





Abstract
Background. A prospective multicentre study was initiated in HCV‐infected haemodialysis patients to assess the tolerance and efficacy of α‐2b interferon.
Methods. We had planned to include 120 patients with HCV RNA detectable by polymerase chain reaction (PCR) (Amplicor Roche) and histologically documented chronic hepatitis. The dose of α‐interferon was 3 million units (MU) three times weekly (TTW), to be reduced to 1.5 MU TTW in case of side‐effects. Tolerance was evaluated monthly; virological efficacy was evaluated by PCR. A liver biopsy was performed at month 18 (M18).
Results. (a) Tolerance. After 37 patients had been included, the study was discontinued by the promoting institution because of severe side‐effects requiring that treatment be stopped in 19 patients. The side‐effects were: cardiac (4) neuropsychiatric (2), digestive (3), acute necrosis of the graft (1), severe asthenia (9), minor side‐effects were observed in 22 patients. A complete 12‐month course was completed in 12 patients for the 3 MU TTW dose and in six patients for the 1.5 MU TTW reduced dose. Normal ALT level (OR, 0.16; CI 95%, 0.03–0.89) at inclusion was associated with interruption of treatment (univariate analysis). (b) Efficacy. Sustained virological response was observed in only seven (18.9%), of the 18 patients who completed the treatment (38%). Increased ALT at inclusion (OR, 1.04; CI 95%, 1.01–1.09) and cumulated doses of interferon (OR, 1.01; CI 95%, 1.004–1.026) were jointly associated with a sustained response, while positive PCR at M2 was strongly predictive of treatment failure.
Conclusion: Tolerance of interferon is poor in haemodialysis patients. Sustained response is fairly high in patients who have 12 months of treatment and seems to be based on the immune status of the patients (ALT) and the cumulative doses of interferon.
Introduction
Hepatitis C virus (HCV) infection is a serious problem in the outcome of kidney transplant recipients and hemodialysis patients. Other studies have described a prevalence of anti HCV antibodies in approximately 20% in both situations [1,2], and the evolution of chronic hepatitis due to HCV is a matter of prime concern in the long‐term follow‐up of kidney transplant patients [3–5]. Although the treatment of HCV infection is now based on combined ribavirin and α‐interferon therapy [6], this cannot be routinely used in transplant patients, because of the immunostimulant effect of α interferon and its risk of graft rejection [7]. Therefore these patients must be treated before transplantation, during the haemodialysis period. Unfortunately, ribavirin is metabolized by the kidney and cannot be used in patients with renal failure. Indeed, haemolytic anaemia is associated with ribavirin in up to 80% of treated patients and the risk is markedly increased by dialysis, which contra‐indicates its use [6]. Therefore interferon is the only possible drug in these cases. Several small series of patients have reported the experience of treatment with interferon in these patients with controversial results, on both the efficacy and the tolerance of the drug [8–12]. We and others have studied the pharmacokinetics of interferon in haemodialysis patients, and showed a high concentration of the drug in serum 48 h after administration, with a possible storage effect [13–15]; this could explain the poor tolerance of the drug, but might also allow the administration of lower doses of interferon than in immunocompetent patients.
Therefore we decided to evaluate the efficacy and tolerance of α‐interferon in a prospective multicentre cohort study in HCV‐infected dialysis patients.
Subjects and methods
Inclusion criteria
Haemodialysis patients were considered for treatment if they met the following criteria: (i) infection with HCV documented by positive anti‐HCV test in serum, evidence of viral replication before inclusion with positive HCV RNA by polymerase chain reaction (PCR) (Amplicor Roche), and (ii) chronic hepatitis assessed by a liver biopsy (performed at most 3 years before inclusion) regardless of the Knodell score [16]. Transaminase activity was noted but could be either increased or normal at inclusion.
Patients were excluded if they met at least one of the following criteria: (i) age <18 or >60 years, (ii) receiving immunosuppressive therapy or other treatments, namely anti‐histaminics, omeprazole, non‐steroidal anti‐inflammatory drugs, aciclovir, or amiodarone, (iii) presence of co‐infection with hepatitis B virus or human immunodeficiency virus, (iv) active drug addiction, (v) alcohol consumption >40 g/day, (vi) previous treatment for HCV infection, (vii) evidence of hepatocellular carcinoma (α fetoprotein >100 ng/ml), (viii) haemophiliacs, and (ix) patients with contra‐indication to interferon. All patients provided an informed consent form. The study was approved by the Ethics Committee of Pitié Salpetrière, Paris, France.
Study design and protocol
Patients were scheduled to receive 3 MU s.c. of interferon α‐2b (Schering Plough, Kenilworth, USA) at the end of the haemodialysis procedure session thrice weekly for 48 weeks.
The follow‐up of the patients was in the charge of both nephrologists and hepatologists. The patients were carefully followed up three times a week by the nephrologists, during haemodialysis procedures with weekly haematological and biochemical tests, and key points of the protocol were decided together with the referent hepatologist; a visit was planned with the hepatologist at week 4 to evaluate the treatment tolerance. If important asthenia (auto‐evaluated on visual analogue scale, VAS) or other side‐effects were noted, the dose was reduced to 1.5 MU TTW. Interruption of treatment was considered in case of severe side‐effects or continued side‐effects with the reduced dose (Figure 1). Scheduled follow‐up was 72 weeks (18 months) for each patient. Epidemiological data were recorded before inclusion, and patients were followed both by nephrologists during haemodialysis procedures, and by a hepatologist at weeks 4, 8, and every 2 months until the 18th month. Blood pressure, pulse, body weight and asthenia (by VAS) were evaluated at each visit, and side‐effects were recorded. The study started in October 1995, the last patient was included in May 1997, and inclusion of further patients was interrupted by the promoting institution (DRRC, Assistance Publique‐Hopitaux de Paris) in June 1997, due to the frequency (30%) and the severity of side‐effects. We therefore report our experience on the treatment of the 37 included patients, followed‐up prospectively for 18 months.
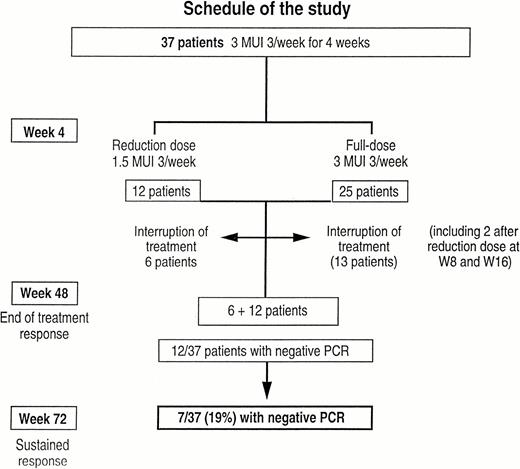
Design and results of the treatment.
Methods
Biological analysis
Blood cell counts and ALT levels were performed and expressed according to the upper limit normal value of each centre.
Virological analysis
Serum samples for virological studies were collected in each centre, and centrally stored at −80°C.
Serological assays
Serum antibodies to HCV (anti‐HCV) were detected with third‐generation HCV enzyme‐linked immunosorbent assay (ELISA associated to analytic RIBA3 test (Ortho Diagnosis Systems, Raritan, NJ, USA) according to the manufacturer's instructions.
Quantitative detection of serum HCV RNA
HCV RNA in the serum was quantified with the Amplicor‐HCV Monitor assay version 1.0 (Roche SA, Neuilly/ Seine, France). To do this longitudinal study, all sequential samples from each patient were tested with the same run of the technique. The Roche test 1.0 was constantly used along this study so that the results could be compared to the initial values previously obtained with the same methodology. Before RNA extraction, 100 μl of serum was added to 900 μl of RPMI and ultracentrifuged at 23500 g for 1 h at 4°C, to avoid PCR inhibitors that are frequently present in the sera of haemodialysis patients. The supernatant was discarded and the pellet was lysed with lysis buffer according to the manufacturer's instructions. The Roche Monitor assay was based on reverse transcription and amplification of the HCV RNA with primers from the 5′NCR in the presence of an internal control that shared primers with HCV. Both competitor and sample DNAs were detected by hybridization to a biotin‐labelled probe, followed by incubation with enzymatically labelled streptavidin and detection in a colorimetric assay.
Genotyping of HCV strains
Genotyping was performed using a commercially available line probe assay (Innolipa, Innogenetics, Gent, Belgium).
Histological analysis
All patients underwent at least one liver biopsy. Liver biopsy samples were embedded in paraffin. After staining by H&E and Perls' stain, biopsies were blindly reviewed by the same pathologist. Chronic liver disease was classified as chronic hepatitis or cirrhosis. Liver disease activity was evaluated according to the Knodell score [16]. The Knodell score provides semi‐quantification of four indices: periportal necrosis (range 0–10), intralobular degeneration and focal necrosis, portal inflammation and fibrosis (range 0–4). Liver biopsies were performed before treatment and 18 months after entering the study. Specimens were analysed in each centre, and graded using the Knodell scoring system by histology activity index according to the degree of periportal necrosis, portal and lobular inflammation (total activity), and fibrosis.
Endpoints
The main endpoint was classically defined as negative HCV RNA by PCR at 18 months, defining long‐term response [6]. Responders and non‐responders were defined as patients with negative or positive PCR by the end of treatment respectively. Relapse was defined as negative PCR at the end of treatment but positive PCR at the end of follow‐up. Sustained response was defined as negative PCR at the end of treatment and at the end of follow‐up (18 months). Histological response was based on a liver biopsy at month 18.
Statistical analysis
Actuarial occurrences of dose reduction, dose discontinuation and severe side‐effects over time were separately analysed using the Kaplan–Meier method.
Predictive factors for treatment discontinuation and for sustained response were analysed separately, using univariate then multivariate modelling, both based on the logistic regression model. The association between covariates and outcome were represented by estimated odds ratio (OR) with 95% confidence intervals (CI 95%). Two‐sided tests were computed with P value <0.05 defining statistical significance. SAS software package (SAS Inc, Cary, NC, USA) was used for the statistical analysis.
Results
The epidemiological, clinical, biological, virological and histological data at inclusion are given in Table 1.
Safety
The legal promoter had to interrupt the study prematurely due to the frequency and severity of side‐effects. Dose reduction was required in 21 patients (56.7%), after a median time to dose reduction of 24 weeks, with 12 patients at week 4, and the other nine before week 32. The figures of dose reduction according to the duration of treatment are displayed in Figure 2a.
Treatment was stopped in 19 patients (51%) (six patients who had had a previous dose reduction at week 4, and 13 patients who had had a full dose at week 4, but had dose reduction at week 8 and 16 respectively). The figures for interruption of treatment are displayed in Figure 2b. The reasons for interruption of treatment were severe neurological disorders (seizures in one patient and psychiatric disorders in another patient), major asthenia (nine patients, including seven after a reduced dose), cardiovascular disorders (four patients), severe anorexia (one patient), acute pancreatitis (one patient), severe diarrhoea (one patient), acute necrosis of a previously non‐functional renal allograft (one patient). (This patient was previously treated by renal transplantation, had chronic renal failure leading to haemodialysis, but the non‐functional graft had not been removed at the time of interferon administration.) Normal ALT activity was associated with interruption of treatment in univariate analysis but no factor was found as predictive of treatment discontinuation by multivariate analysis (Table 2).
Side‐effects were divided into severe and mild effects, and occurred in 89% of the patients. Severe side‐effects were defined as life‐threatening or death, or occurrence of cancer. They were observed in 12 patients (32%): treatment was discontinued in nine patients (pulmonary oedema, cerebral haemorrhage, acute pancreatitis, dilated cardiomyopathy, cervical adenopathies linked to an HTLV1 lymphoma that developed during interferon administration, diplopia; while treatment was continued in three patients (acute transient abdominal pain, acute necrosis of the kidney graft, and septic shock). The median time until occurrence of side‐effects was 29.5 weeks (Figure 2c). Mild side‐effects were observed in 22 patients (60%). In addition, an increase of the weekly dose of erythropoietin was needed in 11 patients. Finally, 18 patients reached week 48 on treatment (six patients with a reduced dose and 12 patients with a full dose) (Figure 1).
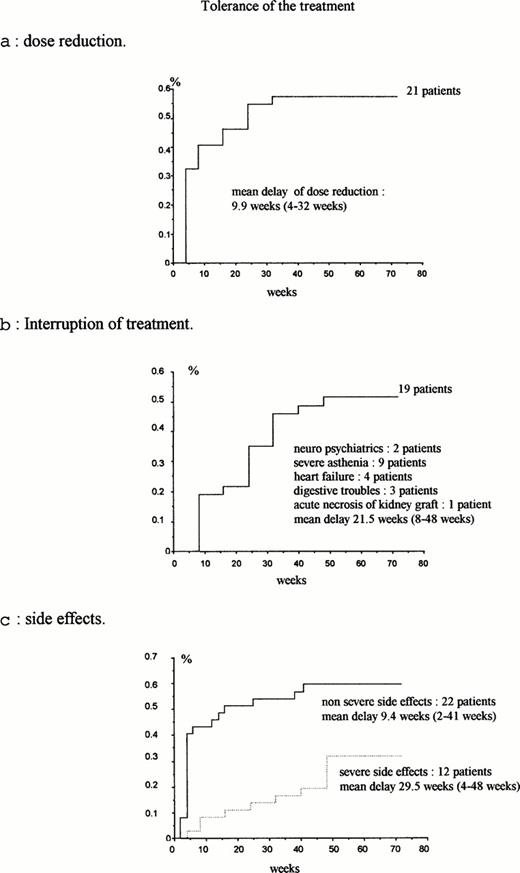
(a) Dose reduction; (b) interruption of treatment; (c) side‐effects.
Characteristics of the 37 patients at inclusion
| Males/females | 25/12 (68/32%) |
| Age (years) | 45±12 |
| Weight (kg) | 62±9 |
| Geographic origin: | |
| France | 38% |
| North Africa | 32% |
| Other | 30% |
| Route of contamination: | |
| Blood transfusion | 24 patients (64%) |
| Unknown | 12 patients (32%) |
| Drug addiction | 1 patient (3%) |
| Duration of the disease (years) | 7.8±6.8 |
| Transaminase activity: | |
| normal | 26 patients (70%) |
| >normal | 11 patients (30%) |
| HCV genotype: | |
| 1a+1b | 29 patients (78%) |
| 2 | 5 patients (13%) |
| 3 | 1 patient (3%) |
| Median viraemia at initiation of treatment | 5.12 (2.5–5.8) |
| (log copies/ml) | |
| PCR positive (Amplicor 100 copies) (patients) | 36*/37 |
| Knodell score: | |
| Fibrosis | 1.1±0.8 |
| Activity | 4±2 |
| Asthenia (VAS: 0–100)** | 37±29 |
| Males/females | 25/12 (68/32%) |
| Age (years) | 45±12 |
| Weight (kg) | 62±9 |
| Geographic origin: | |
| France | 38% |
| North Africa | 32% |
| Other | 30% |
| Route of contamination: | |
| Blood transfusion | 24 patients (64%) |
| Unknown | 12 patients (32%) |
| Drug addiction | 1 patient (3%) |
| Duration of the disease (years) | 7.8±6.8 |
| Transaminase activity: | |
| normal | 26 patients (70%) |
| >normal | 11 patients (30%) |
| HCV genotype: | |
| 1a+1b | 29 patients (78%) |
| 2 | 5 patients (13%) |
| 3 | 1 patient (3%) |
| Median viraemia at initiation of treatment | 5.12 (2.5–5.8) |
| (log copies/ml) | |
| PCR positive (Amplicor 100 copies) (patients) | 36*/37 |
| Knodell score: | |
| Fibrosis | 1.1±0.8 |
| Activity | 4±2 |
| Asthenia (VAS: 0–100)** | 37±29 |
*One patient was PCR positive at selection and PCR negative at initiation of treatment. **VAS, visual analogue scale grading the intensity of the symptoms from 0 to 10.
Characteristics of the 37 patients at inclusion
| Males/females | 25/12 (68/32%) |
| Age (years) | 45±12 |
| Weight (kg) | 62±9 |
| Geographic origin: | |
| France | 38% |
| North Africa | 32% |
| Other | 30% |
| Route of contamination: | |
| Blood transfusion | 24 patients (64%) |
| Unknown | 12 patients (32%) |
| Drug addiction | 1 patient (3%) |
| Duration of the disease (years) | 7.8±6.8 |
| Transaminase activity: | |
| normal | 26 patients (70%) |
| >normal | 11 patients (30%) |
| HCV genotype: | |
| 1a+1b | 29 patients (78%) |
| 2 | 5 patients (13%) |
| 3 | 1 patient (3%) |
| Median viraemia at initiation of treatment | 5.12 (2.5–5.8) |
| (log copies/ml) | |
| PCR positive (Amplicor 100 copies) (patients) | 36*/37 |
| Knodell score: | |
| Fibrosis | 1.1±0.8 |
| Activity | 4±2 |
| Asthenia (VAS: 0–100)** | 37±29 |
| Males/females | 25/12 (68/32%) |
| Age (years) | 45±12 |
| Weight (kg) | 62±9 |
| Geographic origin: | |
| France | 38% |
| North Africa | 32% |
| Other | 30% |
| Route of contamination: | |
| Blood transfusion | 24 patients (64%) |
| Unknown | 12 patients (32%) |
| Drug addiction | 1 patient (3%) |
| Duration of the disease (years) | 7.8±6.8 |
| Transaminase activity: | |
| normal | 26 patients (70%) |
| >normal | 11 patients (30%) |
| HCV genotype: | |
| 1a+1b | 29 patients (78%) |
| 2 | 5 patients (13%) |
| 3 | 1 patient (3%) |
| Median viraemia at initiation of treatment | 5.12 (2.5–5.8) |
| (log copies/ml) | |
| PCR positive (Amplicor 100 copies) (patients) | 36*/37 |
| Knodell score: | |
| Fibrosis | 1.1±0.8 |
| Activity | 4±2 |
| Asthenia (VAS: 0–100)** | 37±29 |
*One patient was PCR positive at selection and PCR negative at initiation of treatment. **VAS, visual analogue scale grading the intensity of the symptoms from 0 to 10.
Prognostic factors of interruption of treatment (univariate analysis)
| OR (95% CI) | P value | |
| Genotype non 1 | 1.07 (0.19–6.22) | 0.94 |
| ALT | 0.97 (0.94–1.00) | 0.057 |
| ALT >normal | 0.16 (0.03–0.89) | 0.04 |
| VAS | 1.03 (0.99–1.07) | 0.14 |
| Body mass index | 1.27 (0.96–1.68) | 0.10 |
| Age | 1.04 (0.98–1.11) | 0.18 |
| Duration of the disease | 0.96 (0.86–1.07) | 0.48 |
| High viraemia (>3×106) | 0.35 (0.09–1.37) | 0.13 |
| OR (95% CI) | P value | |
| Genotype non 1 | 1.07 (0.19–6.22) | 0.94 |
| ALT | 0.97 (0.94–1.00) | 0.057 |
| ALT >normal | 0.16 (0.03–0.89) | 0.04 |
| VAS | 1.03 (0.99–1.07) | 0.14 |
| Body mass index | 1.27 (0.96–1.68) | 0.10 |
| Age | 1.04 (0.98–1.11) | 0.18 |
| Duration of the disease | 0.96 (0.86–1.07) | 0.48 |
| High viraemia (>3×106) | 0.35 (0.09–1.37) | 0.13 |
Prognostic factors of interruption of treatment (univariate analysis)
| OR (95% CI) | P value | |
| Genotype non 1 | 1.07 (0.19–6.22) | 0.94 |
| ALT | 0.97 (0.94–1.00) | 0.057 |
| ALT >normal | 0.16 (0.03–0.89) | 0.04 |
| VAS | 1.03 (0.99–1.07) | 0.14 |
| Body mass index | 1.27 (0.96–1.68) | 0.10 |
| Age | 1.04 (0.98–1.11) | 0.18 |
| Duration of the disease | 0.96 (0.86–1.07) | 0.48 |
| High viraemia (>3×106) | 0.35 (0.09–1.37) | 0.13 |
| OR (95% CI) | P value | |
| Genotype non 1 | 1.07 (0.19–6.22) | 0.94 |
| ALT | 0.97 (0.94–1.00) | 0.057 |
| ALT >normal | 0.16 (0.03–0.89) | 0.04 |
| VAS | 1.03 (0.99–1.07) | 0.14 |
| Body mass index | 1.27 (0.96–1.68) | 0.10 |
| Age | 1.04 (0.98–1.11) | 0.18 |
| Duration of the disease | 0.96 (0.86–1.07) | 0.48 |
| High viraemia (>3×106) | 0.35 (0.09–1.37) | 0.13 |
Efficacy
End of treatment response was evaluated in 12/18 of those patients who completed treatment (66%). The sustained response was evaluated 6 months after treatment discontinuation and occurred in 7/37 patients (18.9%), among whom five of the 29 patients had genotype 1 (17%). However, in patients who underwent 12‐month treatment, the sustained virological response was observed in 7/18 (38%). All patients with a sustained response had negative PCR at month 2, while no end of treatment or sustained response was observed in patients with positive PCR at month 2.
Iterative 6‐month post‐treatment liver biopsy (performed at the end of follow‐up, 72 weeks) could be obtained in 20/37 patients, (including seven sustained responders). The Knodell fibrosis scores were stable in 14/20 (70%) and improved in 2/20 (10%). By contrast, increase of the fibrosis score (one point) was noted in 4/20 cases, none of the four patients being a sustained responder.
Knodell activity scores were stable or improved in most cases (12/20 (60%) and 2/20 (10%) respectively). Iron overload was minor in all patients.
Body mass index, age, transaminase activity >normal, and cumulated doses of interferon were related to sustained response in the univariate analysis (Table 3). Due to the small number of events (seven patients with sustained response), we did not perform multivariate analysis.
Prognostic factors of sustained response: univariate analysis
| OR | CI 95% | |
| Viraemia at inclusion | 0.81 | (0.33–1.95) |
| Genotype 1 | 2.40 | (0.34–16.9) |
| Body mass index | 0.62 | (0.40–0.97) |
| Age | 0.92 | (0.86–1.01) |
| Duration since contamination | 0.98 | (0.84–1.14) |
| ALT >normal | 1.04 | (1.01–1.09) |
| Knodell fibrosis score* | 0.86 | (0.37–2.03) |
| Knodell histological activity score* | 1.15 | (0.80–1.67) |
| Cumulative dose of interferon | 1.01 | (1.004–1.026) |
| OR | CI 95% | |
| Viraemia at inclusion | 0.81 | (0.33–1.95) |
| Genotype 1 | 2.40 | (0.34–16.9) |
| Body mass index | 0.62 | (0.40–0.97) |
| Age | 0.92 | (0.86–1.01) |
| Duration since contamination | 0.98 | (0.84–1.14) |
| ALT >normal | 1.04 | (1.01–1.09) |
| Knodell fibrosis score* | 0.86 | (0.37–2.03) |
| Knodell histological activity score* | 1.15 | (0.80–1.67) |
| Cumulative dose of interferon | 1.01 | (1.004–1.026) |
*Initial biopsies, before initiation of treatment.
Prognostic factors of sustained response: univariate analysis
| OR | CI 95% | |
| Viraemia at inclusion | 0.81 | (0.33–1.95) |
| Genotype 1 | 2.40 | (0.34–16.9) |
| Body mass index | 0.62 | (0.40–0.97) |
| Age | 0.92 | (0.86–1.01) |
| Duration since contamination | 0.98 | (0.84–1.14) |
| ALT >normal | 1.04 | (1.01–1.09) |
| Knodell fibrosis score* | 0.86 | (0.37–2.03) |
| Knodell histological activity score* | 1.15 | (0.80–1.67) |
| Cumulative dose of interferon | 1.01 | (1.004–1.026) |
| OR | CI 95% | |
| Viraemia at inclusion | 0.81 | (0.33–1.95) |
| Genotype 1 | 2.40 | (0.34–16.9) |
| Body mass index | 0.62 | (0.40–0.97) |
| Age | 0.92 | (0.86–1.01) |
| Duration since contamination | 0.98 | (0.84–1.14) |
| ALT >normal | 1.04 | (1.01–1.09) |
| Knodell fibrosis score* | 0.86 | (0.37–2.03) |
| Knodell histological activity score* | 1.15 | (0.80–1.67) |
| Cumulative dose of interferon | 1.01 | (1.004–1.026) |
*Initial biopsies, before initiation of treatment.
Discussion
This study provides prospective results on both the tolerance and efficacy of interferon in haemodialysis patients with HCV infection. Results in previous studies are controversial [8–12], showing either the treatment to be effective when possible, but with serious side‐effects, or poor results.
The poor tolerance of interferon in haemodialysis patients may be related to the pharmacokinetics of the drug. Prior pharmacokinetic studies performed with natural interferon showed that the clearance of interferon was not modified by haemodialysis [17]. However, we and others have performed pharmacokinetic studies of recombinant interferon and shown that the serum concentrations of interferon were significantly higher in dialysis than in patients with normal renal function [13–15]; then providing a possible explanation of the poor tolerance of the drug. Side‐effects were noted at all periods of the drug administration of the drug and occurred in many cases. However, this frequency should be considered in relation to the spontaneous occurrence of cardiac and neoplastic events in these patients, who have an annual mortality rate of approximately 15% [18]. Special attention must be paid to the possible acute necrosis of the non‐functional kidney graft, which is immunologically reactivated by interferon, and nephrectomy of the graft must be discussed prior to treatment when necessary. Moreover, our results did not provide predictive factors of the interruption of treatment, except ALT activity above the upper limit of normal and normal body mass index, both factors that probably reflect a good condition in the patients.
The sustained effect of therapy is higher, even if it is still low, in haemodialysis patients than that in patients with normal renal function and genotype 1 [6], not only if the results are analysed on an intent‐to‐treat basis (18.9%) but more strikingly if the patients reaching the end of treatment period are taken into account (7/18, 38%). Moreover these patients were mostly infected by genotype 1, which is related to a lower response (from 3 to 18%) in immunocompetent patients [6].
Results of univariate analysis
Increased ALT activity was found to be predictive of sustained response. This probably reflects the immune response of the host to HCV infection, and patients with chronic hepatitis and normal ALT probably have important immune response impairment. All patients with a sustained response had negative PCR at month 2, while positive PCR at month 2 was never associated with a sustained response. However, we did not include a negative PCR at month 2 in the logistic model since negative PCR was already included in the definition of the end‐point (sustained response). If, due to marked side‐effects, a dose reduction, or interruption of treatment is discussed in patients treated with interferon, the efficacy of treatment should be evaluated in relation to negative PCR after 2 months, so that therapy will only be continued in responders, since this subgroup has better chance of long‐term sustained response. Treatment would then be interrupted in patients with positive PCR and side‐effects.
The consequences of HCV infection are a matter of concern during haemodialysis and the transplantation periods. The prevalence of HCV infection in haemodialysis patients is nearly 20%, due to either blood transfusion, with a markedly reduced risk due to administration of erythropoietin, nosocomial transmission, or the transplanted kidney [19]. Nosocomial transmission of HCV is still observed, even if rarely, in haemodialysis units [2]. HCV infection is an important issue in the long‐term survival of patients treated by renal transplantation [3–5]. Antiviral therapy with interferon has been shown to be potentially dangerous, inducing severe rejection episodes [7], and is no longer used in transplanted patients. Therefore, interferon administration must be undertaken before transplantation.
In conclusion, this prospective study showed the poor tolerance of interferon in haemodialysis patients. Therefore this treatment should only be offered to patients who are waiting for kidney transplantation, to avoid the severe evolution of liver disease under immunosuppressive therapy, or in those with severe liver disease to limit development of cirrhosis. When the treatment has been decided, the physician and the patient must be aware that there is a risk of severe side‐effects, leading to the interruption of treatment, and that occurs at any time during the course of treatment. However, these limitations are counterbalanced by a fairly high chance of sustained response in patients reaching 12 month of treatment.
Correspondence and offprint requests to: Dr Françoise Degos, Service d'Hépatologie, Hôpital Beaujon, 100 Bd du Gl Leclerc, F‐92110 Clichy, France.
We thank Mrs Emmanuelle Andre for her helpful assistance in the monitoring of the multicentre trial. This work was supported by a grant from the ‘Programme Hospitalier de Recherche Clinique’, AOA 94022 Ministère de la Santé, Paris France.
References
Zeldis JB, Depner TA, Kuramoto IK, Gish RG, Holland PV. The prevalence of hepatitis C virus antibodies among hemodialysis patients.
Jadoul M, Cornu C, Van Ypersele de Strihou C and the UCL collaborative group. Incidence and risk factors for hepatitis C virus seroconversion in hemodialysis. A prospective study.
Pereira BJ, Natov SN, Bouthot BA et al. Effects of hepatitis C infection and renal transplantation on survival in end‐stage renal disease.
Legendre C, Garrigue V, Le Bihan C et al. Harmful long‐term impact of hepatitis C virus infection in kidney transplant recipients.
Mathurin P, Mouquet C, Poynard T et al. Impact of hepatitis B and C virus on kidney transplantation outcome.
Rostaing L, Izopet J, Baron E, Duffaut M, Puel J, Durand D. Treatment of chronic hepatitis C with recombinant interferon alpha in kidney transplant recipients.
Koenig P, Vogel W, Umlauft F et al. Interferon treatment for chronic hepatitis C virus infection in uremic patients.
Pol S, Thiers V, Carnot F et al. Efficacy and tolerance of alpha‐2b interferon therapy on HCV infection of hemodialyzed patients.
Izopet J, Rostaing L, Moussion F et al. High rate of hepatitis C virus clearance in hemodialysis patients after interferon‐alpha therapy.
Chan TM, Wu PC, Lau JY, Lok AS, Lai CL, Cheng IK. Interferon treatment for hepatitis C virus infection in patients on haemodialysis.
Huraib S, Tanimu D, Romeh SA et al. Interferon‐alpha in chronic hepatitis C infection in dialysis patients.
Buisson C, Degos F, Daniel F et al. Pharmacokinetics of interferon alpha 2b in haemodialysis.
Uchihara M, Izumi N, Sakai Y et al. Interferon therapy for chronic hepatitis in hemodialysis patients: increased serum levels of interferon.
Rostaing L, Chatelut E, Payen JL et al. Pharmacokinetics of alpha interferon‐2b in chronic hepatitis C virus patients undergoing chronic hemodialysis or with normal renal function: clinical implications.
Knodell RG, Ishak KG, Black WC et al. Formulation and application of a numerical scoring system for assessing histological activity in asymptomatic chronic active hepatitis.
Hirsch MS, Tolkoff‐Rubin NE, Kelly AP, Rubin RH. Pharmacokinetics of human and recombinant leucocyte interferon in patients with chronic renal failure who are undergoing hemodialysis.
Wolfe RA, Ashby VB, Milford EL et al. Comparison of mortality in all patients on dialysis, patients on dialysis awaiting transplantation and recipients of a first cadaveric transplant.


{kind=link}
{kind=link}
Comments