





Abstract
Background. Chronic metabolic acidosis may increase alkali mobilization from the bone and thus promote the development of osteoporosis. The objective of the current study was to compare urinary acidification in patients with reduced bone mineral content with that in control subjects with normal bone density.
Methods. Forty‐six subjects (41 females, 5 males) with osteopenia or osteoporosis were studied. In none of the subjects were overt metabolic acidosis, derangement of potassium homeostasis, or renal insufficiency present. Distal tubular acidification was studied by means of oral ammonium chloride loading test (0.1 g/kg body weight) and the oral frusemide test (40 mg). In addition the frusemide test was performed in 20 healthy age‐ and sex‐matched controls (17 females, 3 males).
Results. In all control subjects a urinary pH <5.5 was observed following the ingestion of 40 mg frusemide. In contrast, in patients with reduced bone mineral density incomplete renal tubular acidosis type I (RTA I) was diagnosed in 10 of 46 subjects (22%) by oral ammonium chloride loading test. Disorders possibly related to RTA I were detected in eight of these 10 patients. Thirty‐six patients had a normal urinary pH response following oral ammonium chloride loading. Oral frusemide, 40 mg, failed to lower urinary pH <5.5 in sixteen patients (35%), these included 10 subjects with incomplete RTA I, and six subjects with a normal oral ammonium chloride loading test. An abnormal frusemide test was found in 35% of patients with reduced bone mass and in none of the normal controls (χ2=7.39; P<0.01). With the ammonium chloride test as the gold standard for diagnosis of distal RTA, the frusemide test showed a sensitivity of 1.0 (95% CI, 0.69–1.0) and a specificity of 0.89 (95% CI, 0.78–0.96) for the diagnosis of distal RTA. Patients with incomplete RTA I were younger than those without incomplete RTA I (42±16 vs 54±14 years; P=0.025; mean±SD). Basal serum bicarbonate concentrations and capillary pH did not differ between the groups.
Conclusion. Incomplete RTA I may be prevalent in a significant proportion of patients suffering from osteopenia or osteoporosis. The outcome of the frusemide test suggests either a defect of the H+ATPase in the cortical collecting tubule (CCT) or a defective Na+ reabsorption in the CCT. Prospective studies are needed to further elucidate the impact of incomplete RTA I on the development of reduced bone mineral content.
Introduction
In Western industrialized countries an average 70‐kg adult ingests a diet that generates approximately 70 mEq of acid per day. Physiologically, this acid challenge is dealt with by the kidneys, which excrete 70 mEq of acid, 40 mEq/day as ammonium ions and 30 mEq as titratable acid [1]. In steady state the renal acid excretion equals the endogenous acid production and thus acid–base balance is neutral. A positive body acid balance may be induced either by a significant rise of endogenous acid production that exceeds the renal acid excretion capacity, or by defects of the renal acid excretion [1]. In response to a positive acid balance, serum bicarbonate (
Subjects with a reduced renal acid excretory reserve, e.g. in the setting of incomplete distal renal tubular acidosis (renal tubular acidosis type I (RTA I)), may be at extra risk of bone mineral loss especially when ingesting the usual mixed Western diet. Recently we observed a high prevalence of incomplete distal tubular acidosis and ‘primary’ osteoporosis [3]. The current study was undertaken to estimate the prevalence of incomplete RTA I in subjects with a reduced bone mineral content as compared to subjects with a normal bone density and to further define the mechanism of RTA I in this group of patients. A secondary goal was to study the test characteristics of the frusemide test.
Subjects and methods
Study population and biochemical methods
Sixty‐six subjects, 58 females and 8 males, were studied. Forty‐six subjects (41 females, 5 males) suffered from osteoporosis or osteopenia, 20 subjects were matched healthy controls (age 46±7 years; 17 females, 3 males) with a normal bone mineral density. The patients were referred by primary care physicians to our hospital for suspected osteoporosis. The subjects do not resemble a cohort of patients with osteoporosis and incomplete RTA I described recently [3]. All subjects were on standard hospital diet, which provides on average 120 mmol sodium chloride, 1000 mg of calcium, and 70 g of protein per day. No dietary extremes (e.g. high‐ or low‐protein diet, low‐sodium diet) were given.
In all subjects (patients and controls) bone mineral density was assessed at the lumbar site (L2–L5) using dual energy X‐ray absorptiometry on a Sophos XLA machine. According to the criteria proposed by the World Health Organization (WHO), osteopenia and osteoporosis were diagnosed by a reduced bone mineral density (t‐score −1.0 to −2.5, or t‐score <−2.5 respectively) measured by DXA [4]. Any radiological evidence of vertebral fractures was also taken as a diagnostic criterion of osteoporosis. Applying these diagnostic criteria, 20 patients (43%) had osteopenia and 26 patients (57%) had osteoporosis. The baseline characteristics of the study population are shown in Table 1.
Sixteen patients had disorders possibly associated with impaired renal distal tubular acidification, namely hypercalciuria with (n=2) and without nephrolithiasis (n=2), past history of nephrolithiasis without current hypercalciuria (n=3), habitual analgesic abuse with a cumulative amount >1500 g (n=4), autoimmune thyroid disease (n=3), nephrocalcinosis due to medullary sponge kidney (n=1), and a familial history of incomplete RTA I (n=1). The patients with thyroid disease had been prescribed thyroid hormone to achieve normal basal TSH levels. Hypercalciuria was defined as a daily urinary excretion of calcium >4 mg/kg body weight. Patients with hypercalciuria had not been advised to follow a Ca‐restricted diet.
The classical causes of secondary osteoporosis (glucocorticoid excess, hyperthyroidism, primary hyperparathyroidism) were ruled out by measurement of thyroid hormone (free T3, free T4, basal TSH), serum calcium and iPTH, and on the basis of clinical examination and patient history.
In none of the patients were renal insufficiency or overt metabolic acidosis present. The presence of RTA type IV (due to hyporeninaemic hypoaldosteronism) was considered unlikely because of normal serum potassium concentrations. Urinary pH was measured with a KCl electrode (pH‐meter E 512, Metrohm, Herisau, Switzerland). Urinary tract infection was ruled out by urinalysis.
In all 46 patients both an oral ammonium chloride (NH4Cl) loading test and an oral frusemide test was performed; in 20 controls the frusemide test was done. In addition, three patients with a positive frusemide test and a negative NH4Cl loading test consented to a fludrocortisone‐modified frusemide test [5].
Prior to the frusemide loading test patients were off diuretics and non‐steroidal anti‐inflammatory drugs (NSAID) for at least 2 days. The patients were given 40 mg of frusemide orally and urinary pH was measured 0, 2, 4, and 6 h after the frusemide dose [6]. In case of the fludrocortisone‐modified frusemide test, oral frusemide was preceded on the previous evening by 0.5 mg of fludrocortisone. At least 2 days after the frusemide test the standardized short course oral NH4Cl test was performed [7]. Briefly, patients ingested 0.1 g/kg body weight of NH4Cl in gelatin capsules. Blood gas analysis was performed from capillary blood with an AVL 995 Hb automatic blood gas system (AVL, Graz, Austria) on an hourly basis. The metabolic acidosis induced by the NH4Cl load was documented by a fall of serum
Baseline characteristics of the patients with reduced bone mass
| No RTA I | Incomplete | |
| RTA I | ||
| Number | 36 | 10 |
| Gender (females/males) | 33/3 | 8/2 |
| Osteopenia/osteoporosis | 14/22 | 6/4 |
| Age (years) | 54±14 | 42±16a |
| Blood pH | 7.41±0.03 | 7.40±0.03 |
| Serum bicarbonate (mmol/l) | 22.9±2.2 | 21.7±1.9 |
| Capillary pCO2 (mmHg) | 36.9±3.3 | 36.1±3.5 |
| Serum creatinine (mg/dl) | 0.8±0.1 | 0.9±0.2 |
| Serum potassium (mmol/l) | 4.4±0.4 | 4.4±0.3 |
| Serum calcium (mmol/l) | 2.4±0.1 | 2.4±0.1 |
| Serum phosphate (mg/dl) | 3.4±0.5 | 3.2±0.5 |
| Serum iPTH (pg/ml) | 33±24 | 23±7 |
| Urinary calcium | 0.07±0.05 | 0.08±0.03 |
| (mg/kg body weight/day) |
| No RTA I | Incomplete | |
| RTA I | ||
| Number | 36 | 10 |
| Gender (females/males) | 33/3 | 8/2 |
| Osteopenia/osteoporosis | 14/22 | 6/4 |
| Age (years) | 54±14 | 42±16a |
| Blood pH | 7.41±0.03 | 7.40±0.03 |
| Serum bicarbonate (mmol/l) | 22.9±2.2 | 21.7±1.9 |
| Capillary pCO2 (mmHg) | 36.9±3.3 | 36.1±3.5 |
| Serum creatinine (mg/dl) | 0.8±0.1 | 0.9±0.2 |
| Serum potassium (mmol/l) | 4.4±0.4 | 4.4±0.3 |
| Serum calcium (mmol/l) | 2.4±0.1 | 2.4±0.1 |
| Serum phosphate (mg/dl) | 3.4±0.5 | 3.2±0.5 |
| Serum iPTH (pg/ml) | 33±24 | 23±7 |
| Urinary calcium | 0.07±0.05 | 0.08±0.03 |
| (mg/kg body weight/day) |
Mean±standard deviation; aP=0.025 (two‐sided).
Baseline characteristics of the patients with reduced bone mass
| No RTA I | Incomplete | |
| RTA I | ||
| Number | 36 | 10 |
| Gender (females/males) | 33/3 | 8/2 |
| Osteopenia/osteoporosis | 14/22 | 6/4 |
| Age (years) | 54±14 | 42±16a |
| Blood pH | 7.41±0.03 | 7.40±0.03 |
| Serum bicarbonate (mmol/l) | 22.9±2.2 | 21.7±1.9 |
| Capillary pCO2 (mmHg) | 36.9±3.3 | 36.1±3.5 |
| Serum creatinine (mg/dl) | 0.8±0.1 | 0.9±0.2 |
| Serum potassium (mmol/l) | 4.4±0.4 | 4.4±0.3 |
| Serum calcium (mmol/l) | 2.4±0.1 | 2.4±0.1 |
| Serum phosphate (mg/dl) | 3.4±0.5 | 3.2±0.5 |
| Serum iPTH (pg/ml) | 33±24 | 23±7 |
| Urinary calcium | 0.07±0.05 | 0.08±0.03 |
| (mg/kg body weight/day) |
| No RTA I | Incomplete | |
| RTA I | ||
| Number | 36 | 10 |
| Gender (females/males) | 33/3 | 8/2 |
| Osteopenia/osteoporosis | 14/22 | 6/4 |
| Age (years) | 54±14 | 42±16a |
| Blood pH | 7.41±0.03 | 7.40±0.03 |
| Serum bicarbonate (mmol/l) | 22.9±2.2 | 21.7±1.9 |
| Capillary pCO2 (mmHg) | 36.9±3.3 | 36.1±3.5 |
| Serum creatinine (mg/dl) | 0.8±0.1 | 0.9±0.2 |
| Serum potassium (mmol/l) | 4.4±0.4 | 4.4±0.3 |
| Serum calcium (mmol/l) | 2.4±0.1 | 2.4±0.1 |
| Serum phosphate (mg/dl) | 3.4±0.5 | 3.2±0.5 |
| Serum iPTH (pg/ml) | 33±24 | 23±7 |
| Urinary calcium | 0.07±0.05 | 0.08±0.03 |
| (mg/kg body weight/day) |
Mean±standard deviation; aP=0.025 (two‐sided).
Statistical analysis
The t‐test of untransformed data was used for group comparison of patients with and without a normal response in the NH4Cl test. A P value <0.05 was considered as significant. Data are shown as mean and ±SD. The 95% confidence interval (CI) of relative frequencies was derived from the binomial distribution. Absolute frequencies were compared by the χ2 test.
With the NH4Cl test as the gold standard for the diagnosis of distal RTA, the diagnostic performance of the frusemide test was evaluated in terms of sensitivity and specificity. The failure to lower urinary pH <5.5 both in the NH4Cl and the frusemide test was considered as a true positive (TP) result, whereas an appropriate urinary acidification in both tests was scored as a true negative (TN) outcome. False positive (FP) and false negative (FN) frusemide tests were defined accordingly. Sensitivity was defined as TP/(TP+FN), specificity was defined as TN/(TN+FP) [8].
Results
In all control subjects (100%; 95% CI, 83–100%) the administration of oral frusemide resulted in a fall of the urinary pH below 5.5 (Figure 1).
Overt metabolic acidosis, impaired renal function, or disturbances of potassium metabolism were not observed in the patients studied. In 30 patients (65%) both NH4Cl testing and the oral frusemide test revealed normal responses, i.e. a fall of urinary pH <5.5 (Figure 2a).
Ten patients (22%) failed to lower urinary pH below 5.5 (Table 1, Figure 2c) despite the induction of systemic metabolic acidosis following the ingestion of NH4Cl. In these 10 patients the diagnosis of incomplete RTA I was made. All of these patients with incomplete RTA I showed also a urinary pH above 5.5 during the frusemide test (Figure 2c). Additionally, six subjects (13%) with a normal NH4Cl test failed to lower urinary pH <5.5 during the frusemide test (Figure 2b). In three patients of the latter group a fludrocortisone‐modified frusemide test resulted in essentially the same outcome, i.e. the failure to lower urinary pH below 5.5 (nadir urinary pH, 6.5, 6.0, and 6.1 respectively). An abnormal frusemide test was significantly more frequent in osteopenic/osteoporotic patients as compared to the control group (χ2=7.39; P<0.01).
Serum bicarbonate, capillary pH, pCO2, serum creatinine, and potassium were within the normal range and did not differ between the two groups (Table 1).
In the group of patients with normal NH4CI testing but an abnormal frusemide test (n=6) serum bicarbonate (23.5±1.2 mmol/l), capillary pH (7.4±0.02), and capillary pCO2 (38.3 mmHg), serum creatinine (0.9±0.15 mg/dl), and serum potassium (4.2± 0.39 mmol/l) were also not different from those in subjects (n=30) with normal responses in either diagnostic procedure.
Patients with incomplete RTA I presented on average 12 years younger as compared to patients without RTA I (95% CI, 1.6–22.4 years; P=0.025; Table 1). Four of 26 (15%) patients with osteoporosis and six of 20 patients with osteopenia (30%) had incomplete RTA I.
In eight of the 10 subjects with incomplete RTA I, the following disorders possibly related to incomplete RTA I were present: nephrolithiasis and hypercalciuria (n=2), autoimmune thyroid disease (n=2), long‐term habitual analgesic abuse (n=2), nephrocalcinosis due to medullary sponge kidney disease (n=1), and a familial history of RTA I in one patient. In two patients with incomplete RTA I, no associated disorders were revealed. In those six subjects with a normal NH4Cl test but an abnormal urinary pH response to oral frusemide, a past history of nephrolithiasis without current hypercalciuria (n=1), and autoimmune thyroid disease (n=1) were present. In the 30 patients with both a normal frusemide and NH4Cl test, hypercalciuria (n=2), habitual analgesic (n=2), and a past history of nephrolithiasis without current hypercalciuria (n=2) were recognized.
With the NH4Cl test as the gold standard for diagnosis of distal RTA the frusemide test showed a sensitivity of 1.0 (95% CI, 0.69–1.0) and a specificity of 0.89 (95% CI, 0.78–0.96).
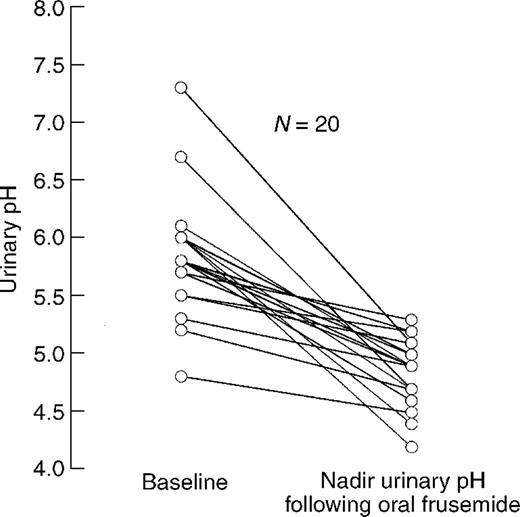
Urinary pH in response to oral frusemide in 20 healthy controls. All subjects were given an oral dose of 40 mg frusemide. Data represent the baseline and the nadir urinary pH during a period of 6 h following oral administration of frusemide.
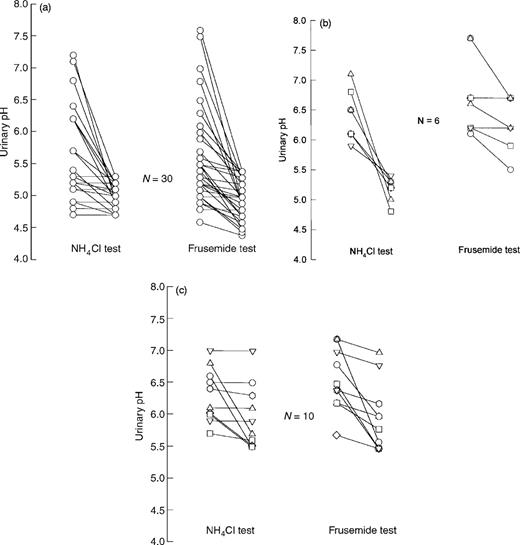
Urinary pH in response to a short‐course oral NH4Cl loading and to frusemide. All subjects were given 0.1 g/kg body weight of encapsulated NH4Cl and an oral dose of 40 mg frusemide. Data represent the base line and the nadir urinary pH during a period of 6 h following the NH4Cl ingestion and frusemide respectively. (a) Results obtained in patients with both normal NH4Cl and frusemide testing (n=30). Note that in nine subjects the individual urinary pH responses to NH4Cl loading were similar. (b) Results in six patients with a normal NH4Cl test but an insufficient fall of urinary pH following the frusemide test. (c) Individual urinary pH data in 10 patients with incomplete RTA I.
Discussion
Bone is critically involved in buffering during the chronic stages of metabolic acidosis [9,10]. A chronically positive acid balance, e.g. due to an impairment of renal acid excretion, may trigger the release of alkali and calcium from the bone and eventually lead to reduction of bone mass and osteoporosis. In the past, the idea of chronic retention of acid as a possible cause of osteoporosis, particularly in postmenopausal women, was put forward by Sebastian et al. [11]. Sanchez and Libman [12] described both proximal and distal renal tubular acidosis in eight patients with osteoporosis. Recently we described a high prevalence of incomplete RTA I in patients with ‘primary’ osteoporosis [3].
The RTA syndromes encompass a disparate group of disorders that have in common an inability to excrete acid that is out of proportion to any reduction in glomerular filtration rate. RTA type I involves defects of acid excretion confined to the distal tubule. The hallmark of overt RTA type I is a hyperchloraemic metabolic acidosis accompanied by a reduced net acid excretion and the inability to lower urinary pH below 5.5 in the face of spontaneous acidaemia or after acid loading. In the complete form of RTA I plasma
Unfortunately, the NH4Cl test is frequently accompanied by abdominal discomfort, even when encapsulated NH4Cl is used. None of the patients studied by us agreed to a prolonged NH4Cl loading test over 3 days.
RTA I resembles a diagnosis with a variety of underlying aetiologies. Despite a recognized association of RTA I with disorders such as medullary sponge kidney, hypercalciuria, analgesic nephropathy, and immuno‐mediated diseases such as lupus nephritis, Sjögren syndrome, and Graves disease [14], the cause of RTA I in some patients remains obscure. Recently, in some of these idiopathic forms of RTA I the absence of H+ATPase in the intercalated cells has been shown [15]. In all but two patients of the RTA I group a thorough work‐up revealed disorders with a recognized association with RTA I. In addition, in two of the six patients with an abnormal frusemide test and a normal NH4Cl test disorders possibly related to distal tubular acidification disturbances were present. One may speculate that in some of these patients these disorders may have contributed to the blunted urinary pH response following oral frusemide.
Acidification in the collecting tubules (CT) is primarily achieved via H+ secretion by a luminal H+ATPase [16]. This pump is located in the intercalated cells of the cortex and in the medulla. The H+ secretion by intercalated cells of the cortical collecting tubule (CCT) is indirectly influenced by the Na+ reabsorption of the adjacent principal cells, since the removal of Na+ from the tubular fluid generates a lumen‐negative potential which facilitates net H+ secretion by minimizing the degree of passive proton back‐diffusion. Any reduction of Na+ reabsorption in the CCT diminishes the degree of lumen negativity (a condition called voltage‐dependent defect) and results in a reduced H+ net secretion and an inability to lower the urinary pH. Application of frusemide provides a means of increasing the delivery of sodium to the sites of distal Na+ reabsorption. Following a rise in intraluminal sodium the Na+ reabsorption in the CCT increases [6] followed by a decrease of tubular fluid pH. In RTA I a common problem is thought to be a defect in the H+ATPase pump that may be present in the cortex and/or the medulla. Patients with this type of RTA I may or may not respond normally to the administration of frusemide. In situations where the H+ATPase defects are limited to the medullary collecting duct, an acidification of the urine may be expected, since the medullary H+ secretion is essentially Na+ dependent [17]. In contrast, a defective H+ secretory pathway in the CCT is expected to cause an inability to lower the urinary pH following frusemide application. The response of urinary pH to frusemide may be blunted by the presence of amiloride‐type diuretics [18], NSAIDs, and volume contraction. Therefore we took care to perform the tests in clinically euvolaemic patients being off diuretics and NSAIDs for at least 2 days. The outcome of the frusemide test in the 10 patients with incomplete RTA I studied by us suggests either a defect of the H+ATPase in the CCT or a defective Na+ reabsorption in the CCT, whereas a defective H+ secretion in the medullary collecting tubules seem to be unlikely.
A dose of 40 mg frusemide given orally was chosen since none of the patients studied had renal insufficiency (the maximum serum creatinine concentration in the study population was 1.1 mg/dl) and since we wanted to avoid significant volume depletion. In patients with renal insufficiency a dose of 80 mg frusemide has been advocated in order to overcome the accompanying resistance to frusemide [19]. Circumstantial evidence from a non‐osteoporotic female with a body weight of 111.5 kg and normal renal function showed that the oral dose of 40 mg may be too low in obese subjects; in this subject, 80 mg of frusemide were necessary to produce a normal urinary acidification. None of the patients studied was overweight. It has been suggested recently that the administration of a mineralocorticoid such as fludrocortisone on the evening preceding the frusemide test may reduce the frequency of false positive results of the frusemide test [5]. In our hands, a fall of urinary pH <5.5 was not observed in the three frusemide‐positive and NH4Cl‐negative subjects who agreed to a fludrocortisone‐modified frusemide test, supporting the view that some form of tubular dysfunction is present. In future studies, the additional measurement of urinary citrate and
A distal acidification defect was found in 10 of 46 patients (22%; CI, 11–36%) with reduced bone mineral content patients when tested by the NH4Cl test and in as many as 16 of 46 (35%; CI, 21–50%) by the frusemide test. When interpreting these frequencies one has to appreciate that the primary aim of the study was not to estimate the prevalence rates of RTA I in patients with osteoporosis or osteopenia. However, it is noteworthy that renal acidification was normal in all 20 subjects with normal bone density. Since the patients studied were referred to the hospital because of suspected osteoporosis, data are not controlled for referral bias. Studies in a randomly selected group of patients with osteopenia or osteoporosis as compared to matched controls with normal bone densities are needed to obtain an unbiased estimation of the prevalence of renal acidification defects.
It is interesting to note that subjects with incomplete RTA I presented at an earlier age (Table 1). Admittedly, this observation may arise from patient selection due to referral bias, but it is attractive to speculate that subjects with incomplete RTA I are prone to a more rapid loss of bone mineral and therefore present at a younger age. Clearly, an additional prospective study in an age‐ and sex‐matched group of patients with and without incomplete RTA I is needed to define the rate of bone loss in patients with incomplete RTA I.
In conclusion, incomplete RTA I, both primary and secondary, is prevalent in patients suffering from reduced bone mineral content. Because of a reduced renal acid excretory reserve in these subjects the ingestion of nutrients stimulating the endogenous acid production (e.g. a diet rich in proteins) may induce a positive acid balance which eventually promotes the release of alkali and calcium from the bone.
Correspondence and offprint requests to: Peter Kotanko MD, Department of Internal Medicine, Krankenhaus der Barmherzigen Brüder, Marschallgasse 12, A‐8020 Graz, Austria.
This study was supported by the Fonds zur Forderung der wissenschaftlichen Forschung, Austria, SFB 007 ‘Biomembranes’.
References
Alpern RJ, Sakhaee K. The clinical spectrum of chronic metabolic acidosis: homeostatic mechanisms produce significant morbidity.
Myburgh KH, Noakes TD, Roodt M, Hough FS. Effects of exercise on the development of osteoporosis in adult rats.
Weger M, Deutschmann H, Weger W, Kotanko P, Skrabal F. Incomplete renal tubular acidosis in ‘primary’ osteoporosis.
Assessment of fracture risk and its application to screening for postmenopausal osteoporosis. Report of WHO study group.
Walter SJ, Shirley DG, Unwin RJ, Wrong OM. Assessment of urinary acidification.
Batlle D. Segmental characterisation of defects in collecting tubule acidification.
Garella S, Salem MM. Clinical acid‐base disorders. In: Davison AM, Cameron JS, Grunfeld JP, Kerr DNS, Ritz E, Winearls CG, ed.
Griner PF, Mayewski RJ, Mushlin AI, Greenland P. Selection and interpretation of tests and procedures.
Green J, Kleeman CR. Role of bone in regulation of systemic acid–base balance.
Sebastian A, Harris ST, Ottaway JH, Todd KM, Morris RC jr. Improved mineral balance and skeletal metabolism in postmenopausal woman treated with potassium bicarbonate.
Sanchez A, Libman J. Trastornos de los mecanismos renales de acidificacion en pacientes osteoporoticos.
Konishi K, Hayashi M, Saruta T. Renal tubular acidosis with autoantibody directed to renal collecting‐duct cells.
Joo KW, Jeon US, Han JS et al. Absence of H+ATPase in the intercalated cells of renal tissues in classical distal renal tubular acidosis.
Levine DZ, Jacobson HR. The regulation of renal acid excretion: new observations from studies of distal nephron segments.
Lombard WE, Kokko JP, Jacobson HR. Bicarbonate transport in cortical and outer medullary collecting tubules.
Batlle D, Flores G. Underlying defects in distal renal tubular acidosis: new understandings.


{kind=link}
{kind=link}
Comments