





Abstract
Background. The treatment approaches to antineutrophil cytoplasmic autoantibody (ANCA) small vessel vasculitis expose patients to the risks associated with long-term use of corticosteroids and cytotoxic agents. In an effort to explore approaches to minimize risks, we conducted a pilot efficacy and safety study of mycophenolate mofetil (MMF) in the treatment of subjects with nonlife-threatening recurrent or cyclophosphamide-resistant ANCA-vasculitis.
Methods. MMF was initiated at 500 mg orally twice daily and gradually increased to a target dose of 1000 mg twice daily for a duration of 24 weeks. Concomitant therapy with corticosteroids was allowed. The Birmingham Vasculitis Activity Score (BVAS) was used to assess disease activity and treatment efficacy. ANCA titres, serum creatinine and adverse events were secondary measures of efficacy and/or toxicity.
Results. Twelve subjects were enrolled in the study. Treatment with MMF led to an improvement in disease activity as measured by the BVAS at 24 weeks (P = 0.0013) and 52 weeks (P = 0.0044) as compared to baseline. The BVAS decreased from an average of 9.1±3.5 at baseline (range, 3–17) to an average of 2.8±1.9 (range, 1–6) at 24 weeks and to 2.8±4.3 (range, 0–13) at 52 weeks. Early and sustained reductions in BVAS occurred in subjects initially classified as disease relapses vs those with treatment resistance. Side effect profile was consistent with the mechanism of action and pharmacokinetic disposition of MMF.
Conclusions. MMF is a reasonable option in the treatment of non-life-threatening recurrent or resistant vasculitis and may obviate the immediate need for recurrent use of cytotoxic agents.
Introduction
Antineutrophil cytoplasmic autoantibodies (ANCA) are associated with a group of pauci-immune small vessel vasculitides (SVV), which includes microscopic polyangiitis, Wegener's granulomatosis and the Churg–Strauss syndrome (CS). The main treatment of SVV has been the combination of corticosteroids and cyclophosphamide as initially suggested by Fauci et al. [1]. ANCA-positive patients who achieve a complete remission within 6 months of therapy usually have cyclophosphamide discontinued with close patient follow-up with or without maintenance immunosuppression (with azathioprine, for example) [2]. In patients with persistently active disease at 6 months, cyclophosphamide is usually continued for a total of 12 months. Although the optimum length of therapy with cyclophosphamide has not been determined, its long-term use is prohibited by its associated risks, which include reduced bone mineral density, bone marrow suppression, infection, infertility and secondary malignancies. Chronic glucocorticoid therapy also has significant toxicity including osteoporosis, increased risk of infection, glucose intolerance, and changes in body habitus.
It has been recognized for years that cyclophosphamide is a necessary adjunct to prednisone therapy for the treatment of ANCA-associated vasculitis [1]. Treatment with cyclophosphamide is beneficial over the use of corticosteroids alone for achieving a remission as well as for patient survival [3,4]. Patients treated with either intravenous or oral cyclophosphamide have a long-term remission rate between 60 and 85% [3,5]. The risk of relapse after an initial response to cyclophosphamide treatment is ∼30% [4]. Relapse typically occurs in the same organ system that was initially affected by the disease, although new organ system involvement can occur as well [4]. Fortunately, a similar rate of response is achieved in the treatment of relapse and initial disease [4]. Re-treatment is therefore an important and beneficial option. While only rare patients exhibit complete resistance to treatment with cyclophosphamide, a minority of patients maintain some degree of disease activity despite prompt institution of immunosuppressive therapy.
The best treatment approach for relapsing ANCA-associated vasculitis is currently a matter of substantial investigation. Therapies that have been used include trimethoprim-sulfamethoxazole [6], methotrexate, azathioprine and cyclosporine [7]. Mycophenolate mofetil (MMF) is a newer immunosuppressant that is gaining interest as a treatment for ANCA-associated vasculitis. Other therapies under recent investigation include TNF-α blockade with infliximab and etanercept, B-cell depletion with rituximab, and deoxyspergualin.
Based on the mechanism of action and suggested efficacy in several immunologically derived diseases, we conducted a feasibility, dose-escalating pilot study of MMF in the treatment of ANCA-associated SVV in subjects with relapsing disease or with disease that was resistant to cyclophosphamide treatment with the goal of reducing disease activity. Treatment with MMF was offered to subjects with recurrent or persistent non-life-threatening vasculitis and a history of prior treatment with cyclophosphamide.
Subjects and methods
This study was a feasibility study to assess the safety and effectiveness of MMF in inducing remission in subjects with ANCA-associated SVV who were resistant to cyclophosphamide therapy or had relapsing disease. Our study was conducted using an open-label, dose-escalating approach to treatment. It was approved by the institutional Committee on the Protection of the Rights of Human Subjects. At baseline, subjects were defined as being either treatment resistant or as having a relapse. Subjects were defined as being treatment resistant if they had a progressive decline in renal function with persistence of active urine sediment or the persistence or new appearance of any extra-renal manifestation of vasculitis despite immunosuppressive therapy. Subjects were deemed to have a relapse if, after a period of remission at least one of the following conditions were present: (i) rapid rise in serum creatinine accompanied by an active urine sediment; (ii) a renal biopsy demonstrating active necrosis or crescent formation; (iii) haemoptysis, pulmonary haemorrhage, new or expanding infiltrates or nodules without evidence for infection or any other active vasculitis of the respiratory tract; (iv) vasculitic involvement of the gastrointestinal tract as demonstrated by endoscopy; (v) iritis or uveitis; (vi) new mononeuritis multiplex; or (vii) necrotizing vasculitis identified by biopsy in any tissue. To be eligible, subjects also had to meet at least one of the following additional criteria: (i) treatment with at least two prior courses of cyclophosphamide (a course is defined as a 6 month cycle of once monthly intravenous or daily oral cyclophosphamide); (ii) development of intolerance to cyclophosphamide or azathioprine (intolerance is defined as severe gastrointestinal side effects, severe bone marrow suppression, or infectious complications); (iii) treatment with a complete course of cyclophosphamide and refusal to be re-treated with this drug because of concerns about malignancies or gonadal failure; (iv) current disease activity despite a full course of cyclophosphamide.
After enrolment, subjects were followed longitudinally, and formal measurements of disease activity were determined using the Birmingham Vasculitis Activity Score (BVAS) [8]. The BVAS is a clinical index measurement of disease activity based on the signs and symptoms in nine separate organ systems and is weighted to reflect the clinical importance of specific organ involvement. The BVAS includes two scores. BVAS.1 quantifies present/persistent organ-specific signs or symptoms, and BVAS.2 quantifies signs of new or worsening disease activity. By measuring abnormalities that have developed within the 4 weeks that precede each scoring, it is devised to be a reflection of disease activity only, without regard to the degree of cumulative damage (whether from the disease itself or its treatment). The BVAS assessments were to be performed at least at study entry, midpoint of therapy, and study conclusion (after 24 weeks of therapy). BVAS assessment at 52 weeks (6 months after completion of the active study) was performed if feasible. Effectiveness of MMF therapy was defined by the changes in BVAS scores. Outcomes including remission, relapse and poor response to therapy were also documented. Treatment response (disease remission) was defined as a stabilization or improvement of renal function (as measured by serum creatinine), resolution of haematuria, and resolution of extra-renal manifestations of systemic vasculitis. A disease relapse was defined as an increase in vasculitis disease activity following a period of remission. A poor response to treatment was defined as a reduced or delayed response to therapy. Persistence of proteinuria was not considered to be indicative of persistence of disease activity.
Secondary measures of efficacy included assessment of ANCA titres and ability to reduce the dose or withdraw therapy with corticosteroids. ANCA titres were measured during the study with a frequency that was indicated by the nephrologist caring for the individual subjects. ANCA titres were assessed by indirect immunofluorescence microscopy on ethanol fixed neutrophils and by antigen-specific ELISA.
The exclusion criteria for this study included: (i) women who were pregnant or lactating, or who had child-bearing potential and were not willing to employ effective contraception during the trial; (ii) renal insufficiency caused by obstruction, infection or nephrotoxic drugs; (iii) ANCA-associated vasculitis with glomerulonephritis only (no extra-renal manifestation of vasculitis) in the setting of advanced renal failure requiring dialysis or evidence of advanced scarring on renal biopsy (>75% globally sclerotic glomeruli, or advanced interstitial fibrosis); (iv) acute or chronic infections that required antimicrobial therapy, or serious viral infections (e.g. hepatitis, herpes zoster), or tuberculosis; (v) serologic evidence for HIV infection; (vi) history of diabetes mellitus; (vii) leukopaenia, with white blood cell counts <3000 cells/mm3; (viii) history of lymphoproliferative disease or a recent malignancy within the previous 5 years; or (ix) history of drug sensitivity to MMF or mycophenolic acid.
Treatment protocol
Subjects with relapse
Subjects entering with relapsing disease could receive oral prednisone at a dose of 1 mg/kg per day (not to exceed 80 mg per single dose) for 30 days. Corticosteroids were then tapered to an alternate day regimen during the second month, then tapered by 25% per week for 4 weeks and discontinued by the end of the third month. Subjects with a rapid loss of renal function, or evidence of severe disease activity on renal biopsy could also receive initial treatment with intravenous pulse methylprednisolone at a dose of 7 mg/kg per day for 3 days, followed by a regimen of daily oral corticosteroids.
MMF (CellCept®, Roche Laboratories, NJ) 500 mg twice daily was instituted on the first study day and increased to 1000–1500 mg twice daily by increments of 250 mg twice daily every 2 weeks. The target dosage was defined as 1000 mg twice daily. A maximal dose of 1500 mg twice daily was to be attempted only in subjects who failed to show evidence of response to treatment at the dose of 1000 mg twice daily. Dosage titration was dependent on the preservation of a leukocyte count >3000 cells/mm3 and the absence of severe side effects. All subjects received supportive care, including blood pressure control (blood pressure <140/90 mmHg), and calcium and vitamin D for the prevention of osteoporosis in subjects receiving corticosteroids. MMF was continued at the maximum tolerated dose for a total of 24 weeks. After achieving the target dose, leukocyte counts were measured monthly, and the dose of MMF was adjusted to maintain a leukocyte count >3000 cell/mm3. Medication counts were conducted to determine whether subjects were compliant with the prescribed MMF therapy.
Subjects with resistance to cyclophosphamide
Subjects failing to improve after at least 6 months of treatment with cyclophosphamide were eligible for this study after discontinuing the cyclophosphamide. MMF was instituted following the same dosing regimen as for the treatment of relapse. These subjects were not to receive pulse methylprednisolone or an increase in corticosteroids to 1 mg/kg/day. If the patient was receiving corticosteroids, the same dose was initially maintained, and subsequently tapered according to the protocol described above, as permitted by the degree of disease activity.
Subjects had study visits in the nephrology clinic at baseline, and weeks 4, 8, 12, 16, 20, 24, 32 and 52 (study termination) which included assessments of disease activity, adverse effects and compliance, dosage titration, and laboratory analysis (including electrolytes, blood urea nitrogen, serum creatinine and transaminases). Spot urine collections were performed monthly to assess protein to creatinine ratios. Stop points for ending participation in this study were: (i) the progression of renal disease to end-stage renal failure requiring the need for dialysis or transplantation; (ii) active gastrointestinal bleeding; (iii) serious infection that caused the subject to interrupt therapy for more than 1 month; (iv) activation of tuberculosis; (v) psychosis not responsive to a reduction in the dose of corticosteroids; (vi) persistent leukopaenia defined as a white blood cell count of <2000/mm3 despite a lowered dose of MMF; and (vi) pregnancy.
The active phase of participation in this study concluded at week 24. From week 24 to 52, subjects could either continue MMF therapy or begin another medication in addition to or in place of MMF. The principal investigator made the decision regarding follow-up treatment based on the clinical response of each subject to therapy.
The safety of MMF in the treatment of ANCA-associated vasculitis was assessed by analysis of adverse events reported during the study. Individual complaints that were attributable to vasculitis were considered unrelated to MMF therapy. Only adverse events that were likely to be related to MMF therapy were included in the analysis.
Descriptive statistics for baseline subject demographics were computed and presented as means and standard deviations for continuous measures and as absolute numbers and percentages for categorical measures. Graphs for total and individual subject changes in BVAS scores were generated. Comparisons between baseline and 6 month BVAS scores (and ANCA titres) and between baseline and 12 month BVAS scores (and ANCA titres) were analysed by the Mann–Whitney U test at an α value of 0.05.
Results
A total of 12 adult subjects with ANCA-SVV relapsing disease (n = 6) or disease resistant to cyclophosphamide treatment (n = 6) consented to participate.
The medical diagnosis of their SVV included Wegener's granulomatosis (WG) (n = 7), microscopic polyangiitis (MPA) (n = 2), necrotizing and crescentic glomerulonephritis alone (NCGN) (n = 2) and CS (n = 1). The laboratory analysis of ANCA serology showed a PR3-ANCA and a MPO-ANCA pattern in nine (75%) and three (25%) subjects, respectively. Six subjects were enrolled with persistent or worsening disease despite treatment with prednisone, and cyclophosphamide or azathioprine. Two patients had not received prior cyclophosphamide therapy. Patient no. 4 was a frail elderly patient with a prior history of colon cancer. Patient no. 10 was a young female with mild recurrent nephritis who was reluctant to receive cyclophosphamide because of the associated risk of infertility.
The remaining six subjects were enrolled with relapsing disease. The average age for participants was 45.3±18.4 years. The gender of the subjects was equally divided. All participating subjects were Caucasian. At study entry, eight (66%) subjects had renal disease activity while six (50%) had upper respiratory tract, sinus or ear involvement (Table 1).
Summary of subject's characteristics at study entry
| Subject | Age | Gender | Diagnosis | ANCA | Prior therapy | Organ involvement at initial presentation | Organ involvement at study enrolment | Relapse/resistance |
|---|---|---|---|---|---|---|---|---|
| 1 | 34 | F | WG | PR3 | P, CyP | ENT, MS, R | ENT | Resistance |
| 2 | 36 | F | WG | PR3 | P, CyP | ENT, L, R | ENT, N | Relapse |
| 3 | 53 | F | CS | PR3 | P, CyP | ENT, L, MS,R | ENT, R | Resistance |
| 4 | 78 | F | MPA | MPO | P, AZA | N, C, R | N, R | Resistant/intolerant |
| 5 | 46 | M | MPA | PR3 | P, CyP | ENT, L, R | R | Relapse |
| 6 | 41 | F | NCGN | MPO | P, CyP | R | R | Resistance |
| 7 | 75 | M | WG | PR3 | P, CyP | ENT, R | ENT, R | Relapse |
| 8 | 22 | F | NCGN | MPO | P, AZA | ENT, N, R | R | Relapse |
| 9 | 26 | M | WG | PR3 | P, CyP | L, GI, MS, N, C, R | C | Relapse |
| 10 | 25 | M | WG | PR3 | P, CyP | ENT, L, MS, C, R | ENT, C, L, R | Resistance |
| 11 | 54 | M | WG | PR3 | P, CyP, AZA | ENT, L | ENT | Resistance |
| 12 | 54 | M | WG | PR3 | P, CyP | ENT, L, MS, R | R, MS | Relapse |
| Subject | Age | Gender | Diagnosis | ANCA | Prior therapy | Organ involvement at initial presentation | Organ involvement at study enrolment | Relapse/resistance |
|---|---|---|---|---|---|---|---|---|
| 1 | 34 | F | WG | PR3 | P, CyP | ENT, MS, R | ENT | Resistance |
| 2 | 36 | F | WG | PR3 | P, CyP | ENT, L, R | ENT, N | Relapse |
| 3 | 53 | F | CS | PR3 | P, CyP | ENT, L, MS,R | ENT, R | Resistance |
| 4 | 78 | F | MPA | MPO | P, AZA | N, C, R | N, R | Resistant/intolerant |
| 5 | 46 | M | MPA | PR3 | P, CyP | ENT, L, R | R | Relapse |
| 6 | 41 | F | NCGN | MPO | P, CyP | R | R | Resistance |
| 7 | 75 | M | WG | PR3 | P, CyP | ENT, R | ENT, R | Relapse |
| 8 | 22 | F | NCGN | MPO | P, AZA | ENT, N, R | R | Relapse |
| 9 | 26 | M | WG | PR3 | P, CyP | L, GI, MS, N, C, R | C | Relapse |
| 10 | 25 | M | WG | PR3 | P, CyP | ENT, L, MS, C, R | ENT, C, L, R | Resistance |
| 11 | 54 | M | WG | PR3 | P, CyP, AZA | ENT, L | ENT | Resistance |
| 12 | 54 | M | WG | PR3 | P, CyP | ENT, L, MS, R | R, MS | Relapse |
P, prednisone; CyP, cyclophosphamide; Aza, azathioprine; C, skin; GI, gastrointestinal; L, lung; MS, musculoskeletal; N, nervous; R, renal.
Summary of subject's characteristics at study entry
| Subject | Age | Gender | Diagnosis | ANCA | Prior therapy | Organ involvement at initial presentation | Organ involvement at study enrolment | Relapse/resistance |
|---|---|---|---|---|---|---|---|---|
| 1 | 34 | F | WG | PR3 | P, CyP | ENT, MS, R | ENT | Resistance |
| 2 | 36 | F | WG | PR3 | P, CyP | ENT, L, R | ENT, N | Relapse |
| 3 | 53 | F | CS | PR3 | P, CyP | ENT, L, MS,R | ENT, R | Resistance |
| 4 | 78 | F | MPA | MPO | P, AZA | N, C, R | N, R | Resistant/intolerant |
| 5 | 46 | M | MPA | PR3 | P, CyP | ENT, L, R | R | Relapse |
| 6 | 41 | F | NCGN | MPO | P, CyP | R | R | Resistance |
| 7 | 75 | M | WG | PR3 | P, CyP | ENT, R | ENT, R | Relapse |
| 8 | 22 | F | NCGN | MPO | P, AZA | ENT, N, R | R | Relapse |
| 9 | 26 | M | WG | PR3 | P, CyP | L, GI, MS, N, C, R | C | Relapse |
| 10 | 25 | M | WG | PR3 | P, CyP | ENT, L, MS, C, R | ENT, C, L, R | Resistance |
| 11 | 54 | M | WG | PR3 | P, CyP, AZA | ENT, L | ENT | Resistance |
| 12 | 54 | M | WG | PR3 | P, CyP | ENT, L, MS, R | R, MS | Relapse |
| Subject | Age | Gender | Diagnosis | ANCA | Prior therapy | Organ involvement at initial presentation | Organ involvement at study enrolment | Relapse/resistance |
|---|---|---|---|---|---|---|---|---|
| 1 | 34 | F | WG | PR3 | P, CyP | ENT, MS, R | ENT | Resistance |
| 2 | 36 | F | WG | PR3 | P, CyP | ENT, L, R | ENT, N | Relapse |
| 3 | 53 | F | CS | PR3 | P, CyP | ENT, L, MS,R | ENT, R | Resistance |
| 4 | 78 | F | MPA | MPO | P, AZA | N, C, R | N, R | Resistant/intolerant |
| 5 | 46 | M | MPA | PR3 | P, CyP | ENT, L, R | R | Relapse |
| 6 | 41 | F | NCGN | MPO | P, CyP | R | R | Resistance |
| 7 | 75 | M | WG | PR3 | P, CyP | ENT, R | ENT, R | Relapse |
| 8 | 22 | F | NCGN | MPO | P, AZA | ENT, N, R | R | Relapse |
| 9 | 26 | M | WG | PR3 | P, CyP | L, GI, MS, N, C, R | C | Relapse |
| 10 | 25 | M | WG | PR3 | P, CyP | ENT, L, MS, C, R | ENT, C, L, R | Resistance |
| 11 | 54 | M | WG | PR3 | P, CyP, AZA | ENT, L | ENT | Resistance |
| 12 | 54 | M | WG | PR3 | P, CyP | ENT, L, MS, R | R, MS | Relapse |
P, prednisone; CyP, cyclophosphamide; Aza, azathioprine; C, skin; GI, gastrointestinal; L, lung; MS, musculoskeletal; N, nervous; R, renal.
Ten subjects completed the 6 month treatment phase of the study and were evaluated for an additional 6 months. Two subjects were withdrawn early from the study. Subject no. 9 was withdrawn within 6 weeks because of a febrile illness (treated at another institution, information not available) during which MMF was discontinued. In addition, this subject had deterioration in vasculitic symptoms and was unable to come to the study visits. Subject no. 10 was withdrawn within 2 weeks because of rapid deterioration of pulmonary disease. Nine of the remaining 10 subjects attained the MMF target dose of 1000 mg twice daily, of whom three subjects required the maximal dose of 1500 mg twice daily because of mild persistent disease activity. One subject received a maximal dose of 750 mg twice daily due to mild neutropenia. Medication counts indicated that subjects were compliant with MMF therapy.
During the 6 months of follow-up (months 7–12), all 10 study subjects initially remained on MMF. Subject no. 11 eventually had the dose of MMF decreased and subsequently discontinued therapy by the end of the eighth month. This subject later suffered a significant disease relapse while taking prednisone and was subsequently treated with azathioprine. Subjects nos 7 and 8 had their doses of MMF tapered slowly, and eventually discontinued the drug by the end of month 12. Subject no. 2, suffered a flare of disease activity while on MMF, and was treated by the addition of etanercept therapy.
When compared to baseline, the total BVAS (BVAS.1+BVAS.2) decreased from an average of 9.1±3.5 (range, 3–17) to 2.8±1.9 (range, 1–6), representing a mean change of 7.8±5.4 at the 24 week time point (P = 0.0013) and to 2.8±4.3 (range, 0–13), representing a mean change of 7.1±5.4 at the 52 week time point (P = 0.0044) (Figure 1). When compared to baseline, the BVAS.1 (for present/persistent signs or symptoms) decreased by an average of 2.6±2.1 (range −1 to – 6) (P = 0.0007), and BVAS.2 (for new/worse signs or symptoms) decreased by an average of 5.2±3.6 (range 0 to −10) (P = 0.0280) at the 24 week time point. At the 52 week time point, the BVAS.1 decreased by an average of 2.5±2.8 (range +2 to −7) (P = 0.0044); and the BVAS.2 decreased by an average of 4.6±3.7 (range +2 to −10) (P = 0.0117).
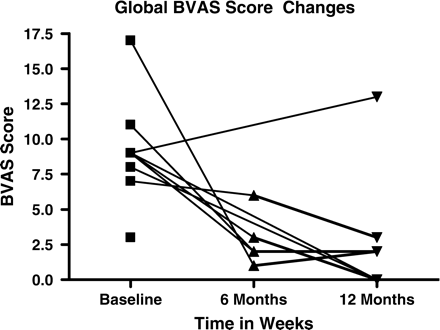
Global changes in BVAS scores. Changes in total BVAS scores for all evaluable subjects at baseline, 24 and 52 weeks are graphically represented. Changes between baseline and 24 weeks (P = 0.0013) and baseline and 52 weeks (P = 0.0044) were statistically significant.
The response to treatment with MMF was varied between subjects. Figure 2A–C reflects a grouping of subjects based on the documented response to treatment. Three subjects experienced an early and sustained decrease in disease activity (Figure 2A). Three subjects experienced an early decrease in disease activity, which was then followed by one or more flares of disease activity (Figure 2B). Five subjects had a delayed or poor response to treatment (including subject nos 9 and 10 who were withdrawn from the study) (Figure 2C). Subject no. 5 was not included in Figure 2 because of insufficient BVAS time points.
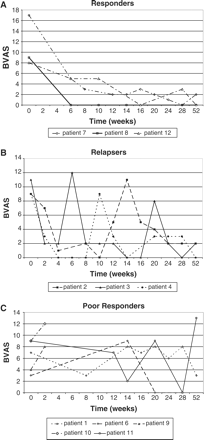
(A) Changes in BVAS scores in subjects classified as ‘Responders’ to MMF therapy. (B) Changes in BVAS scores in subjects classified as ‘Relapsers’ to MMF therapy. (C) Changes in BVAS scores in subjects classified as ‘Poor Responders’ to MMF therapy.
In the 6 months following the 6 month initial treatment period, one subject had a significant increase in disease activity (BVAS increased by 13 points; subject no. 6) and two other subjects had a mild recurrence of symptoms (BVAS increased by 2 points). Six of 10 subjects achieved remission during some part of the active treatment phase (baseline to 6 months) as evidenced by the absence of disease activity and a BVAS score of 0. The group of subjects with an early and sustained decrease in BVAS (Figure 2A) were all classified as having a disease relapse at the time of enrolment into the study.
Available ANCA titres were compared at baseline and 24 weeks and at the baseline and 52 week time points. The mean ANCA titres at baseline, 24 and 52 weeks were 57±29.2, 43.3±35.0 and 50.7±28, respectively. The changes in ANCA titres between baseline and 24 weeks (P = 0.3736) and baseline and 52 weeks (P = 0.8392) did not reach statistical significance. A decrease in ANCA titres was documented in six subjects whereas titres increased in three subjects at the 24 week time point as compared to baseline. During the period from 6 to 12 months, ANCA titres increased in five subjects and decreased in three subjects. Four subjects exhibited concordance between the direction of change in BVAS score and ANCA titres when comparing 6 months to baseline data. When the changes in renal laboratory parameters were evaluated at baseline vs 24 and 52 weeks, there were trends toward an improvement in serum creatinine over the course of the study. Serum creatinine values were 2.3±2.2 mg/dl at baseline, 1.8±1.2 mg/dl at 24 weeks and 1.4±0.6 mg/dl at 52 weeks. Comparisons between serum creatinine at baseline vs 24 weeks (P = 0.4679) and baseline vs 52 weeks (P = 0.2409) failed to show statistical significance.
Corticosteroids were successfully withdrawn in two of five subjects who were on prednisone therapy at study entry. Three additional subjects were prescribed prednisone therapy during the course of study participation because of increased disease activity. Only one subject was receiving prednisone therapy (20 mg daily) at week 52. The baseline (mean and range) prednisone dose (42 mg; 20–60 mg) was four-fold higher than the 24 week dose (12.5 mg; 10–20 mg).
MMF demonstrated a reasonable safety profile. Adverse events deemed possibly or probably related to MMF therapy (and their event numbers) included: upper respiratory tract infection (five), urinary tract infection symptoms (one), Herpes Zoster infection (one), diarrhoea/loose stools (four), abdominal cramping (three), nausea/vomiting (two), constipation (one), leukopaenia (two), insomnia (two), mid-epigastric pain (one) and increased serum amylase (one). These adverse events were transient in nature and resolved spontaneously or with MMF dosage reductions. None of the drug-related adverse events required the subjects to be prematurely removed from the study.
Discussion
The initial treatment of de novo ANCA-associated small vessel vasculitis with high dose corticosteroids and cyclophosphamide is currently considered to be the standard therapy. The use of cyclophosphamide in this setting decreases the risk of death 5-fold and relapse 3-fold when compared to treatment with corticosteroids alone [3,4]. Debate continues as to whether or not to use concomitant pulse corticosteroids and there is disagreement about the most effective dose, duration, and route of administration (oral vs intravenous) of cyclophosphamide. However, most groups have reported similar rates of remission, despite the variations in cyclophosphamide treatment regimen. Although the reported relapse rates among patients with ANCA-associated small vessel vasculitis are also comparable (∼30%), these are not measured uniformly. Furthermore, the risk of relapse may vary substantially depending on disease phenotype, initial organ involvement and ANCA specificity [9]. It is clear from clinical experience that whereas some patients will attain a long-term remission after treatment with cyclophosphamide and corticosteroids, others are prone to a course of waxing and waning disease activity and multiple relapses. The therapy of the latter group of patients can be particularly challenging in light of the short and long-term toxicities of corticosteroids and cyclophosphamide, including infection, bone marrow suppression, malignancies and gonadal failure. Unfortunately, the alternative or adjunctive therapies that have been used for the treatment of recurrent disease are limited in efficacy and have toxicities of their own. In addition, much of the available data have not resulted from large controlled clinical trials, thus limiting applicability. The efficacy of trimethoprim-sulfamethoxazole appears to be limited to the treatment and prevention of isolated mild to moderate upper airway disease only and its efficacy in the treatment of subglottic stenosis is uncertain [6]. The use of concomitant weekly methotrexate and daily corticosteroids was found to be efficacious with reported remission rates of 60–90% [10,11]. However, this treatment approach was associated with an elevated rate of relapse [12] and the clinical experience with methotrexate is limited to patients with predominantly extrarenal manifestations of vasculitis and well preserved renal function (serum creatinine <2.5 mg/dl). There are a few reports of beneficial response to treatment with cyclosporine, including frequently relapsing patients, but no systematic evaluation of the induction of remission has been done. Cyclosporine does not seem to be sufficient in the prevention of relapses as evidenced by the significant rate of relapses in the post transplant population (10–25%) [13]. The role of azathioprine in the treatment of ANCA vasculitis has primarily been limited to that of ‘maintenance therapy’ after remission is achieved with corticosteroids and cyclophosphamide [14].
Preliminary data on the effectiveness of MMF in the treatment of rheumatoid arthritis [15] and historical data on the effectiveness of mycophenolic acid in the treatment of psoriasis [16] suggest therapeutic benefits in the treatment of autoimmune disease. This is supported by the growing data in favour of its use in the management of systemic lupus erythematosus both for induction [17] and maintenance therapy [17–19].
It is against this background that we were interested in assessing the potential use of MMF as induction treatment for active ANCA-associated small vessel vasculitis. MMF offers theoretical safety advantages over the available therapeutic agents mentioned above. Unlike cyclophosphamide and azathioprine, MMF so far shows no evidence of carcinogenicity [16]. The risk of pancytopaenia is lower with MMF than with cyclophosphamide, azathioprine or methotrexate, and MMF has no documented effect on gonadal function or fertility and is not nephrotoxic. Unlike alkylating agents and antimetabolites, the immunosuppressive effects of MMF are promptly reversible after discontinuation of the drug, which is advantageous in the setting of a serious infectious complication of immunosuppression.
In this pilot study, treatment with MMF was generally well tolerated with only transient adverse effects related mostly to gastrointestinal intolerance and shingles. These adverse effects were expected. The gastrointestinal intolerance may be related to hepatobiliary recycling and glucuronidation status, while the activation of herpes zoster is related to the immunosuppressive activity. Because the effective immunosuppressive doses of MMF may hypothetically be different in the setting of an autoimmune vasculitis than in organ transplantation, this study was designed as a dose escalation trial, starting at a dose of 500 mg twice daily and increasing progressively to 1000–1500 mg twice daily. There are potential differences in the pharmacokinetic disposition of MMF between patients with vasculitis and solid organ transplant recipients. First, the concomitant medications may differ and may predispose toward drug interactions (i.e. use of cyclosporine and tacrolimus in transplant patients). Second, patients with glomerulonephritis may have more highly variable serum protein concentrations secondary to the nephrotic syndrome and thus may have altered binding of drugs such as MMF that are highly protein bound [20,21]. Although the number of subjects involved in this pilot study was small, most subjects tolerated ‘full’ target doses of MMF for the control of vasculitis. Only one subject required a reduced dose (750 mg twice daily) because of leukopaenia.
The subjects selected for this induction feasibility trial all had relapsing or persistent disease activity despite prior immunosuppression with corticosteroids and cyclophosphamide. In this selected patient population, induction treatment with MMF led to improvement in vasculitis disease activity in all but one of 10 evaluable subjects. It afforded at least transient complete remission (defined as a BVAS score of 0) in six of 10 subjects while on therapy with MMF and resulted in overall improvement in disease activity (as measured by BVAS). Clinically the therapeutic benefit of MMF was evidenced by an overall reduction in persistent organ specific signs and symptoms (BVAS.1) and new or worsening disease activity (BVAS.2) at 24 and 52 weeks compared to baseline, even though the persistence of some disease activity could not be eliminated. Although only a minority of subjects achieved a long-lasting remission, treatment with MMF obviated the recurrent use of a cytotoxic agent in 10 of 12 subjects.
Most subjects clearly had a favourable long-term response to treatment with MMF, as defined as those who responded or those with relapse who had only minimal residual (‘grumbling’) symptoms. However, several had a poor response (poor responders), which raises several issues with regard to the treatment of ANCA-vasculitis in general, and the role of MMF in particular. Of note, only four patients received concomitant therapy with trimethoprim/sulfamethoxazole and none of these patients were classified as MMF responders. It is intriguing that the subjects who responded to treatment with MMF had relapsing disease, as opposed to being treatment resistant at baseline. Although limited by the small number of subjects and the uncontrolled nature of our study, this result suggests that MMF alone is unlikely to be an effective rescue therapy for subjects resistant to induction therapy with cyclophosphamide.
Publications regarding the use of MMF in ANCA-associated small vessel vasculitis are limited to case reports of responses to induction therapy and small series evaluating maintenance regimens (once remission is attained with cyclophosphamide) in 5–14 subjects [22–24]. The induction case reports (a total of four subjects) describe improved clinical symptoms, serum creatinine and ANCA titres in subjects treated with a MMF dose of 1000 mg daily [24]. The adverse events and their frequencies seen in our induction study were similar to those reported in the MMF maintenance studies [22,23].
It is conceivable that the degree of immunosuppression required for the control of vasculitis may vary significantly from one patient to another based on phenotype and genotype variations. The current approach for the treatment of ANCA-vasculitis is still primarily based on a ‘one-size-fits-all’ concept. Determining risk factors for progressive or relapsing disease should allow us in the future to better tailor the treatment to each individual's needs, thus using agents such as MMF for patients with certain disease phenotypes or genotypes, while reserving the more potent (and toxic) regimens for patients with the propensity toward a more aggressive disease. Although the concept of a ‘staged’ approach to the treatment of vasculitis is being more commonly used (e.g. induction therapy with cyclophosphamide and corticosteroids followed by ‘maintenance’ treatment with azathioprine [14], methotrexate or MMF [22]), most regimens currently used remain primarily based on the use of single immunosuppressive agents (with or without the concurrent use of corticosteroids). While this ‘monotherapy’ approach is needed for the initial assessment of the efficacy and toxicity profiles of a new agent in the treatment of vasculitis, it is likely that the combination of two or more agents would confer effective disease control by slightly different mechanisms of action and potentially decreased toxicity. Thus, the role of MMF in the therapy of ANCA-vasculitis may be as part of a combination regimen with another immunosuppressive agent, with or without low-dose corticosteroids.
Conclusions
MMF is a reasonable option in the treatment of mild-to-moderate non-life threatening relapsing or resistant vasculitis that obviates the use of cytotoxic drugs. Improvements in both persistent organ-specific signs and symptoms as well as new or worsening disease activity was demonstrated. The long-term relapse rate associated with MMF therapy is not currently defined. The outcomes associated with MMF as a single or combination agent in the treatment of more severe de novo or recurrent disease, with long-term therapy, and by ANCA phenotype and genotype requires further study.
We are indebted to Jean C. Brown for assistance in the preparation of the manuscript. This study was presented in part at the 11th International ANCA Workshop, May 2000, Groningen, the Netherlands, and the 33rd Annual Meeting of the American Society of Nephrology, October 2000, Toronto, Canada. This study was supported by a grant from Roche Pharmaceuticals.
Conflict of interest statement. None declared.
References
Fauci AS, Katz P, Haynes BF et al. Cyclophosphamide therapy of severe systemic necrotizing vasculitis.
Jayne D, Rasmussen N, Andrassy K et al. A randomized trial of maintenance therapy for vasculitis associated with antineutrophil cytoplasmic autoantibodies.
Hogan SL, Nachman PH, Wilkman AS et al. Prognostic markers in patients with antineutrophil cytoplasmic autoantibody-associated microscopic polyangiitis and glomerulonephritis.
Nachman PH, Hogan SL, Jennette JC et al. Treatment response and relapse in antineutrophil cytoplasmic autoantibody-associated microscopic polyangiitis and glomerulonephritis.
Falk RJ, Hogan S, Carey TS et al. Clinical course of anti-neutrophil cytoplasmic autoantibody-associated glomerulonephritis and systemic vasculitis. The Glomerular Disease Collaborative Network.
Stegeman CA, Tervaert JW, De Jong PE et al. Trimethoprim-sulfamethoxazole (co-trimoxazole) for the prevention of relapses of Wegener's granulomatosis. Dutch Co-Trimoxazole Wegener Study Group.
Ghez D, Westeel PF, Henry I et al. Control of a relapse and induction of long-term remission of Wegener's granulomatosis by cyclosporine.
Luqmani RA, Bacon PA, Moots RJ et al. Birmingham Vasculitis Activity Score (BVAS) in systemic necrotizing vasculitis.
Hogan SL, Falk RJ, Chin H et al. Predictors of relapse and treatment resistance in ANCA small vessel vasculitis.
Langford CA, Talar-Williams C, Barron KS et al. A staged approach to the treatment of Wegener's granulomatosis: induction of remission with glucocorticoids and daily cyclophosphamide switching to methotrexate for remission maintenance.
Langford CA, Talar W, Sneller MC. Use of methotrexate and glucocorticoids in the treatment of Wegener's granulomatosis. Long-term renal outcome in patients with glomerulonephritis.
Furst DE. Practical clinical pharmacology and drug interactions of low-dose methotrexate therapy in rheumatoid arthritis.
Nachman PH, Segelmark M, Westman K et al. Recurrent ANCA-associated small vessel vasculitis after transplantation: A pooled analysis.
Jayne D. Update on the European Vasculitis Study Group trials.
Goldblum R. Therapy of rheumatoid arthritis with mycophenolate mofetil.
Epinette WW, Parker CM, Jones EL et al. Mycophenolic acid for psoriasis. A review of pharmacology, long-term efficacy, and safety.
Contreras G, Pardo V, Leclercq B et al. Sequential therapies for proliferative lupus nephritis.
Chan TM, Li FK, Tang CS et al. Efficacy of mycophenolate mofetil in patients with diffuse proliferative lupus nephritis. Hong Kong-Guangzhou Nephrology Study Group.
Chan TM, Tse KC, Tang CS et al. Long-term study of mycophenolate mofetil as continuous induction and maintenance treatment for diffuse proliferative lupus nephritis.
Shipkova M, Armstrong VW, Kuypers D et al. Effect of cyclosporine withdrawal on mycophenolic acid pharmacokinetics in kidney transplant recipients with deteriorating renal function: preliminary report.
van Gelder T, Klupp J, Barten MJ et al. Comparison of the effects of tacrolimus and cyclosporine on the pharmacokinetics of mycophenolic acid.
Nowack R, Gobel U, Klooker P et al. Mycophenolate mofetil for maintenance therapy of Wegener's granulomatosis and microscopic polyangiitis: a pilot study in 11 patients with renal involvement.
Langford CA, Talar-Williams C, Sneller MC. Mycophenolate mofetil for remission maintenance in the treatment of Wegener's granulomatosis.
Author notes
Department of Medicine and 1Department of Pathology, University of North Carolina, Chapel Hill, NC, USA


{kind=link}
{kind=link}
Comments