoplasmic (31) but surprisingly DcpS accumulates in the nucleus of Schizosaccharomyces pombe and human cells (10,34). The latter observation could suggest that it is mostly involved in degradation of cap structures released by exosome-mediated degradation of capped nuclear RNA such as CUTs (20). Alternatively, m7GpppN dinucleotides generated by cytoplasmic 3′-5′ degradation diffuse to the nucleus before being hydrolyzed by DcpS. Despite its clear activity catalyzing m7GpppN hydrolysis, deletion of the dcs1 gene in yeast does not result in strong growth phenotype (23,30). This suggests that accumulation of m7GpppN dinucleotide in cells is not sufficient to impact on cell growth in laboratory conditions or that an alternate pathway to eliminate m7GpppN dinucleotide exists. However beside Dcs1, only the Gag proteins from the virus-like L-A and L-BC elements were reported to metabolize m7GpppN in yeast (35). The facultative presence of the L-A and L-BC elements argues against a critical role of this mechanism in mediating m7GpppN elimination. DcpS activity appears to be more relevant in higher eukaryotes as demonstrated by the embryonic lethal phenotype in mice harboring a homozygous disruption of the DcpS gene (36). An inhibitor of DcpS was also reported to improve mice conditions in a model of Spinal Muscular Atrophy (37), suggesting that altering cap degradation may be a mean to modulate pathological conditions. Quantitative estimations of transcript abundances and half-lives indicate that roughly 300–1200 molecules of m7GDP or m7GpppN are generated every minute in exponentially growing yeast cells (38,39) and 250 in mammalian cells (40,41). Accumulation of such compounds could have deleterious effects on cells as they have been reported to impact on many cellular processes (36). Hence, high levels of m7GpppG inhibits in vitro splicing of reporter RNA (42) as well as export of nuclear RNA in vivo in Xenopus oocytes (43). m7GpppG also activates deadenylation mediated by the PARN nuclease (44). Similarly, high levels of m7GDP were reported to inhibit in vitro translation and polysome formation (45). Moreover, m7GDP could be converted into m7GTP or m7dGTP that may potentially be incorporated in RNA or DNA. While such residues are unlikely to alter the coding information stored in nucleic acids, their presence may activate degradation or repair processes that could impact on cell fitness. Altogether, these observations suggest that cells must actively eliminate cap derivatives liberated by RNA decay to avoid the potential impact of such molecules. This issue has been little explored. In Drosophila and human, 5′ nucleotidases, CG3362 and cytosolic nucleotidase III-like respectively, were shown to hydrolyze specifically m7GMP into orthophosphate and m7G (46,47). These proteins have no homologs in yeast, however, suggesting that alternative processes exist. In yeast, m7GMP was also reported to be dephosphorylated by an unknown enzyme (23). However, incubations of m7GMP in yeast extracts demonstrated that it is transformed into by-products of unknown structures (30). Because the eukaryotic pathways involved in elimination of cap derivatives are poorly understood and because the role of DcpS in some steps of this process is controversial, we decided to investigate the fate of m7GpppN and m7GDP generated by RNA decay. We identified and characterized new enzymes involved in cap degradation in vitro and in vivo. Our results indicate that the process of elimination of cap structure derivatives generated by RNA turnover is a complex process.
MATERIALS AND METHODS
Strains and plasmids
Oligonucleotides and strains used in this study are listed respectively in Supplementary Tables S1 and S2. Deletion of YNK1 or APH1 genes in the BMA64 strain was carried out by homologous recombination. The YNK1 gene was replaced by HIS3MX6 marker that was amplified with oligonucleotides OBS6090-OBS6091 carrying, a 45-nt long region at their 5′ end, homologous to, respectively, the 5′ or 3′ termini of the YNK1 open reading frame (ORF). APH1 gene was replaced by LEU2 marker that was amplified with oligonucleotides OBS6240-OBS6241 carrying respectively a 45-nt long region, at their 5′ end, homologous to the 5′ or 3′ termini of the Aph1 ORF. The resulting polymerase chain reaction (PCR) fragments were transformed in the wild-type yeast strain and the resulting colonies, growing on the appropriate selective medium, were verified for disruption by PCR screening. The double or triple deletion mutants were constructed by crosses and tetrads dissection. pBS2312 was used for expression of glutathione-S-transferase (GST)-hDcp2–6His (13) and pBS2497 for GST-DcpS6His (30). The GST-FHIT (fragile histidine triad protein) expression plasmid was kindly provided by Y.H. Wong, (48). The pBS5051 expression plasmid contains the Aph1 coding sequence PCR-amplified from plasmid p19 kindly provided by Plateau (49). The PCR product, obtained by using oligonucleotides OBS6510 and OBS6518 was digested with EcoRI and XhoI and cloned in EcoRI-XhoI digested pBS5026, thus replacing the FHIT ORF. Absence of mutations was verified by sequencing. The catalytic mutations were introduced using the Quickchange Mutagenesis strategy. Oligonucleotides OBS6505-OBS6506 were used for FHIT H96N mutation and OBS6507-OBS6508 for Aph1 H109N mutation. Recombinant proteins were expressed and purified on Glutathione Sepharose 4B beads essentially following the supplier recommendation (GE Healthcare).
Cell extracts preparation
For yeast total extracts, cells were collected in late exponential growth phase (OD around 0.81). The amount of cells corresponding to a total of 200 optical density (OD) were harvested and washed twice respectively in 50 ml of cold water and 3 ml buffer A (10-mM Hepes KOH pH 7.6, 10-mM KCl, 1.5-mM MgCl2, 0.5-mM dithiothreitol (DTT), 0.5-mM phenylmethanesulfonylfluoride (PMSF), 2-mM Benzamidine, 1-μM Leupeptin, 2-μM Pepstatin A, 4-μM Chymostatin, 2,6-μM Aprotinin). Cells were resupended in 1 ml buffer A and cold glass beads (around 750 μl; 0.5 mm diameter BioSpec Products Cat. No. 11079105) were added. Cell disruption was performed by vortexing at high speed four times, each time 30 s at 4°C. After disruption, cells were centrifuged at 3000 g (Beckman JA-25.50 rotor) for 5 min at 4°C, 900 μl of the supernatant were collected, mixed with 100 μl of KCl 2M and centrifuged at 20 000 g in a Tabletop Eppendorf centrifuge for 2 min at 4°C. The supernatant was further centrifuged at 110 000 g for 20 min at 4°C in TLA 120.2 rotor in Beckman TL-100 Tabletop Ultracentrifuge. Supernatants were transferred in dialysis tubing (molecular weight cut-off 12–14 000) and dialyzed two times, each time for 60 min against 2 L cold buffer D (20-mM Hepes KOH pH 7.6, 50-mM KCl, 0.2-mM ethylenediaminetetraacetic acid (EDTA), 0.5-mM DTT, 20% glycerol, 0.5-mM PMSF, 2-mM Benzamidine). After dialysis, extracts were aliquoted and stored at −80°C. Human HEK293 cells were resuspended in buffer A and lysed in a dounce homogenizer. After cell disruption, the extract preparation followed the protocol described above. Protein concentrations of cleared lysates were measured using the Bradford assay.
In vitro cap conversion assays
Cap-labeled RNA synthesis and invitro decapping reactions were done as described (13). Reaction products were separated by PEI cellulose thin layer chromatography (TLC). The developing solution contained 0.3 M LiCl, 1 M formic acid (except if otherwise specified). To obtain radiolabeled m7GDP, and m7GpppG, cap-labeled RNA was incubated with recombinant hDcp2 and nuclease P1 (Sigma-Aldrich) respectively. For m7GMP synthesis, m7GpppG was incubated with recombinant DcpS while to obtain m7GTP, m7GDP was incubated with nucleoside diphosphate kinase (NDK) (Sigma-Aldrich) in presence of adenosine triphosphate (ATP). The reaction products were separated by TLC and visualized with a PhosphorImager. m7GMP, m7GDP, m7GTP or m7cap spots were scraped off the TLC plate and eluted in decapping buffer (45-mM Tris–HCl, pH 8, 27-mM (NH4)2SO, 49-mM MgAc) for 40 min at room temperature with constant shaking. Insoluble TLC material was removed by centrifugation. Product yield was determined by counting. For activity assays 800 cpm of m7GMP, m7GDP, m7GTP or m7cap were incubated with 500 ng of recombinant protein or 6 μg of cellular extract for 60 min at 30°C in a 10 μl final volume. Where specified ATP (final concentration 2 mM), m7GpppG (final concentration 10 μM), 1 unit of NDK (Sigma-Aldrich), and/or glucose (final concentration 5 mM) with 1 unit of hexokinase (Sigma-Aldrich) were added to the reactions. Reactions were stopped by phenol-chloroform extraction. Five microliters of such supernatants were analyzed by TLC using the conditions described above.
RNA electroporation
RNA was electroporated into yeast cell as described previously (50) with slight modification. Briefly, cap-labeled RNA (106 cpm/0.2 μg) generated as indicated above was electroporated into 100 μl of yeast spheroblasts (108 cells/ml) resuspended in 1 M sorbitol. Following discharge (800V/25 faraday/1000 Ohm; Genepulser Bio-rad), cells were washed twice with yeast extract/bactopeptone/dextrose (YPD) containing 1 M sorbitol at room temperature, then resuspended in the same medium and incubated at 30°C with gentle swirling. Forty-five minutes post-electroporation, cells were harvested, washed and resuspended in decapping buffer, and lysed by freezing–thawing in liquid nitrogen. The supernatant was extracted twice with an equal mixture of phenol and chloroform, and once with chloroform. Where indicated 2 μg of DcpS or 1 unit of NDK with ATP (final concentration 2 mM) where added to the lysate. The decapping products fractionated by TLC and detected by autoradiography.
RESULTS
ATP- and Dcs1-dependent conversion of m7GDP into m7GMP and generation of m7GDP from m7GpppG in yeast extracts
To determine the fate of m7GDP and m7GpppG, we incubated these compound radiolabeled on the γ-phosphate of the cap (i.e. the phosphate linked directly to the ribose carrying the m7guanine group) in extracts derived from wild-type or Δdcs1 mutant yeast cells. Reaction products were fractionated by TLC alongside markers and detected by autoradiography (Figure 1A). Consistent with a previous report (30), m7GDP incubated in wild-type extract was efficiently transformed into m7GMP, an uncharacterized m7GMP derivative named product X and free phosphate as well as a few minor species (Figure 1A, lane 5). Conversion of m7GDP into m7GMP and product X was abolished in extracts prepared from Δdcs1 cells (Figure 1A, lane 6) confirming the requirement for this factor in the transformation process (30). Interestingly, beside high levels of m7GDP, we noticed the appearance of a slow-migrating species in such reactions (Figure 1A, lane 6, compare with control reaction lane 4). Co-migration with markers in different TLC systems identified this compound as m7GTP (Supplementary Figure S1). Parallel reactions performed in buffer lacking ATP demonstrated that a source of energy was required for the conversion of m7GDP into m7GMP and product X in extracts from wild-type cells and of m7GDP into m7GTP in extracts prepared from Δ dcs1 cells (Figure 1A, lanes 1–3). Reactions with m7GpppG as substrate, originally designed as controls, provided interesting insights. Hence, m7GpppG was converted into m7GMP and product X both in the presence and absence of ATP (Figure 1A, lanes 8 and 11). This indicated that DcpS was active in both conditions and that the lack of conversion of m7GDP into m7GMP observed in the absence of ATP (Figure 1A, lane 2) did not result from the lack of DcpS activity. Unexpectedly, however, we observed that m7GpppG incubated in Δdcs1 extracts was partly converted into m7GDP (Figure 1A, lanes 9 and 12). In the presence of ATP, a fraction of this compound was also converted into m7GTP (Figure 1A, lane 12). Previous analyses had indicated that product X was a metabolic derivative of m7GMP (30). This conclusion was confirmed as purified m7GMP was converted in product X in the absence and presence of ATP in extracts from wild-type yeast cells (Figure 1B). Traces of downstream by-products were detected in the presence of ATP (Figure 1B, lane 4). As product X could be an intermediate in the recycling of m7GMP, we tested whether enzymes involved in nucleotide degradation and ribose salvage metabolism (Apt1, Apt2, Npt1, Xpt1, Hpt1, Sdt1, Pnp1, Urh1, Phm8, Prm15, Gud1, Lsn1) were implicated in its formation (51–53). Incubation of m7GMP in extracts prepared from cells lacking the proteins, mentioned above, did not block product X formation (data not shown). We also performed reactions using m7GMP labeled on its m7 group with tritium (Supplementary Figure S2A). Counting the radioactivity present in the different product bands revealed that this methyl group was no more present in product X while control reactions with P32 labeled substrates demonstrated that phosphorus was efficiently transferred from m7GMP to product X (Supplementary Figure S2B). Altogether, these results confirmed that m7GDP is converted in m7GMP in a Dcs1-dependent manner and that m7GMP is itself converted in product X. Our experiments demonstrated further that the transformation of m7GDP into m7GMP requires ATP and that in extracts from Δdcs1 mutant cells m7GTP is formed in the presence of ATP (Figure 1C). Remarkably, we also observed that m7GpppG, the end product of 3′-5′ mRNA decay, was converted into m7GDP in extracts (Figure 1D).
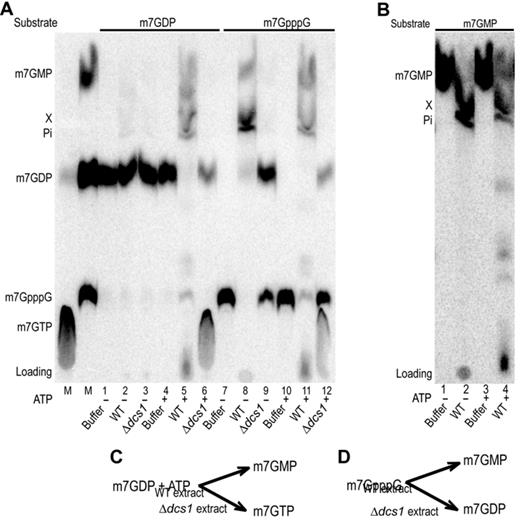
m7GDP, m7GpppG and m7GMP conversion in yeast extracts. (A) Products resulting from the incubation of m7GDP or m7GpppG in various yeast extracts were fractionated by TLC and detected by autoradiography. Purified radiolabeled m7GDP and m7GpppG were incubated with buffer (lanes 1, 4, 7, 10), or whole cell extracts from wild-type yeast (lanes 2, 5, 8, 11) or a Δdcs1 mutant (lanes 3, 6, 9, 12). In lanes 1 to 3 and 7 to 9 addition of ATP was omitted. Lanes labeled M on the left contains molecular markers. Positions of migration of the various cap derivatives are indicated on the left. (B) TLC analysis of m7GMP conversion products. Purified radiolabeled m7GMP was incubated with buffer (lanes 1 and 3), or whole cell extracts from wild-type yeast (lanes 2 and 4). In lanes 1 and 2 addition of ATP was omitted. (C and D) Schemes indicating the fate of m7GDP and m7GpppG in extracts from different strains.
Conversion of m7GDP into m7GMP requires the NDK enzyme to form an m7GTP intermediate
The requirement for ATP to convert m7GDP into m7GMP and the accumulation of m7GTP in extracts lacking Dcs1 were consistent with the idea that m7GTP could be an intermediate in this pathway. Consistently, m7GDP is known to be phosphorylated by the NDK into m7GTP in vitro (13,23) while the latter was shown to be hydrolyzed into m7GMP by recombinant Dcs1 (33). This suggested that NDK could participate to the conversion of m7GDP in extract by phosphorylating it. To quickly provide support for this idea, we tested whether various nucleoside triphosphate supported this reaction as NDK is known to display little specificity (54). The eight common (d)NTPs (nucleoside triphosphate) promoted the conversion of m7GDP into m7GMP in extract from wild-type cells and the formation of m7GTP in extracts lacking Dcs1 (Supplementary Figure S3A) supporting the involvement of NDK. To further demonstrate the implication of NDK in cap degradation, we constructed a yeast strain lacking the dispensable ynk1 gene (55). By crosses, we also built a strain lacking both ynk1 and dcs1. Extracts prepared from these cells were tested for their ability to metabolize m7GDP. Consistent with our hypothesis, conversion of m7GDP into m7GMP did not occur in extracts lacking NDK (Figure 2A, compare lanes 1, 2 and 4). Moreover, the accumulation of m7GTP detected in extracts deprived of Dcs1 (Figure 2A, lane 3) was abolished when NDK was simultaneously absent (Figure 2A, lane 5). Importantly, addition of purified NDK to these extracts restored the conversion of m7GDP into m7GMP in Δynk1 extract and the accumulation of m7GTP in Δdcs1Δynk1 extracts while it had no effect on extracts prepared from Δdcs1 or wild-type cells (Supplementary Figure S3B). By incubating m7GTP in yeast extracts, we confirmed that it was converted into m7GMP (Figure 2B, lane 2). This process was blocked in extracts lacking Dcs1 (lane 3 and 5, some m7GDP accumulates probably owing to the action of some phosphatase present in the extract) but was unaffected by the absence of ynk1 (Figure 2B, lane 4). Altogether, these results demonstrate that NDK is required for the conversion of m7GDP into m7GMP. These results reveal a biochemical pathway for m7GDP elimination in extracts that involves first its conversion in m7GTP mediated by NDK in the presence of (d)NTPs followed by hydrolysis of the m7GTP intermediate into m7GMP catalyzed by DcpS (Figure 2C). This scheme is consistent with the accumulation of m7GTP detected in the absence of DcpS (Figure 2A, lane 3).
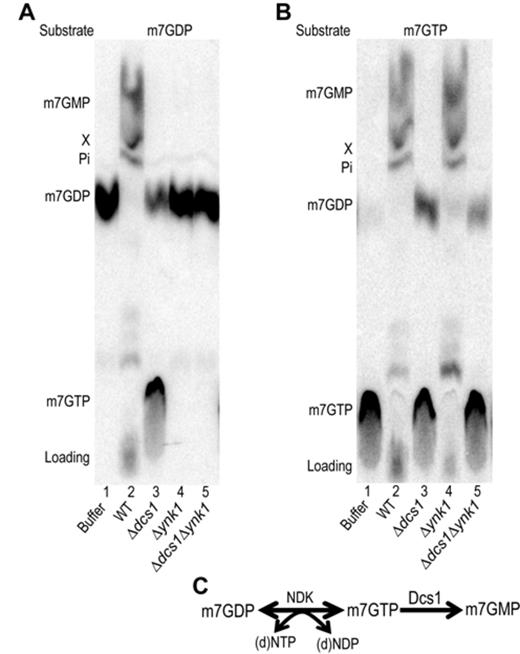
Role of NDK encoded by the YNK1 gene if cap conversion (A). TLC analysis of m7GDP conversion products. Purified radiolabeled m7GDP was incubated with buffer (lanes 1), or whole cell extracts from wild-type yeast (lane 2), a Δdcs1 mutant (lane 3), a Δynk1 mutant (lane 4) or a Δdcs1Δynk1 double mutant (lane 5). (B) Same as in (A) but the substrate is m7GTP. All reactions were performed in presence of ATP. (C) Scheme describing the biochemical pathway involved in m7GDP elimination in extracts.
The m7GDP elimination pathway is conserved in human
It was previously reported that m7GDP incubated in extracts from human cells or Xenopus oocytes was efficiently converted into m7GMP with no evidence for further conversion in product X (30). We thus tested whether the reaction occurring in mammalian cell extract followed the steps that we identified for yeast by incubating m7GDP in HEK293 cell extract under various conditions. In presence of ATP, the human cell extract efficiently transformed m7GDP into m7GMP (Figure 3, lane 8) while when no ATP was added only a partial conversion was detected, some m7GDP remaining after the reaction (Figure 3, lane 4). This residual activity results from the presence of traces of ATP in the extract as it was abolished when an ATP depleting system was used (Supplementary Figure S4). This indicated that like for yeast, ATP was required for the conversion of m7GDP into m7GMP. To test whether DcpS was also involved in this process, we relied on the observation that DcpS can be inhibited by high levels of m7GpppG (56). We incubated m7GDP in HEK293 cell extract in presence of inhibitory concentration of unlabeled m7GpppG. This treatment prevented the conversion of m7GDP and lead to the accumulation of m7GTP (Figure 3 lane 12). A similar result was obtained with extracts prepared from wild-type yeast (Figure 3 lane 10) mimicking an extract prepared from a Δdcs1 strain (Figure 3 lane 11). Altogether, these results show that elimination of m7GDP in extracts from human cells occurs with the same characteristics as its degradation in yeast extract, including requirement for ATP and DcpS activity, and accumulation of m7GTP in the absence of DcpS function. These data strongly argue that the pathway identified in yeast is conserved in mammals and likely other eukaryotes.
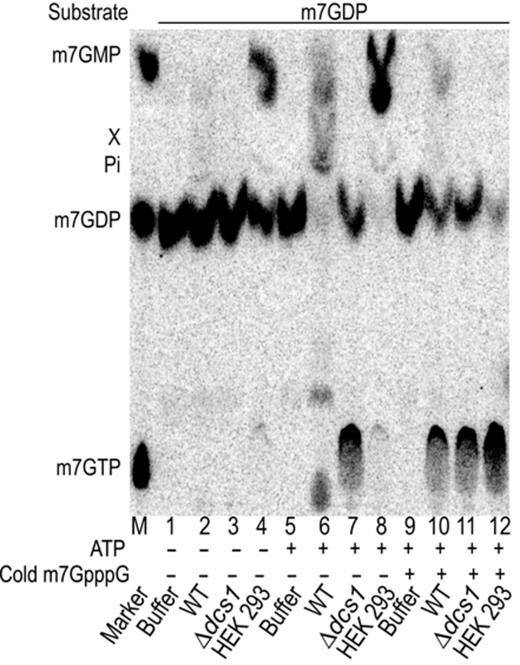
m7GDP conversion in human cell extracts. Products resulting from the incubation of m7GDP in an HEK293 whole-cell extract, or as control yeast extracts, were fractionated by TLC and detected by autoradiography. Purified radiolabeled m7GDP was incubated with buffer (lanes 1, 5 and 9), whole cell extracts from wild-type (lanes 2, 6 and 10) and Δdcs1 (lanes 3, 7 and 11) yeast strains, or HEK293 cell extract (lanes 4, 8, 12). Reactions contained ATP at a final concentration of 2 mM except in lanes 1–4. In lanes 9–12, cold m7GpppG was added to a final concentration of 10 μM to inhibit DcpS. The positions of cap derivatives are indicated on the left side and the left lane labeled M contains molecular markers.
Aph1, a new scavenger decapping enzyme, hydrolyzes m7GpppG into m7GDP
By incubating radiolabeled m7GpppG in yeast extract prepared from Δ dcs1 cells we unexpectedly observed the formation of a product that we identified as m7GDP (Figure 1A, lanes 9 and 12). The absence of ATP prevented the conversion of the latter into m7GTP confirming further the nature of this product and leading to a higher accumulation (Figure 1A lane 9). A low level of m7GDP was also present when a complete extract was used and this was enhanced when Dcs1 was inhibited by the addition of m7GpppG, indicating that this reaction also occurs in wild-type yeast cells (Figure 1A, lane 8; Supplementary Figure S5). This activity was not reported earlier, possibly owing to the fact that m7GDP comigrates with m7GpppG in the TLC system originally used (23) (Supplementary Figure S1). The activity converting m7GpppG into m7GDP was sensitive to sodium dodecyl sulphate, EDTA and heat inactivation (data not shown), implicating an enzyme. Bibliography searches identified some HIT family members, namely Apa1, Apa2 and Aph, as likely candidates to perform this reaction. Among them, Aph1 previously shown to hydrolyze ApppN but also other dinucleotide preferentially with a 5′-5′ triphosphate linkage (49) was a primary candidate. Testing for the fate of m7GpppG in extracts from strains carrying individual or multiple deletion of these genes revealed that in conditions were Dcs1 was inhibited, formation of m7GDP did not occur in extracts prepared from cells lacking Aph1 (Supplementary Figure S6). To study the role of Aph1 in m7GpppG degradation, we constructed a Δaph1 strain in our strain background. By crosses, we also built an isogenic strain lacking both aph1 and dcs1. Extracts prepared from these cells were tested for their ability to metabolize m7GpppG. Consistent with our first result, in extracts prepared from Δaph1Δdcs1 cells, m7GpppG was no further hydrolyzed into m7GDP and remained stable (Figure 4A, lane 5). In extracts prepared from Δaph1 cells, m7GpppG was converted into m7GMP and product X due to the activity of Dcs1 and downstream enzymes (Figure 4A, lane 4). m7GpppG hydrolysis was also prevented in extracts lacking Aph1 when Dcs1 was simultaneously inhibited by a high level of m7GpppG (Figure 4B, lane 4) while under the same conditions formation of m7GMP was strongly inhibited in an extract from wild-type cells leading to increased accumulation of m7GDP (Figure 4B, lane 2). To demonstrate that Aph1 is sufficient to perform this reaction, we expressed recombinant glutathione-S-transferase (GST)-tagged Aph1 in Escherichia coli (Figure 5B). Incubation of purified Aph1 with m7GpppG resulted in the formation of m7GDP with only traces of m7GMP present (Figure 5A, lane 5). As control, no m7GDP production was detected when the substrate was incubated with purified GST (Figure 5A, lane 2). To exclude that the reaction was due to a contaminant, we constructed and purified a Aph1 catalytic site mutant by exchanging histidine 109 with asparagine (Figure 5B). m7GpppG incubated with the mutant factor did not get significantly hydrolyzed into m7GDP (Figure 5A, lane 6). Altogether, these results demonstrate that in yeast m7GpppG is degraded not only by DcpS but also by a second scavenger decapping enzyme, Aph1 (Figure 5C). In contrast to the former, the latter catalyzes the production of m7GDP.
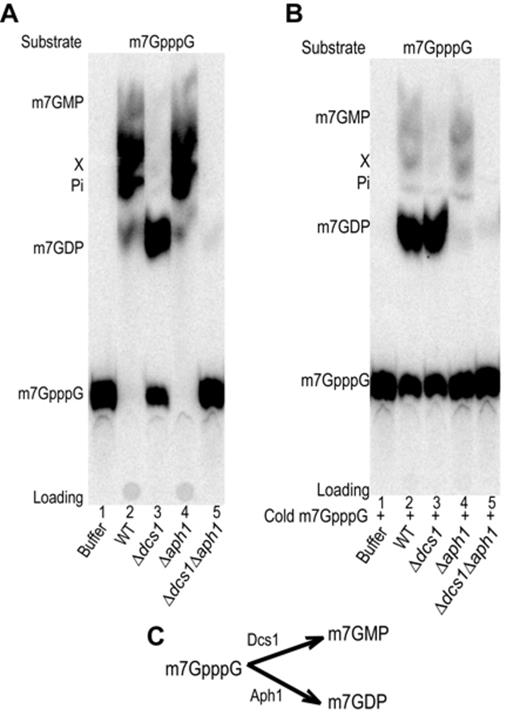
m7GpppG conversion in yeast extracts. A TLC analysis of m7GpppG conversion products is presented. (A). Purified radiolabeled m7GpppG was used as substrate. Products resulting from incubation with buffer (lanes 1), or whole cell extracts from wild-type yeast (lane 2), a Δdcs1 mutant (lane3), a Δaph1 mutant (lane 4) or a Δdcs1Δaph1 mutant (lane 5) were analyzed. (B) Same as in (A) but cold m7GpppG was added to a final concentration of 10 μM to inhibit DcpS.
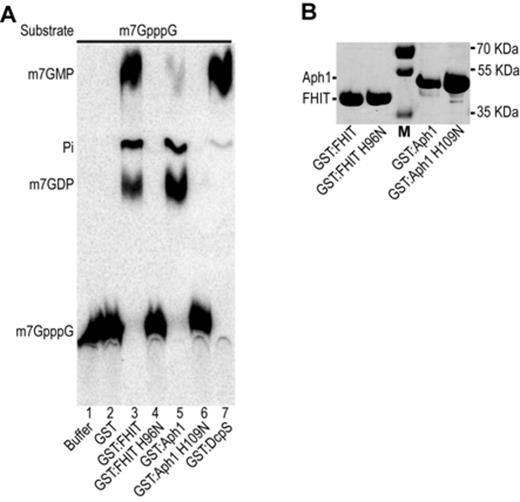
m7GpppG cleavage by recombinant Aph1 and FHIT. (A) Products resulting from the incubation of m7GpppG with recombinant proteins were fractionated by TLC and detected by autoradiography. Purified radiolabeled m7GpppG was incubated with reaction buffer (lane 1), or GST (lane 2), GST-FHIT (lane 3), a GST-FHIT catalytic mutant H96N (lane 4), GST-Aph1 (lane 5), a GST-Aph1 catalytic mutant H109N (lane 6) or as a control human DcpS (lane 7). All reactions contained 0.5 μg of the respective recombinant protein. (B) Purified protein profiles. Recombinant GST:Aph1 and GST:FHIT and the respective catalytic mutants were purified and separated on sodium dodecyl sulphate-polyacrylamide gel electrophoresis. Positions of migration of molecular weight markers are indicated on the right side.
The product of the human tumur suppressor gene FHIT catalyzes cap cleavage
FHIT, the human homolog of Aph1 (49), is known as tumor suppressor and its gene is deleted in many types of cancers (57). Although many efforts were done to uncover the molecular mechanisms leading to cancer suppression, its molecular function remains poorly understood (25). This member of the HIT family also harbors a dinucleoside polyphosphate hydrolase activity (58). To test whether FHIT could also be involved in cap metabolism, we expressed GST-tagged recombinant FHIT (Figure 5B). Incubation of the purified factor with m7GpppG leads to the production of both m7GDP and m7GMP (Figure 5A, lane 3). We again verified that FHIT was directly involved in catalyzing this hydrolysis by substituting the histidine 96 of the FHIT catalytic site with an asparagine. The purified recombinant mutant protein (Figure 5B) was unable to cleave m7GpppG (Figure 5A, lane 4). These results indicate that, like its yeast homologue Aph1, FHIT is likely to be involved in cap degradation.
In vivo analysis of m7G cap metabolism
The results presented above indicate that Dcs1 and Aph1 are involved in cap degradation in yeast extract. To test whether these proteins affect the fate of cap structures released by RNA degradation in vivo in cells, we transformed a 49-nt RNA in the different yeast strains by electroporation. This artificial RNA was radiolabeled on the phosphate in position γ of the cap structure. Forty-five minutes post-electroporation, we extracted the cellular content and fractionated it by TLC after protein inactivation and removal. Several products, some comigrating with m7-containing nucleotides and some of unknown identity, were detected by autoradiography (Figure 6, lane 1). Control reactions demonstrated that these products arose as a consequence of RNA introduction in cells, as they were not detected when no cells were present and when the radiolabeled RNA was added after the electric pulse (data not shown). Interestingly, in absence of Dcs1 (Δdcs1 and Δ dcs1/Δynk1 strains), we observed the accumulation of m7GDP and, to a higher extent, of a second product migrating slightly faster (labeled Ypp), as well as traces of m7GTP and m7GpppG (Figure 6, lanes 3 and 4). Product Ypp is a nucleotide diphosphate as in the presence of ATP it is a substrate of NDK (Figure 6 compare lanes 3–4 and 11–12). Moreover, product Ypp is likely to be a derivative of m7GDP as it accumulates when the m7GDP level rises (Figure 6) and as it is present after electroporation of m7GDP in Δdcs1Δynk1 cells (data not shown). The accumulation of m7GDP and a downstream derivative in Δdcs1 cells provides a direct evidence for the role of Dcs1 in cap metabolism in vivo. Deletion of ynk1 encoding NDK had little effect on the product profile (Figure 6, compare lanes 1 and 2), probably because other enzymes are redundant with Ynk1. The same applied to a Δ aph1 strain, consistent with the limited impact of the absence of Aph1 alone on m7GpppG breakdown in vitro (Figure 6, lane 5). Importantly, however, the simultaneous elimination of Aph1 and Dcs1 lead to strong accumulation of m7GpppG in cells (Figure 6, lane 6) with a slight reduction in m7GDP levels. No further enhancement was detected when NDK was simultaneously absent (Figure 6, lane 8). To ascertain our identifications, we incubated purified recombinant Dcs1 protein in the lysate. This reaction confirmed that m7GpppG accumulated in Δdcs1/Δaph1 electroporated cells as this product was hydrolyzed into m7GMP (Figure 5, compare lanes 6–8 and 22–24).
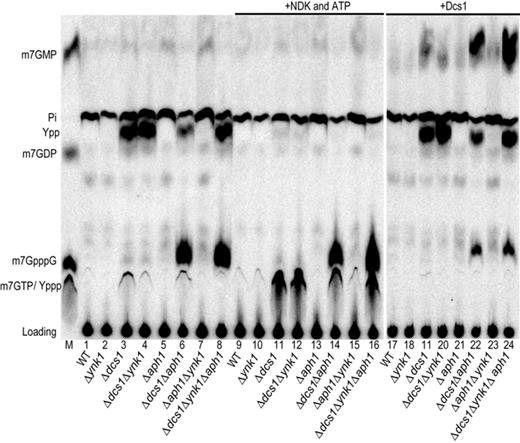
m7G cap metabolism in living cells. An artificial 49 nt long capped RNA radiolabeled on the cap phosphate group in position γ was electroporated in cells: wild-type (lane 1), Δynk1 (lane 2), Δdcs1 (lane 3), Δdcs1Δynk1 (lane 4), Δaph1 (lane 5), Δdcs1Δaph1 (lane 6) Δaph1Δynk1 (lane 7) or Δdcs1Δaph1Δynk1 (lane 8) strains. After 45 min of incubation, the resulting metabolites were extracted, fractionated by TLC and detected by autoradiography. Products recovered from the strains above were incubated with 1 unit of NDK in presence of ATP (lanes 9–16) or 2 μg of purified Dcs1 (lanes 17–24). The product resulting from the conversion of species Ypp by NDK (Yppp) is not resolved from m7GTP. The two TLC were developed in parallel and exposed for the same duration.
Taken together with our in vitro data, these results demonstrate that Dcs1 and Aph1 are scavenger decapping enzymes with overlapping actions mediating the elimination of m7GpppN residues and other cap by-products generated by mRNA decay in eukaryotic cells.
DISCUSSION
The process of mRNA decay, driven by different enzymatic activities, generates as endproduct nucleotides, that may directly serve for numerous cellular functions, as well as m7G cap (di-)nucleotides. Quantification of the number of mRNAs per cells and of their average half-lives indicate that hundreds of molecules of m7GDP and m7GpppN are generated every minute in yeast and mammalian cells (38–41). The fate of cap structures has been only partly elucidated with the identification of DcpS as an enzyme promoting the conversion of m7GpppN in m7GMP (19,23) and the identification of a nucleotidase that converts the latter in m7guanosine in metazoans (46,47). Analyzing the fate of cap (di)nucleotide, we identified and characterized new scavenger decapping enzymes, Aph1/FHIT, that actively convert m7GpppG into m7GDP (Figure 7) and demonstrated that DcpS participates to the conversion of m7GDP produced by the 5′-3′ pathway into m7GMP through an m7GTP intermediate, both in yeast and mammalian cells. As DcpS was reported to be essential for the degradation of m7GpppG cap in extracts, we were surprised to observe an efficient conversion of m7GpppG in m7GDP in lysates from yeast cells lacking the DCS1 gene. This result was highly reproducible and we explain that this pathway was not detected earlier by a technical limitation of the original TLC system used to follow the DcpS activity (19,23). Indeed, in these conditions, m7GDP co-migrates with m7GpppG (Supplementary Figure S1). Hence, inter-conversion of these two products could not be detected. Testing yeast strains mutant for several HIT family members revealed that Aph1 is required to cleave m7GpppG into m7GDP. This is consistent with its ability to cleave dinucleotide triphosphate reported in the literature. Further analyses performed with the recombinant Aph1 protein demonstrate that this enzyme cleaves m7GpppG to generate m7GDP. In vivo analyses, importantly, confirm the implication of both Aph1 and DcpS in cap breakdown in yeast cells. The synergic effect of APH1 and DCS1 deletions shows further that they have overlapping role in the degradation of m7GpppG under normal physiological conditions. The cap scavenging activities of Aph 1 and Dcs1 are not shared with other HIT family members. Indeed, in the double mutant Δaph1 Δdcs1 m7GpppG accumulates indicating that other HIT protein still present in the extract do not efficiently hydrolyze this product. The observation that the double mutant strains grow relatively well may be accounted for by the fact that 3′-5′ mRNA decay is not the major route to eliminate mRNAs. Consistently, mutations abolishing the activity of the Ski complex (Ski2/3/8) or of Ski7, factors that are specifically involved in the 3′-5′ degradation of mRNAs, also result in no or limited growth phenotypes. Interestingly, the product of the human tumor suppressor gene FHIT, a homolog of Aph1, also cleaves m7GpppG but produces a mixture of roughly 50% m7GMP and 50% m7GDP. The difference in product composition can be explained by the ability of FHIT to recognize both bases of the m7GpppG dinucleotide, namely m7guanine and guanine, thus being able to bind this pseudo-symmetrical substrate in two orientations. In contrast, yeast Aph1 appears to productively bind m7GpppG essentially in a single orientation, thus leading to the formation of an essentially homogeneous products. The observation that Aph1 participates to the elimination of cap structures in vitro and in vivo may provide some hints about the function of its human homolog FHIT. The gene encoding for FHIT is located in the fragile site FRA3B on the chromosome 3 and was found deleted in many type of cancers (59). FHIT acts as a proapoptotic factor inhibiting uncontrolled cell proliferation through an elusive mechanism (25). Like some other HIT family members, FHIT hydrolyses diadenosine polyphosphates (58). The observation that Aph1 contributes to the clearance of cap structures combined with ability of FHIT to hydrolyze m7GpppG, suggest that FHIT may contribute to cell growth control by binding to m7GpppG, mediating the formation of downstream products and/or impacting on mRNA decay. It will be important to investigate these possibilities in the future. Our analyses of the fate of m7GDP incubated in extracts revealed that it is converted into m7GMP in a two-step process that appears to be conserved between yeast and human cells. m7GDP is first transformed into m7GTP that is itself metabolized into m7GMP (Figure 7). Traces of ATP (or any other (d)NTP) present in extracts may have impaired the detection of the first of these steps previously (30). Under our conditions, NDK appears to be the main enzyme catalyzing m7GTP formation in yeast extracts. However, the absence of growth phenotype of yeast cells lacking the gene encoding NDK indicates the existence of other enzyme(s) converting nucleoside diphosphate into triphosphate. Those may also catalyze the conversion of m7GDP into m7GTP in living cells. m7GTP is then hydrolyzed into m7GMP by DcpS. These observations reconcile previous studies that had argued for, or against, the involvement of DcpS in m7GDP turnover (23,30,31,33). Consistent with the two-step pathway that we identified, we observed the accumulation of products co-migrating with m7GTP and m7GDP (and the Ypp derivative) in Δdcs1 cells transfected with cap-labeled RNA. Previous analyses following electroporation identified m7GpppG and m7GpppGp as products accumulating in Δdcs1 cells (23). This difference may be accounted for by the fact that in these earlier analyses, metabolites present in the cells were analysed 6 h postelectroporation and may thus represent indirect or late conversion products. It remains nevertheless difficult to reconcile them with the presence of active Aph1. An alternative interpretation is that their identity was incorrectly assigned. Our observation of the specific presence of m7GTP and m7GDP (and its Ypp derivative) at early time points after electroporation in Δdcs1 cells argues for a physiological role of this factor in cap metabolism in vivo in a manner consistent with its biochemical activities. The m7GMP originating from the 3′-5′ or 5′-3′ pathways is converted into a product of uncharacterized structure (X) in yeast extracts, as previously reported. Using radioactive labels on different groups of m7GMP, we observed that the methyl group located on position 7 of the guanosine is not present in product X. This suggests that transformation of m7GMP in product X could minimally be a demethylation. However, more complex reaction such as those severing of the base-ribose bond or the transfer of the phosphate group to a recipient molecule can't yet be excluded. Altogether, our results reveal the existence of a complex multistep pathway mediating the elimination of cap structures arising as a consequence of mRNA decay (Figure 7) and suggest that it may impact in human cell physiology. Further analyses will be necessary to uncover how altered cap metabolism affect cell metabolism and how this drives disease development.
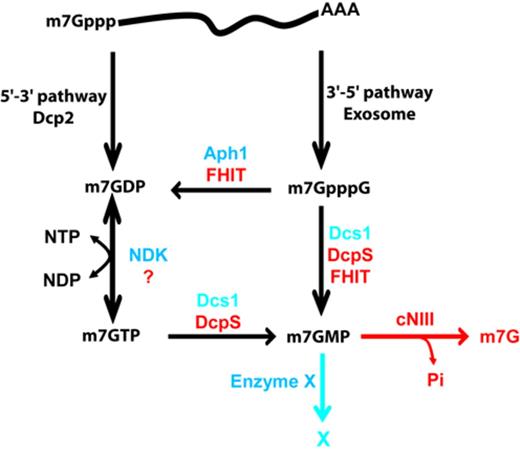
RNA decay pathways and cap (di-)nucleotide elimination mechanisms in yeast and mammalian cells. Enzymes involved in the generation of cap (di-)nucleotide in the 5′-3′ and 3′-5′ mRNA decay pathways are indicated as well as the resulting products. Mechanisms mediating the elimination of these compounds identified in this study are indicated with yeast enzymes indicated in blue and human factors in red. Degradation of the end-product m7GMP by cNIII was reported earlier (46).
We are indebt to J.D. Rabinowitz and B. Daignan-Fornier for providing yeast mutant strains impaired in nucleotide metabolism, to P. Plateau for the aph1, apa1 and apa2 mutant strains and a APH1 cDNA, and to Y.H. Wong for the FHIT expression construct. We thank the members of our group for discussion and advice and IGBMC services for support.
Author contribution: B.S. designed the research project, V.T. designed and performed the experiments, B.S. and V.T. analyzed the data and wrote the manuscript.
FUNDING
Ligue Contre le Cancer (Equipe Labellisée 2014); Centre National pour la Recherche Scientifique; CERBM-IGBMC; Decapping from Agence Nationale pour la Recherche [ANR 11 BSV8 009 02]; Investissements d'Avenir ANR-10-IDEX0002-02 [ANR-10LABX-0030-INRT]. Funding for open access charge: Ligue Nationale contre la Cancer.
Conflict of interest statement. None declared.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments